- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Human treponemal infections are caused by a family of closely related _Treponema pallidum_ that give rise to the diseases yaws, bejel, pinta and, most notably, syphilis1. Debates on
a common origin for these pathogens and the history of syphilis itself have weighed evidence for the ‘Columbian hypothesis’2, which argues for an American origin, against that for the
‘pre-Columbian hypothesis’3, which argues for the presence of the disease in Eurasia in the Medieval period and possibly earlier. Although molecular data has provided a genetic basis for
distinction of the typed subspecies4, deep evolution of the complex has remained unresolved owing to limitations in the conclusions that can be drawn from the sparse palaeogenomic data that
are currently available. Here we explore this evolutionary history through analyses of five pre- and peri-contact ancient treponemal genomes from the Americas that represent ancient
relatives of the _T. pallidum_ subsp. _pallidum_ (syphilis), _T. pallidum_ subsp. _pertenue_ (yaws) and _T. pallidum_ subsp. _endemicum_ (bejel) lineages. Our data indicate unexplored
diversity and an emergence of _T. pallidum_ that post-dates human occupation in the Americas. Together, these results support an American origin for all _T. pallidum_ characterized at the
genomic level, both modern and ancient. SIMILAR CONTENT BEING VIEWED BY OTHERS REDEFINING THE TREPONEMAL HISTORY THROUGH PRE-COLUMBIAN GENOMES FROM BRAZIL Article Open access 24 January 2024
GLOBAL PHYLOGENY OF _TREPONEMA PALLIDUM_ LINEAGES REVEALS RECENT EXPANSION AND SPREAD OF CONTEMPORARY SYPHILIS Article Open access 24 November 2021 PHYLOGENETIC AND GENETIC CHARACTERIZATION
OF _TREPONEMA PALLIDUM_ STRAINS FROM SYPHILIS PATIENTS IN JAPAN BY WHOLE-GENOME SEQUENCE ANALYSIS FROM GLOBAL PERSPECTIVES Article Open access 04 February 2021 MAIN In 1494 Charles VIII of
France assembled an army from across Western Europe to pursue his claims in Italy. Although he enjoyed brief success, events that followed changed the disease landscape: a disfiguring
illness unknown to medical professionals of the time erupted in the army camps and followed the men to their homelands upon their demobilization in 1495. Within five years’ time, the
epidemic had spread throughout Europe5. A variety of contemporary theories on the origin of the disease circulated, largely reflecting political tensions of the period. In the early
sixteenth century, the noted proximity in timing between the onset of the epidemic and Columbus’ maiden voyage across the Atlantic led to the idea that the disease originated on the other
side of the ocean, and had been introduced to Europe as a consequence of recent contacts5. The range of symptoms recorded in contemporary sources are thought to describe the first prolific
outbreak of syphilis among Europeans, which was observed in both sexually acquired and congenital manifestations5. Today we understand this disease to be part of a group of clinically
similar infections of bacteria of the genus _Treponema_—namely syphilis (_T. pallidum_ subsp. _pallidum_ (TPA)), yaws (_T. pallidum_ subsp. _pertenue_ (TPE)), bejel (_T. pallidum_ subsp.
_endemicum_ (TEN)) and pinta (_Treponema carateum_). Genomic studies reveal that TPA, TPE and TEN share 99% nucleotide sequence identity4. The underlying biological factors that contribute
to phenotypic variation in disease severity in humans and non-human African primates, their uneven distribution across the globe, and differences in transmission potential have remained
poorly understood despite the evaluation of hundreds of genomes1,6,7. It is also uncertain whether syphilis, the only _T. pallidum_ disease type with a predominantly sexual mode of
transmission, is the sole cause of congenital cases8,9. Drawing heavily upon historical documentation, Alfred Crosby2 popularized the Columbian hypothesis, which argues that syphilis emerged
in the Americas and accompanied Columbus’ crew on one of their return voyages from the Caribbean, possibly as early as January 1493. Additional support for this theory comes from
morphological analyses of lesions in archaeological bone associated with chronic treponemal infection that indicate its long history in the Americas (see Supplementary Information, section
1). Others have since revised this hypothesis to accommodate the existence of an ancestral variety in the Americas that rapidly evolved into modern syphilis in response to different
selection pressures in terms of host biology, behaviour and climate10. Since its inception, this narrative has raised controversy. Contrary views have looked beyond syphilis and considered
the evolutionary history of the disease family as a whole. Early theories were presented before the availability of genetic data—for example, Hudson’s unitarian hypothesis11, which proposed
that phenotypic differences within the treponemal subspecies are merely environmentally determined expressions of what is functionally an identical organism. Support for the subspecies
cladal structure in whole-genome phylogenies of TPA, TPE and TEN refutes this hypothesis in the opinion of some discussants12; however, others have cautioned that their degree of similarity
could indicate the theory was dismissed prematurely1. On the basis of early ideas that treponemal disease was globally distributed before the fifteenth century13, models that accommodated
hypothetical evolutionary chronologies of the subspecies3,14 set the stage for the pre-Columbian hypothesis, which, in simple models, supports a broad and historically deep distribution of
infectious treponema in Europe3 or Africa15, and in more specific models supports the presence of sexually transmitted and congenitally acquired syphilis in Europe16 long before Columbus’
first trans-Atlantic expedition. A large body of literature, including two edited volumes, has weighed the skeletal evidence in Europe17 and North America18, but debates continue about the
diagnostic specificity of skeletal pathology associated with chronic or congenital treponemal infection19,20 and the accuracy of radiocarbon dates from archaeological bone that span the
fifteenth and sixteenth centuries12,21. Antibiotic treatments have reduced the frequency of skeletal involvement in treponemal infection today, which further compromises our ability to
establish morphologically diagnostic criteria in archaeological tissues. Ancient DNA is a valuable tool to integrate into the ongoing debates. Although initial expectations were dampened by
indications that treponemal DNA would either not preserve22 or would be recovered only in rare cases23, the number of historic individuals yielding treponemal DNA at the genome level is
increasing9,24,25,26. Genetic studies agree in their conclusion that yaws is much younger than previously thought25,26, and further indicate that both yaws and syphilis underwent a
substantial increase in genetic diversity approximately 500 years ago25,26,27. The small number of ancient genomes yielded from these studies, however, are insufficient to explore the more
pressing questions related to the global distribution of these pathogens prior to 1492, in particular those controversies surrounding the origin of syphilis. Genome-level information from
pre-1492 treponemal infections—whether from Europe, Africa or the Americas, where treponemal disease circulated in the early colonial period—are of high value for refining the evolutionary
history of _T. pallidum_. Here we offer a substantial contribution to the debate through presentation of five treponemal genomes from pre-contact and peri-contact American contexts. Together
these data reveal early relatives for all currently defined _T. pallidum_ sublineages, and point toward an extensive unexplored diversity of ancient forms in the Americas. SCREENING AND
CAPTURING ANCIENT TREPONEMAL DNA As a first approach, we sampled from pathological bone displaying lesions consistent with treponemal infection19 from multiple sites in Chile (145 tissues
from 39 individuals), 41 individuals from Mexico and 1 individual from Argentina (Supplementary Information, section 2 and Supplementary Tables 1 and 2). Teeth and pathological features in
bone were preferentially targeted to generate approximately 50 mg of pulverized tissue for molecular extraction. Powders were processed using established techniques with both
double-stranded28 and single-stranded libraries29 constructed for sequencing and analysis. Datasets of bulk DNA content were generated to a depth of around 5 million reads and were screened
for the presence of _T. pallidum_ DNA via analysis within the MALT/HOPS pipeline30,31,32 (Supplementary Table 3). This process revealed four candidates for genome enrichment: an adult tibial
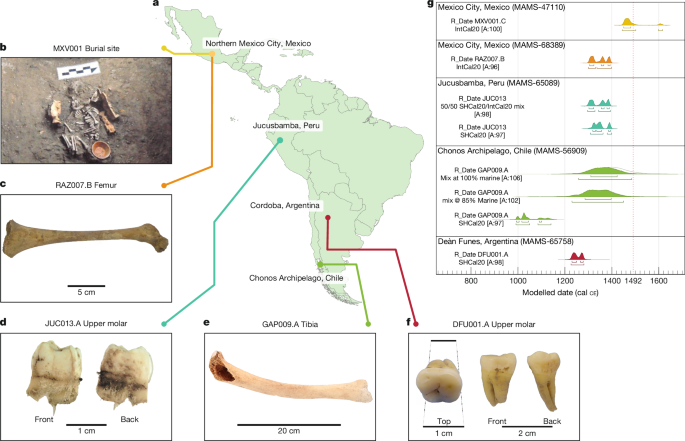
fragment (GAP009) from the Chonos Archipelago of southern Chile, a tibia and humerus (pooled as MXV001) from an infant in Mexico City, a femur and occipital bone (pooled as RAZ007) from a
child in Mexico City, and an upper molar, pelvic bone (iliac crest) and rib (pooled as DFU001) from a young adult in Deán Funes, Córdoba Province, in central Argentina (Fig. 1, Supplementary
Table 4 and Supplementary Figs. 1–5). In a parallel process, a weak _T. pallidum_ signal was identified in one individual from Jucusbamba, Peru (JUC013, an immature molar) via a custom
MALT31-based TPA computational screening of 36,214 sequencing datasets generated for other research applications at the Max Planck Institute for Evolutionary Anthropology (Supplementary
Figs. 6 and 7 and Supplementary Table 4). To maximize chances of DNA recovery, additional libraries were prepared from the extracts and digested products of GAP009, DFU001, RAZ007 and
JUC013. Genome-level enrichment for all five candidates was carried out using established protocols25. Enriched products were sequenced to depths of 4.8 to 108 million reads, mapped to the
TPA Nichols reference genome (RefSeq accession NC_021490.2) in nf-core/eager32 (Supplementary Table 5), and single-nucleotide polymorphism (SNP) profiles were generated on the basis of
positions with a minimum of fourfold read support. Pooled data from all captured libraries unique to each individual yielded average genome coverages of 25.6-fold, 24.7-fold, 7.2-fold,
4.3-fold and 2.3-fold for MXV001, GAP009, DFU001, JUC013 and RAZ007, respectively (Fig. 2 and Supplementary Table 5). Treponemal captured data for JUC013, DFU001 and RAZ007 (no uracil-DNA
glycosylase (UDG) treatment), as well as GAP009 and MXV001 (both partial UDG treatment) showed damage profiles consistent with human DNA, thus supporting their ancient authenticity (Extended
Data Fig. 1, Supplementary Table 6 and Supplementary Figs. 8–10). Negative controls were free of _T. pallidum_ DNA (Supplementary Table 7). DIVERSITY OF _T. PALLIDUM_ GENOMES THAT PREDATE
1492 Placement of these genomes within known treponemal diversity was evaluated with 60 modern genomes (_n_ = 28 TPA, _n_ = 23 TPE, _n_ = 8 TEN and _n_ = 1 _Treponema paraluiscuniculi_ as an
outgroup), along with 9 published genomes of ancient origin (Supplementary Tables 8–10). The threshold for inclusion in phylogenetic analysis was set at a minimum 50% coverage of the genome
with fourfold read support (Extended Data Fig. 2). This eliminated the ancient genomes RAZ007 (Mexico City), KM14-7 (Netherlands; Supplementary Information, section 7.4, Supplementary Fig.
11 and Supplementary Tables 11–15), CHS119 (Finland), SJ219 (Estonia) and 133 (Mexico) from further phylogenetic analysis owing to their low coverage (Supplementary Tables 5 and 16–20).
Genome ZH1540, previously published as TEN from Brazil around 2000 bp, was also not included (see Majander et al.33, as accessed online December 2024). As recombination is commonly observed
amongst _T. pallidum_ subspecies, genomic regions affected by this phenomenon were removed through filtering of the joined recombinant positions identified in both Gubbins34 and
ClonalFrameML35 following a quality evaluation for heterozygosity in all genomes (Supplementary Figs. 12–14 and Supplementary Tables 18–20). Maximum likelihood trees were generated in
RAxML-ng36 with 1,000 bootstrap replicates and 25% permission of missing data (Fig. 3 and Supplementary Figs. 15–17). This process revealed that genomes JUC013 and GAP009 branch together as
a group paraphyletic to TPA and the 94A/B ancient clade. By contrast, genomes MXV001 and RAZ007 branch together from the lineage common to TPE and TEN. DFU001 forms a sister lineage to the
TEN clade. This phylogenetic grouping is maintained when mapping is performed using TPE CDC-2 or TEN Bosnia A as a reference (Supplementary Fig. 16). Where possible, placement of the lower
covered genomes was accomplished through phylogenetic reconstruction based on twofold read support (Supplementary Table 10 and Supplementary Fig. 17). Special attention was given to the
European genome KM14-7 (1494–1631 ce), whose previously published phylogenetic position showed it to branch from the common lineage of TPE and TEN26. Our reanalysis reveals a coverage that
is too low for its resolved phylogenetic placement within any _T. pallidum_ sublineage (Supplementary Information, section 7.4). Branch shortening in phylogenetic reconstruction is a common
feature of ancient genomes, and is reflective of the reduced time over which derived positions have accumulated37. In the case presented here, genomes GAP009 and MXV001 paradoxically do not
display this phenomenon (Fig. 3). Through multiple lines of investigation, we conclude that the longer branch lengths in GAP009 and MXV001 do not result from a currently detectable artefact
(Supplementary Figs. 15–17 and Supplementary Tables 16 and 17). For all positions that are either uniquely derived in the ancient genomes, or where JUC013 and GAP009 carry the ancestral
allele compared with modern syphilis, at least one-third comprise non-synonymous nucleotide changes with potential functional significance (Supplementary Tables 21–24). Given the antiquity
of these genomes, representation of known _T. pallidum_ virulence determinates (Supplementary Tables 25–26) was assessed on the basis of regional coverages. This revealed the presence of all
known virulence genes in the five ancient genomes featured here, with predictable reduced coverage across them for genomes JUC013 and RAZ007 (Extended Data Fig. 3). To explore evolutionary
history of the disease complex, radiometric 14C dates for each individual were considered (Supplementary Information, section 4). At 2_σ_ confidence level, these results securely place
JUC013, DFU001 and RAZ007 in pre-1492 contexts. A modelled 14C age for GAP009 that accommodates an influence of marine protein consumption yielded a pre-Columbian estimated date range that
spans into the late fifteenth century. By contrast, the age of MXV is estimated somewhere within the fifteenth to seventeenth centuries. Analyses of human uniparental markers (mitochondrial
haplogroups C1b1 (MXV001), A2 + (64) (JUC013, DFU001 (A2bc) and RAZ007) and C (GAP009); Y-chromosome haplogroups Q1b1a1a1e1a (Q-CTS10359; MXV001), Q1b1a1a1 (Q-M3; DFU001) and Q1b1a1a1
(Q-CTS2610; JUC013)) and whole-genomic human DNA for JUC013, GAP009, DFU001 and MXV001 (Fig. 4 and Supplementary Table 27), reveal them to be genetically of Native American origin, with no
indications of non-Indigenous admixture. Human DNA recovery in individual RAZ007 was too low for whole-genomic evaluation. Ancient genome representation exceeding fourfold average coverage
(_n_ = 9) provides multiple calibration points for molecular dating across the entire _T. pallidum_ phylogeny. Radiometric data (bounded by 2_σ_ intervals) or historical data from ancient
genomes, along with isolation dates of modern strains, were used with the filtered SNP alignment for a molecular dating analysis using BEAST 2.6.338 (Supplementary Information, section 12
and Supplementary Table 28). Beforehand, the presence of significant temporal signal in our dataset was confirmed using root-to-tip regression with Clockor239 (_R_2 = 0.25; Supplementary
Fig. 18) as well as Bayesian evaluation of temporal signal (BETS40; log Bayes factor (BF) = 11.1; Supplementary Information, section 12). The Clockor2 analysis suggested similar rates for
TPA and TPE/TEN clades (Supplementary Fig. 18). Clock model comparison using path sampling41 showed that an uncorrelated relaxed clock model was largely favoured over a strict clock model
based on the marginal likelihood comparison in path sampling41 (log BF = 167; Supplementary Information, section 12). The uncorrelated relaxed clock model was therefore used and yielded an
estimated mean clock rate of 9.3 × 10−8 substitutions per site per year (95% highest probability density (HPD): 5.9 × 10−8 to 1.3 × 10−7 substitutions per site per year), and a time to the
most recent common ancestor (_T_MRCA) estimate for all _T. pallidum_ of 5048 bp (95% HPD: 7970–2669 bp) (Fig. 5, Supplementary Table 29 and Supplementary Fig. 19). Rates for the lineages
giving rise to genomes MXV001 and GAP009 appeared higher compared with the average, with mean estimates of 1.1 × 10−7 and 3.1 × 10−7 substitutions per site per year, respectively under this
clock model. The use of an alternative epoch model that accounts for potential clock rate time dependency42 (but not inter-lineage rate variation as offered by the relaxed clock) indicated a
detectable negative correlation between time and evolutionary rate (slope coefficient 95% HPD: −0.26 to −0.01) and yielded an older _T_MRCA estimate of 6296 bp (95% HPD: 8758–4179 bp,
Supplementary Table 29 and Supplementary Figs. 20 and 21). Resolution of current dating uncertainties in deep temporal estimates may come from eventual analyses of ancient genomes that
pre-date those presented here and the development of clock models that jointly accommodate multiple types of rate variation. AN AMERICAN ORIGIN FOR _T. PALLIDUM_ This study confirms the
presence in the Americas of deeply derived lineages for all three known _T. pallidum_ subspecies prior to the first Columbian expedition in 1492. Consideration of these broad data in two
different molecular dating approaches has yielded an upper bound of around 9000 bp for emergence of the most recent ancestor common to all _T. pallidum_. This post-dates both current genetic
estimates for the initial divergence of Indigenous American populations from East Asians43 and archaeological estimates for human arrival in the Americas44. Our data thus support a scenario
in which all genomically typed ancient and modern _T. pallidum_ stem from an origin in the Americas during the middle Holocene epoch, possibly as a zoonotic infection from an unidentified
host. Alternatively, post-glacial migrations of Siberian peoples into the Americas over the past 5,000 years45 could have introduced _T. pallidum_ to the continent under the assumption of a
Eurasian emergence; however, the exclusively northern movements of these small populations make it unlikely for them to have both harboured and transmitted the diversity needed to seed
endemic sister lineages of both TPA and the TPE/TEN clade in the time periods and locations we report here. Palaeopathological interpretation also supports an endemic treponematosis in the
Americas that predates these migrations (Supplementary Information, section 1). The divergence estimates presented here, which derive from time-calibrated ancient genomes across all typed
sublineages, provide the strongest support thus far for an American emergence of _T. pallidum_, consistent with the Columbian hypothesis. In light of this assertion, several questions
persist regarding treponemal history such as: (1) the putative cases of Eurasian treponemal pathology that pre-date 149317; (2) the mysterious ‘venereal leprosy’ historically described in
the European Middle Ages46; and (3) the reported intensity of the late fifteenth and sixteenth century European epidemic that is unparalleled in modern contexts of any treponemal disease47.
Although limited specificity in skeletal lesions have led to treponematosis being commonly referred to as ‘the great imitator’, the abundance of data supporting pre-Columbian treponemal
infection outside the Americas should not be quickly dismissed in light of the temporal dating estimates presented here. This study provides additional threads in what is presumably a
complex web of past infectious treponemal diversity both within and outside the currently typed _T. pallidum_ subspecies that yet await discovery. The potential effect of uncharacterized
treponemal diversity is, however, not limited to Eurasia. The Americas house a higher abundance of morphologically diagnosed archaeological cases of treponematosis, and extinct forms could
account for noted pre- and post-colonial epidemiological differences as inferred from analyses of osteological collections from North American contexts48. Such unknown lineages may also
partially account for the unexpectedly long branches we observe in genomes GAP009 and MXV001, given the known frequency of detectable recombination events that occur across multiple lineages
of this pathogen. Although syphilis receives the greatest attention in relation to global human health, the impact and resilience of other _T. pallidum_ members should not be minimized:
Yaws persists in many parts of the developing world and has survived previous eradication efforts49. Further refinements to scenarios of evolutionary history may also be needed once a genome
sequence of the elusive _T. carateum_ (pinta) joins the discussion. On the basis of these new data, let us now consider where and when syphilis, the sexually transmitted disease we know
today, may have first appeared. Our analyses estimate a _T_MRCA of at most around 5000 bp for the divergence between the pre-contact American JUC013/GAP009 lineage and that which has given
rise to all typed modern syphilis (Supplementary Table 29). With regard to syphilis sensu stricto, our analyses yield strongest support for its emergence from an ancestral form within the
Americas just before the arrival of Columbus; however, analytical uncertainty in this date leaves open its possible emergence in Europe or even Africa from an American ancestral strain that
was newly introduced in the late fifteenth or early sixteenth century (Supplementary Table 29). We cannot comment on the frequency or geographic distribution of this ancestral form in the
Americas, and nor can we temporally place its adoption of sexual transmission, the genetic basis for which remains undetermined. Whereas our Bayesian skyline analysis indicates a possible
population expansion for the pathogen in the second half of the fifteenth century (Extended Data Fig. 4), our analysis does not yield enough resolution to confidently date the divergence of
the three main TPA subclades of Nichols, SS14 and Seattle as before or after 1492 ce. Despite the density of morphologically identified treponemal cases in the Americas before European
arrival, few examples of possible congenital infection exist, and the observed palaeopathology is not specific to an endemic sexually transmitted treponematosis across the continents50.
Although the reconstructed genomes presented here indicate accumulation of unique non-synonymous alleles, these changes cannot be linked to phenotypic differences in transmission strategy.
In light of this, we interpret our data as indicating a high degree of treponemal variation in the Americas, which, through a series of bottlenecks and rapid population expansions during
their global dissemination in the early colonial period, responded to a variety of selection pressures imposed by climate, population density, host genetics (both human and non-human) and
cultural activity that favoured transmission of a small number of successful lineages. Their diffusion was subsequently enabled by human trafficking networks and European expansions, where
lineages that are now globally dominant achieved their current distributions and replaced those adapted to the varied regional climates and Indigenous populations across the Americas. All
five of the genomes isolated here, as well as those from historic Mexico (genomes 94A/B)9, may well represent diversity exclusive to the Americas that was lost in the centuries following
contact. Given the rarity of cases recovered from our screening, DNA alone may not permit reconstruction of the treponemal historical narrative to the satisfaction of all discussants.
Although we anticipate tangible contributions from molecular data in the coming years, debates are likely to continue on the basis of balanced review of the available evidence. METHODS
RADIOCARBON DATING Radiocarbon dating was carried out at the Curt-Engelhorn Centre for Archaeometry (Mannheim, Germany). Skeletal fragments were processed using ultra filtrated collagen
(fraction >30kD)55,56 and dated using the MICADAS-AMS of the Klaus-Tschira-Archäometrie Zentrum. Radiocarbon dates are reported with the lab code MAMS. STABLE ISOTOPE MEASUREMENTS AND
MARINE RESERVOIR EFFECTS Radiocarbon and stable isotope measurements were made on the remains from Jucusbamba, Peru (JUC013), the Chonos Archipelago of southern Chile (GAP009) and Deán
Funes, Argentina (DFU001). Prior to calibration, stable isotope measurements were made on extracted collagen from individuals where a potential marine reservoir offset could result in a
radiocarbon age older than a contemporary individual who had consumed a wholly terrestrial-based diet. Data were generated from the collagen extract used for the radiocarbon date (Mannheim,
Germany, laboratory code MA), and were subsequently corroborated via analysis of a second collagen extract of the same bone generated at the Max Planck Institute for Geoanthropology in Jena,
Germany (laboratory code Jena). In the latter process, 1 mg of bone collagen was weighed in duplicate in tin capsules and combusted in a Thermo Scientific Flash 2000 Elemental Analyser
coupled with a Thermo Delta V Advantage Mass Spectrometer at the Max Planck Institute of Geoanthropology. Isotopic values are reported as the ratio of the heavier isotope to the lighter
isotope (13C/12C or 15N/14N) as δ values in parts per thousand (‰) relative to international standards (Vienna Peedee belemnite (VPDB) for δ13C and atmospheric N2 (air) for δ15N). Results
were calibrated against international standards (International Atomic Energy Agency (IAEA)-CH-6: δ13C = −10.80 ± 0.47‰, IAEA-N-2: δ15N = 20.3 ± 0.2‰, and USGS40: δ13C = −26.38 ± 0.042‰, δ15N
= 4.5 ± 0.1‰) and a laboratory standard (fish gelatin: δ13C ≈ −15.1‰, δ15N ≈ 14.3‰). Based on replicate analyses long-term machine error over a year is ±0.2‰ for δ13C and ± 0.2‰ for δ15N.
Overall measurement precision was studied through the measurement of repeats of fish gelatin (_n_ = 80, ±0.2‰ for δ13C and ± 0.2‰ for δ15N). Isotopic data are reported in Supplementary Table
2. All samples met quality control criteria57,58 with a C/N ratio between 2.9–3.6, %C of 15–48%, and %N of 5–17%. The individual from Jucusbamba (JUC013) had stable isotopes indicative of a
fully terrestrial-based C4 diet, thus marine offset was not considered. By contrast, the individual from the Chonos Archipelago (GAP009) had a δ13C value consistent with both C4 and marine
dietary inputs (−10.5‰), but a δ15N value that pointed to a high marine component in the diet (+18.2‰). Chile is dominated by C3 plants59, thus a high input of marine protein in the diet
likely contributed to the shift in stable isotopes. Of interest, the δ13C value for this individual showed more enrichment than high trophic level marine carnivores from the region, such as
sea lion data from Tierra del Fuego summarized by Yesner et al.60. For the individual from Deán Funes (DFU001), only δ13C as measured on the AMS were available, which lacks the precision
needed for a dietary correction. Regardless, this site is located over 600 km from the coast, so a marine dietary component is considered highly unlikely. A modelling approach was necessary
to account for the associated radiocarbon offset in individual GAP009. Because of the lack of isotopic data directly relevant for interpreting this individual (for example, measurements made
on faunal samples from the same site) a literature search was made to approximate the range of expected δ13C and δ15N values for both a wholly terrestrial and marine diet. The δ13C range
for a terrestrial diet is estimated to be −22.5 to −19.5‰ and −16 to −12‰ for a marine diet, while the δ15N was set to +7–11‰ and +14–22‰, respectively. These ranges are estimated from the
archaeological faunal data for Tierra del Fuego and summarized Yesner et al.60 and the Late Pleistocene stable isotope data from horse (_Equus andium_) and llama (_Hemiauchenia paradoxa_)
from northwestern Chilean Patagonia61. The date correction was made following the linear interpolation method62,63 with δ13C endmembers of −20 and −12‰ (100% Terrestrial and Marine,
respectively) and δ15N endmembers of +10 and +22‰ (100% terrestrial and marine, respectively). Undertaking the interpolation using both isotopes allowed for a check on the overall
sensitivity of the dating to this correction. This is especially important as: (1) there were no isotope data on associated faunal material to produce a temporally and spatially relevant
bioavailable baseline; and (2) the δ13C values are enriched beyond expectation for a diet consisting entirely of marine protein. The result of the modelling estimates either a 100% or 85%
marine diet, when using the δ13C and δ15N values, respectively. Since all three individuals (GAP009, JUC013 and DFU001) lived in the Southern Hemisphere, the SHCal20 calibration curve of
Hogg et al.52 was used to calibrate the radiocarbon results to a calendrical scale using OxCal v4.451. Jucusbamba, Peru lies just west of the Intertropical Convergence Zone (ITCZ), a mixing
zone indicated by Marsh et al.64. As such, a second calibration was done with a 50/50 mix between the Northern Hemisphere IntCal2053 and SHCal20, with 1_σ_ error of 25%, as a relatively
vague approximation of a mixed calibration so that the effect could be assessed. With no modelling for ITCZ mixing, the calibrated date is bimodal with ranges of 1308–1363 ce (73.7%
probability) and 1380–1399 ce (21.7% probability). The modelled calibration is also bimodal and slightly earlier, with ranges of 1296–1328 ce (40.0% probability) and 1342–1394 ce (55.5%
probability). For the individual from the Chonos Archipelago in Chile, the two calibrations mixed the SHCal20 and Marine20 calibration curves at 85 ± 10% and 100 ± 10%, respectively. The
Marine20 also had a local ΔR correction of −93 ± 30 years made, which is a weighted average of the eight nearest points in the Calib.org Marine20 database (http://calib.org/marine/). The 85%
modelled correction calibrates to 1226–1451 ce (95.4% probability), and the 100% modelled correction calibrates to 1255–1483 ce (95.4% probability). Finally, Deán Funes is well within the
area designated by Marsh et al.64 as requiring calibration using SHCal20. Therefore, no modelling or other correction was necessary, and so only SHCal20 was used for calibration. This
individual died in 1226–1281 ce (95.4% probability). Additional numerical data for the above can be found in Supplementary Table 2. Modelled dates are shown in Fig. 1g. DNA EXTRACTION,
LIBRARY PREPARATION AND SEQUENCING Processing of archaeological tissues was performed in the ancient DNA clean room facility of the Max Planck Institute for Evolutionary Anthropology.
Pulverised material from bone was generated with a dental drill at low rotation and high torque. For the immature molar of JUC013, powder was drilled through entry to the crown via the
unfused roots to generate two aliquots of approximately 50 mg for dentin each for extraction. Subsampling of Argentinian and Chilean tissues was done on site (by D.A.R., R.B. and T.v.H.),
and material was subsequently processed in the clean room facility. The collection of biological material from Chile included soft tissues, which were dissolved directly in a protein kinase
digestion buffer. For individual GAP009 specifically, powder was obtained both at the site of the subperiosteal lesion and at a location adjacent to the lesion. Individual MXV001 was sampled
at multiple skeletal elements in locations that displayed pathology consistent with treponemal infection. Individual RAZ007 was sampled at the occipital bone where lesions were displayed,
and at the distal diaphysis of the left femur. Finally, individual DFU001 was sampled from the left iliac crest where lesions attributable to unspecific infections were observed. DNA was
extracted with a silica-based method optimized for the recovery of short DNA fragments65. In brief, lysates were prepared by adding 1–1.8 ml extraction buffer (0.45 M EDTA, pH 8.0, 0.25 mg
ml−1 proteinase K, 0.05% Tween-20) to the sample material in 2.0-ml Eppendorf Lo-Bind tubes65,66 and rotating the tubes at 37 °C for approximately 16 h. For GAP009, DFU001 and RAZ007 this
lysate was used directly for library preparation. For MXV001 powders and JUC013, the lysate was further purified and concentrated over a silica membrane65. A double-stranded DNA library was
manually generated from JUC013 with 10 µl of extract28, which later produced the shotgun screening library JUC013.A0101. Libraries with partial UDG treatment67 were manufactured for the
MXV001 DNA isolate using 30 µl of extract. Lysates of 150μl-aliquots from both GAP009 digests were purified via an automated liquid handling system (Bravo NGS Workstation B, Agilent
Technologies) with silica-coated magnetic beads and binding buffer D as described in Rohland et al.66. Elution volume was 30 µl. Single-stranded libraries of RAZ007, DFU001 and GAP009 lysate
from the lesion (GAP009.A0101) and the region adjacent to it (GAP009.A0201), as well as subsequent libraries from 30 µl of the JUC013 extract (JUC013.0102) and additional powder
(JUC013.A0201; extracted as above for GAP009) were manufactured via automation. DNA libraries were prepared from 30 µl extract using an automated version of single-stranded DNA library
preparation68 described in detail in Gansauge et al.29. _Escherichia coli_ UDG and _E. coli_ endonuclease VIII were added during library preparation (together with the phosphatase) to remove
uracils (either partially or in full) in the interior of molecules was applied for generation of libraries GAP009.A0301, GAP009.A0401, GAP009.A0501, JUC013.A0102 and JUC013.A0201. Negative
controls were produced in parallel for all extractions and libraries. Library yields and efficiency of library preparation were determined using two quantitative PCR assays29. COMPUTATIONAL
SCREENING FOR _T. PALLIDUM_ DNA Sequencing data from GAP009 (two libraries), MXV001 (three libraries), DFU001 (one library) and RAZ007 (two libraries) was screened via HOPS30 using a MALT69
database consisting of all ‘complete’ or ‘chromosome’ level bacterial genomes (on 3 November 2017), ‘complete’ viral genomes (without ‘other’ or ‘unknown’ in their names) (on 30 October
2017) and a selection of eukaryotic genomes obtained through the NCBI assembly portal. Read processing, removal of human reads, as well as HOPS analysis to asses MALT output, was performed
through the nextflow (nf)-core/eager v2.4.432 pipeline, built on the nextflow v21.04.170 workflow language. nf-core/eager is a reproducible pipeline for quality control and assessment of
sequencing data, generating as well as analysing mapping data and generating SNP calls, among other features. The nf-core/eager pipline is optimized for aDNA and employs several gold
standard published software packages including AdapterRemoval v271 for removal of adapters and trimming of low-confidence base calls at the 3′ end of reads, bwa v0.7.1772 for mapping,
samtools c1.1273 for the removal of reads with low mapping quality (<37), dedup v0.21.8 for removal of duplicates, and DamageProfiler74 for analysing damage percentages in reads, among
others. A full list of the software used by this nf-core/eager pipeline can be found in Supplementary Table 3. Candidates were assessed based on their HOPS candidate profiles (Supplementary
Fig. 10). JUC013 was identified as a _T. pallidum_ candidate via an alternative computational approach to the one described above (Supplementary Information, section 3). The number of reads
from RAZ007 that were assigned to the genus _Treponema_ in MALT (_n_ = 3 for the femur and _n_ = 2 for the occipital) were too low to permit an assessment in HOPS. ENRICHMENT FOR _T.
PALLIDUM_ DNA AND SEQUENCING Sample and control libraries were enriched for TPA DNA in two rounds of consecutive in-solution capture75 automated on the Bravo NGS workstation. Both
non-enriched and enriched library products were sequenced on an Illumina HiSeq 4000 using single-end 75 bp chemistry. ANALYSES OF _T. PALLIDUM_ CAPTURE DATA See Supplementary Information,
sections 6–8. HUMAN ANCESTRY ANALYSES We analysed the shotgun and human-enriched capture data using nf-core/eager v. 2.3.432. AdapterRemoval v.271 was used to trim adapter sequences and to
remove adapter dimers and low-quality sequence reads (min length = 30; min base quality = 20). Pre-processed sequences were mapped to the human genome assembly GRCh37 (hg19) from the Genome
Reference Consortium76 using BWA v. 0.7.1272 and a seed length of 32. The C to T misincorporation frequencies typical of aDNA were obtained using mapDamage 2.077 to assess the authenticity
of the ancient DNA fragments. Genetic sex of the analysed individuals was assigned using SNP capture data by calculating the ratio of average X chromosomal and Y chromosomal coverage to the
average autosomal coverage normalized by the chromosome length at the targeted SNPs78. Samples with an X rate between 0.35 and 0.55 and a Y rate between 0.4 and 0.7 were confirmed male
(JUC013, DFU001, MXV001 and RAZ007). ANGSD v. 0.935 (as implemented in nf-core/eager) was used to estimate nuclear contamination, since males are expected to be homozygous at each X
chromosome position79. The contamination estimates returned a value of 0.0 ± 0.0 for all individuals analysed. We used samtools mpileup (parameters –q 30 –Q 30 –B) to generate a pileup file
from the merged sequence data of each individual, and used a custom script (pileupCaller v. 8.2.280) to genotype the individuals, using a pseudo-haploid random draw approach. Only
individuals with >10,000 SNPs called (GAP009: 156,533; JUC013: 761,151; MXV001: 1,027,622; DFU001: 43,818 (on the Human Origins panel)) were retained for further population genetics
analyses (RAZ007 was excluded due to a lower number of SNPs called, 2,134). Consensus sequences for the mitochondrial genomes of the individuals were determined via the mapping of the
sequencing reads obtained from shotgun and post-capture enrichment (post-capture enrichment only for MXV001) to the revised Cambridge reference sequence81. The mitochondrial DNA (mtDNA)
sequences were aligned and manually inspected with Geneious Prime 2021.1.1 (https://www.geneious.com). For the resulting sequences, we filtered positions with likelihoods above 30 and used
HaploGrep282 and HAPLOFIND83 to assign and confirm the corresponding mtDNA haplogroups. The aligned mtDNA genomes were compared to other available genomes from the literature. A new
contamination estimation was performed after the processing of the human data, this time using the mtDNA information and the Haplocheck–Contamination Detection v1.0.0 from Mitoverse84. The
results obtained for the five individuals at the contamination status and contamination level are non-detectable except for RAZ007 (average coverage on the mtDNA reference: 549X (MXV001),
1252X (DFU001), 721X (GAP009), 53X (JUC013) and 224 (RAZ007, contamination level 4.5%)). For the Y-haplogroup assignments, we created pileups of reads for each individual which mapped to
Y-chromosome SNPs as listed on the ISOGG Y-DNA Haplogroup Tree (v15.73; https://isogg.org/tree/). We then manually assigned Y-chromosome haplogroups for each individual based on the most
downstream SNP retrieved after evaluating the presence of upstream mutations along the Y-chromosome haplogroup phylogeny as described in Rohrlach et al.85. We merged sequence data for
JUC013, MXV001, DFU001 and GAP009 with sources of published genomic data summarized in Supplementary Table 27. A principal component analysis (PCA) from this worldwide reference set
comprising 7271 ancient and modern individuals (593,115 SNPs), was constructed to investigate admixture with European/African ancestry components (Fig. 4a). RAZ007 was excluded from this
analysis due to its low coverage (fewer than 10,000 resolved SNPs in the 1240 K reference panel). We used a set of ancestral populations from five continental regions to estimate admixture
proportions to test for admixture with non-Native American populations (which would be a clear indication of the individual being born during the post-contact period): we included Huichol43,
Mayan, Karitiana86 and Mixtec87 as part of a genetic pool to assess the Native American contribution to our samples; the African component was estimated by using the genetic data of
individuals from Yoruba86,88, Esan, Mende, and Luo78 groups; Spanish87,89 and French86,89 were used to model the European genetic contribution; the East Asian genetic contribution consisted
of Han and Cambodian88; and the near Oceania component was estimated with genotypes from Papuan88. We then used ADMIXTURE90 and AdmixturePlotter91 to calculate the best _K_ for our model
(that is, the one with the lowest cross validation error (CVE); _K_ = 5) and the admixture proportions of the components that are maximized in each of the parental populations modelled (Fig.
4d,e). To further refine the visualization of the Native American ancestry in the individuals analysed, another ADMIXTURE run was performed, but only with Indigenous populations of the
Americas as ancestral populations: populations from the Arctic, North American, Caribbean, Mesoamerican and South American regions were included as ancestral (Fig. 4f,g). The lowest CVE was
estimated at _K_ = 5. The results yielded admixture proportions expected for the geographical regions and archaeological periods that the contextual information provided for the individuals
analysed. To assess the genetic relationships and admixture events suggested in the ADMIXTURE analysis, we carried out _F_-statistics analyses using the Xerxes CLI software from the Poseidon
framework92. We performed _F_3-tests of the form _F_3(outgroup; test, _X_) to measure the amount of shared genetic drift between Indigenous American test populations and each analysed
individual after their divergence from an African outgroup (Fig. 4a–c), where _X_ refers to each of the individuals analysed, test is each of the Indigenous American populations included in
our analyses, and Outgroup corresponds to Mbuti individuals from Congo88. VIRULENCE FACTOR ANALYSIS To infer putative functional differences among ancient and modern treponemal genomes, we
investigated the presence or absence of genes potentially associated with virulence profiles in the _pallidum_ (syphilis) and _pertenue_ (yaws)/_endemicum_ (bejel) lineages, as well as in
their outgroup _T. paraluiscuniculi_93,94,95 (Supplementary Table 25). For this, BAM files prior to the application of a mapping quality filter with SAMtools73 were used to calculate the
coverage across 68 chromosomal genes annotated on the Nichols reference genome (NC_021490.2) (Supplementary Table 26), as previously performed26,96. Coverage proportions across each
investigated gene were calculated with BEDtools97 and subsequently plotted on a heatmap using ggplot2 of R version 4.2.2 (Extended Data Fig. 3 and Supplementary Table 26). Moreover, we used
the program snpEff version 3.1i [build 2012-12-12]98 to infer the possible functional impact of called SNPs (see Supplementary Information, section 7.3 for details on SNP calling strategy)
across eight ancient and 53 modern treponemal genomes. The inferred SNP effects were integrated into the comparative SNP table of all treponemal genomes generated for our analyses (_n_ = 61)
using MultiVCFAnalyzer v0.85.299. The table was filtered to identify SNPs and their effects present on the three newly reported lineages, which gave rise to ancient genomes MXV001
(Supplementary Table 21), DFU001 (Supplementary Table 22), and JUC013/GAP009 (Supplementary Table 23), as well those derived in all modern TPA (syphilis) diversity (Supplementary Table 24).
MOLECULAR DATING To investigate the potential of a molecular dating analysis, we formally evaluated the presence of temporal signal in our dataset using two different approaches. First, we
performed a root-to-tip regression, as implemented with Clockor239 (Supplementary Fig. 18). This showed a positive correlation between sample date and genetic distance to the root (_R_2 =
0.25). Regressions fitted independently for the TPA and TPE/TEN clades suggested similar evolutionary rates in both lineages (6.7 × 10−8 and 8.4 × 10−8 substitutions per site per year,
respectively; Supplementary Fig. 18), although the local clock model was slightly preferred over a global clock model (Bayesian information criterion (BIC) = −1,250 and −1,254,
respectively). In a second step, we compared the fit of a strict clock and an uncorrelated log-normal relaxed clock using path sampling41, as implemented in the BEAST 2 model selection
package38. In this case, an exponential coalescent tree prior was used due to known issues of the skyline coalescent for model comparison100. A proper uniform prior between 0 and 1 million
was used for the end population size. For these analyses, the dates of ancient genomes were fixed to their median estimates. The marginal likelihood of each model was then estimated using
path sampling40. We used 10 million pre-burn-in iterations and 100 steps initially set to 3 million iterations each (including 50% burn-in), which was then extended by the same length to
ensure that the resulting Bayes factor estimate had converged. The latter showed overwhelming support for the relaxed clock model (log BF = 167). Finally, we performed Bayesian evaluation of
temporal signal (BETS)40. The previously selected relaxed clock model was then compared to an isochronous model in which all samples were set to 0 bp and the clock rate fixed to one, with
all other parameters otherwise identical. To this end, path sampling40 was used as described above. The results indicated strong support for the heterochronous model (log BF = 11.1)
confirming the presence of significant temporal signal in our dataset. Following the assessment of significant temporal signal in our dataset, a time-calibrated phylogenetic tree was
estimated using BEAST 2.6.738 based on the alignment containing 2104 SNPs described above (Fig. 3 and Supplementary Table 20). A filtered alignment was used to account for the number of
constant sites remaining after all filtering steps were applied. The tree was calibrated using sample dating information. Isolation dates were used for modern strains. For ancient strains,
dating information was entered as uniform prior distributions bounded by intervals based on either dietary modelled 2_σ_ 14C date ranges, or historical data (Supplementary Table 28). A
general time reversible (GTR) substitution model was used with a gamma-distributed rate heterogeneity using four discrete categories101, with permission for 25% missing data (ambiguous base
calls) across the dataset for each position. A Bayesian skyline coalescent model with ten groups was used as the tree prior102 to allow estimation of changes in population size through time,
and following Majander et al.26. Following clock model selection, an uncorrelated log-normal relaxed clock model was used103 (Supplementary Data, BEAST2_UCLN.xml), with uniform prior
between 0 and 1 substitutions per site per year for the mean rate. All other parameters were set to default (BEAUti v.2.6.7)38. The analysis was run using an MCMC of 300 million iterations,
with sampling every 30,000 steps. Ten percent of the iterations were discarded as burn-in and convergence was assessed in Tracer v. 1.7.1104, ensuring that all effective sample size (ESS)
values were above 200. We also sampled the prior distribution of parameters using the same MCMC specifications to ensure that clock rate and _T_MRCA estimates were driven by the data
(Supplementary Fig. 19). A maximum clade credibility (MCC; Fig. 4) tree was constructed from the posterior distribution of trees using Treeannotator105. Sampled trees and population
parameters were used to reconstruct a Bayesian skyline plot (Extended Data Fig. 4) using a dedicated R script to visualize estimates of bacterial population size through time. In addition,
to account for the fact that observed evolutionary rates may vary depending on the considered time-scale, we performed a second analysis using the time-dependent-rate (TDR) model implemented
in BEAST 1.10.442,106 (Supplementary Data, BEAST1_TDR.xml). The latter model can, however, not accommodate inter-lineage rate variation as the uncorrelated relaxed clock model does. We used
a three-epoch model with an exponential structure including a transition first at 100 years ago and a second at 1,000 years ago. The midpoint of each period was used as a reference point
(taking 5,000 years ago for the last epoch). We used normal priors with mean = −14.5 and 0, and s.d. = 5 and 5 for the intercept and slope coefficients of the log-linear relationship between
the substitution rate and time, respectively (corresponding to an average of around 5.10–7 substitutions per site per year at time 0 and no prior assumption on the direction of the
relationship). Other model specifications were identical to the main analysis described above (Supplementary Fig. 20). Results of the dating analyses are presented in Supplementary Table 29.
REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Data are accessible via ENA Project
ID PRJEB62879. Data from other public sources utilized in this study can be accessed via the information contained in Supplementary Table 8 (_T. pallidum_ data) or Supplementary Table 27
(human data). Mapping to the human genome was performed against reference GCF_000001405.13. Map figures were generated as follows: ‘America’ was downloaded from:
https://web.archive.org/web/20180625082054/https://tapiquen-sig.jimdo.com/descargas-gratuitas/américa/. Carlos EfraÌn Porto TapiquÈn. OrogÈnesis Soluciones Geogr·ficas. Porlamar, Venezuela
2015. Based on shapes from Enviromental Systems Research Institute (ESRI). Free Distribuition. CODE AVAILABILITY BEAST 1 and BEAST2.xml are included as supplementary files. The custom code
used for the analysis of SNP effects and virulence-gene coverage is available at: https://github.com/mariaspyrou/Treponema-Americas-paper/tree/main/virulence-gene-analysis and
https://github.com/mariaspyrou/Treponema-Americas-paper/tree/main/snpeff. REFERENCES * Giacani, L. & Lukehart, S. A. The endemic treponematoses. _Clin. Microbio. Rev._ 27, 89 (2014).
Article Google Scholar * Crosby, A. W. Jr. The early history of syphilis: a reappraisal. _Am. Anthropol._ 71, 218–227 (1969). Article MATH Google Scholar * Hackett, C. J. On the origin
of the human treponematoses (pinta, yaws, endemic syphilis, and venereal syphilis). _Bull. World Health Organ._ 29, 7–41 (1963). CAS PubMed PubMed Central MATH Google Scholar *
Štaudová, B. et al. Whole genome sequence of the _Treponema pallidum_ subsp. _endemicum_ strain Bosnia A: the genome is related to yaws treponemes but contains few loci similar to syphilis
treponemes. _PLOS Negl. Trop. Dis._ 8, e3261 (2014). Article PubMed PubMed Central Google Scholar * Quétel, C. _History of Syphilis_ (Johns Hopkins Univ. Press, 1990). * Beale, M. A. et
al. Global phylogeny of _Treponema pallidum_ lineages reveals recent expansion and spread of contemporary syphilis. _Nat. Microbiol._ 6, 1549–1560 (2021). Article CAS PubMed PubMed
Central MATH Google Scholar * Janečkocá, K. et al. The genomes of the yaws bacterium, _Treponema pallidum_ subsp. _pertenue_, of nonhuman primate and human origin are not genomically
distinct. _PLOS Negl. Trop. Dis._ 17, e0011602 (2023). Article Google Scholar * Wicher, K., Wicher, V., Abbruscatom, F. & Baugh, R. E. _Treponema pallidum_ subspecies _pertenue_
displays pathogenic properties different from those of _T. pallidum_ subsp. _pallidum_. _Infect. Immun._ 68, 3219–3225 (2000). Article CAS PubMed PubMed Central MATH Google Scholar *
Schuenemann, V. J. et al. Historic _Treponema pallidum_ genomes from colonial Mexico retrieved from archaeological remains. _PLOS Negl. Trop. Dis._ 12, e0006447 (2018). Article PubMed
PubMed Central Google Scholar * Harper, K. N. et al. On the origin of the treponematoses: a phylogenetic approach. _PLOS Negl. Trop. Dis._ 2, e148 (2008). Article PubMed PubMed Central
MATH Google Scholar * Hudson, E. H. _Treponematosis_ (Oxford Univ. Press, 1946). * Harper, K. N., Zuckerman, M. K., Harper, M. L., Kingston, J. D. & Armelagos, G. J. The origin and
antiquity of syphilis revisited: an appraisal of Old World pre-Columbian evidence for treponemal infection. _Am. J. Phys. Anthropol._ 146, 99–133 (2011). Article PubMed MATH Google
Scholar * Holcomb, R. C. _Who Gave the World Syphilis?_ _The Haitian Myth_ (Froben Press, 1930). * Brothwell, D. R. Microevolutionary change in the human pathogenic treponemes: an
alternative hypothesis. _Int. J. Syst. Bacteriol._ 31, 82–87 (1981). Article Google Scholar * Livingstone, F. B. On the origin of syphilis: an alternative hypothesis. _Curr. Anthropol._
32, 587–590 (1991). Article MATH Google Scholar * Striland, A. Pre-Columbian treponematosis in medieval Britain. _Int. J. of Osteoarchaeol._ 1, 39–47 (1991). Article Google Scholar *
Dutour, O. et al. _L’origine de la Syphilis en Europe Avant our Après 1493?_ (Editions Errance, 1994). * Powell, M. L. & Cook, D. C. _The Myth of Syphilis: the Natural History of
Treponematosis in North America_ (Univ. Florida Press, 2005). * Hackett C. J. _Diagnostic Criteria of Syphilis, Yaws, and Treponarid (Treponematoses) and Some Other Diseases in Dry Bone (for
Use in Osteo-archaeology)_ (Springer, 1976). * Hillson, S. C. et al. Dental defects of congenital syphilis. _Am. J. Phys. Anthropol._ 107, 25–40 (1998). Article CAS PubMed MATH Google
Scholar * Baker, B. et al. Advancing the understanding of treponemal disease in the past and present. _Am. J. Phys. Anthropol._ 171, 5–41 (2020). Article PubMed MATH Google Scholar *
Bouwman, A. S. & Brown, T. A. The limits of biomolecular palaeopathology: ancient DNA cannot be used to study venereal syphilis. _J. Archaeol. Sci._ 32, 703–713 (2005). Article MATH
Google Scholar * Kolman, C. J., Centurion-Lara, A., Lukehart, S. A., Owsley, D. W. & Tuross, N. Identification of _Treponema pallidum_ subspecies _pallidum_ in a 200-year-old skeletal
specimen. _J. Infect. Dis._ 180, 2060–2063 (1999). Article CAS PubMed Google Scholar * Barquera, R. et al. Origin and health status of first-generation Africans from early colonial
Mexico. _Curr. Biol._ 30, 2078–2091.e11 (2020). Article CAS PubMed MATH Google Scholar * Giffin, K. L. et al. A treponemal genome from an historic plague victim supports a recent
emergence of yaws and its presence in 15th century Europe. _Sci Rep._ 10, 9499 (2020). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Majander, K. et al. Ancient
bacterial genomes reveal a high diversity of _Treponema pallidum_ strains in Early Modern Europe. _Curr. Biol._ 30, 3788–3803.e10 (2020). Article CAS PubMed MATH Google Scholar * Arora,
N. et al. Origin of modern syphilis and emergence of a pandemic _Treponema pallidum_ cluster. _Nat. Microbiol._ 2, 16245 (2016). Article CAS PubMed Google Scholar * Meyer, M. &
Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. _Cold Spring Harb. Protoc._ 6, pdb.prot5448 (2010). Article MATH Google Scholar *
Gansauge, M.-T. et al. Manual and automated preparation of single-stranded DNA libraries for the sequencing of DNA from ancient biological remains and other sources of highly degraded DNA.
_Nat. Protoc._ 15, 2279–2300 (2020). Article CAS PubMed MATH Google Scholar * Huebler, R. et al. HOPS: automated detection and authentication of pathogen DNA in archaeological remains.
_Genome Biol._ 20, 280 (2019). Article CAS Google Scholar * Vågene, Å. J. et al. _Salmonella enterica_ genomes from victims of a major sixteenth-century epidemic in Mexico. _Nat. Ecol.
Evol._ 2, 520–528 (2018). Article PubMed Google Scholar * Fellows Yates, J. A. et al. Reproducible, portable, and efficient ancient genome reconstruction with nf-core/eager. _PeerJ_ 9,
e10947 (2021). Article PubMed PubMed Central Google Scholar * Majander, K. et al. Redefining the treponemal history through pre-Columbian genomes from Brazil. _Nature_ 627, 184–188
(2024). Article ADS Google Scholar * Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. _Nucleic Acids
Res._ 43, e15 (2015). Article PubMed MATH Google Scholar * Didelot, X. & Wilson, D. J. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. _PLoS Comput.
Biol._ 11, e1004041 (2015). Article ADS PubMed PubMed Central Google Scholar * Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable and
user-friendly tool for maximum likelihood phylogenetic inference. _Bioinformatics_ 35, 4453–4455 (2019). Article CAS PubMed PubMed Central Google Scholar * Slatkin, M. Statistical
methods for analyzing ancient DNA from hominins. _Curr. Opin. Genet. Dev._ 41, 72–76 (2016). Article CAS PubMed PubMed Central MATH Google Scholar * Bouckaert, R. et al. BEAST 2: a
software platform for Bayesian evolutionary analysis. _PLoS Comput. Biol._ 10, e1003537 (2014). Article PubMed PubMed Central MATH Google Scholar * Featherstone, L. A., Rambaut, A.,
Duchene, S. & Wirth, W. Clockor2: inferring global and local strict molecular clocks using root-to-tip regression. _Syst. Biol._ 73, 623–628 (2024). * Duchene, S. et al. Bayesian
evaluation of temporal signal in measurably evolving populations. _Mol. Biol. Evol._ 37, 3363–3379 (2020). Article CAS PubMed MATH Google Scholar * Lartillot, N. & Philippe, H.
Computing Bayes factors using thermodynamic integration. _Syst. Biol._ 55, 195–207 (2006). Article PubMed MATH Google Scholar * Membrebe, J. V., Suchard, M. A., Rambaut, A., Baele, G.
& Lemey, P. Bayesian inference of evolutionary histories under time-dependent substitution rates. _Mol. Biol. Evol._ 36, 17931803 (2019). Article MATH Google Scholar * Raghavan, M. et
al. Genomic evidence for the Pleistocene and recent population history of Native Americans. _Science_ 349, 6250 (2015). Article MATH Google Scholar * Davis, L. G. et al. Late upper
paleolithic occupation at Cooper’s Ferry, Idaho, USA, similar to 16,000 years ago. _Science_ 365, 891–897 (2019). Article ADS CAS PubMed MATH Google Scholar * Skoglund, P. & Reich,
D. A genomic view of the peopling of the Americas. _Curr. Opin. Genet. Dev._ 41, 27–35 (2016). Article CAS PubMed PubMed Central MATH Google Scholar * Buret, F. _Syphilis Today and
Among the Ancient_ (F. A. Davis, 1891). * Knell, R. J. Syphilis in renaissance Europe: rapid evolution of an introduced sexually transmitted disease? _Proc. Biol. Sci._ 271, S174–S176
(2004). Article PubMed PubMed Central MATH Google Scholar * Weaver, D. S. et al. in _The Myth of Syphilis_ (eds Powell M. L. & Cook D. C.) 77–91 (Univ. Florida Press, 2005). *
Asiedu, K. et al. Eradication of yaws: historical efforts and achieving WHO’s 2020 target. _PLOS Negl. Trop. Dis._ 8, e3016 (2014). Article PubMed PubMed Central MATH Google Scholar *
Cook, D. C. & Powell M. L. in _The Myth of Syphilis_ (eds Powell, M. L. & Cook, D. C.) 443–479 (Univ. Florida Press, 2005). * Bronk Ramsey, C. Bayesian analysis of radiocarbon dates.
_Radiocarbon_ 51, 337–360 (2009). Article MATH Google Scholar * Hogg, A. G. et al. SHCal20 Southern Hemisphere calibration, 0–55,000 years cal bp. _Radiocarbon_ 62, 759–778 (2020).
Article CAS Google Scholar * Reimer, P. J. et al. The IntCal20 Northern Hemisphere radiocarbon age calibration curve (0–55 cal kbp). _Radiocarbon_ 62, 725–757 (2020). Article CAS MATH
Google Scholar * Heaton, T. J. et al. Marine20—The marine radiocarbon age calibration curve (0–55,000 cal bp). _Radiocarbon_ 62, 779–820 (2020). * Longin, R. New method of collagen
extraction for radiocarbon dating. _Nature_ 230, 241–242 (1971). Article ADS CAS PubMed MATH Google Scholar * Brock, F., Higham, T., Ditchfield, P. & Bronk Ramsey, C. Current
pretreatment methods for AMS radiocarbon dating at the Oxford Radiocarbon Accelerator Unit (ORAU). _Radiocarbon_ 52, 103–112 (2010). Article CAS Google Scholar * Ambrose, S. H.
Preparation and characterization of bone and tooth collagen for isotopic analysis. _J. Archaeol. Sci._ 17, 431–451 (1990). Article MATH Google Scholar * van Klinken, G. J. Bone collagen
quality indicators for palaeodietary and radiocarbon measurements. _J. Archaeol. Sci._ 26, 687–695 (1999). Article MATH Google Scholar * Powell, R. L. & Still, C. J. Biogeography of
C3 and C4 vegetation in South America. In_ Proc._ _XIV Simpósio Brasileiro de Sensoriamento Remoto_ 2935–2942 (INPE, 2009). * Yesner, D. R., Figuerero Torres, M. J., Guichon, R. A. &
Borrero, L. A. Stable isotope analysis of human bone and ethnohistoric subsistence patterns in Tierra del Fuego. _J. Anthropol. Archaeol._ 22, 279–291 (2003). Article Google Scholar *
González-Guarda, E. et al. Late Pleistocene ecological, environmental and climatic reconstruction based on megafauna stable isotopes from northwestern Chilean Patagonia. _Quat. Sci. Rev._
170, 188–202 (2017). Article ADS Google Scholar * Arneborg, J. et al. Change of diet of the Greenland Vikings determined from stable carbon isotope analysis and 14C dating of their bones.
_Radiocarbon_ 41, 157–168 (1999). Article CAS MATH Google Scholar * Cook, G. T. et al. Best practice methodology for 14C calibration of marine and mixed terrestrial/marine samples.
_Quat. Geochronol._ 27, 164–171 (2015). Article MATH Google Scholar * Marsh, E. J. et al. IntCal, SHCal, or a mixed curve? Choosing a 14C calibration curve for archaeological and
palaeoenvironmental records from tropical South America. _Radiocarbon_ 60, 925–940 (2018). Article CAS Google Scholar * Dabney, J. et al. Complete mitochondrial genome sequence of a
Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. _Proc. Natl Acad. Sci. USA_ 110, 15758–15763 (2013). Article ADS CAS PubMed PubMed Central Google Scholar *
Rohland, N. et al. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. _Nat. Protoc._ 13, 2447–2461 (2018). Article CAS PubMed MATH
Google Scholar * Rohland, N. et al. Partial uracil-DNA-glycosylase treatment for screening of ancient DNA. _Philos. Trans. R. Soc. B_ 370, 20130624 (2015). Article Google Scholar *
Gansauge, M.-T. et al. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. _Nucleic Acids Res._ 45, e79 (2017). CAS PubMed PubMed Central Google Scholar
* Herbig, A. et al. MALT: fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. Preprint at _BioRxiv_ https://doi.org/10.1101/050559 (2016). * di
Tommaso, P. et al. Nextflow enables reproducible computational workflows. _Nat. Biotechnol._ 35, 316–319 (2017). Article PubMed Google Scholar * Schubert, M., Lindgreen, S. & Orlando,
L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. _BMC Res. Notes_ 9, 88 (2016). Article PubMed PubMed Central Google Scholar * Li, H. & Durbin, R.
Fast and accurate short read alignment with Burrows-Wheeler transform. _Bioinformatics_ 25, 1754–1760 (2009). Article CAS PubMed PubMed Central MATH Google Scholar * Li, H. et al. The
Sequence Alignment/Map format and SAMtools. _Bioinformatics_ 25, 2078–2079 (2009). Article PubMed PubMed Central MATH Google Scholar * Neukamm, J., Peltzer, A. & Nieselt, K.
DamageProfiler: fast damage pattern calculation for ancient DNA. _Bioinformatics_ 37, 3652–3653 (2021). Article CAS PubMed MATH Google Scholar * Fu, Q. et al. DNA analysis of an early
modern human from Tianyuan Cave, China. _Proc Natl Acad. Sci._ 110, 2223–2227 (2013). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Lander, E. S. et al. Initial
sequencing and analysis of the human genome. _Nature_ 409, 860–921 (2001). Article ADS CAS PubMed MATH Google Scholar * Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F. &
Orlando, L. MapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. _Bioinformatics_ 29, 1682–1684 (2013). Article PubMed PubMed Central Google Scholar *
Lamnidis, T. C. et al. Ancient Fennoscandian genomes reveal origin and spread of Siberian ancestry in Europe. _Nat. Commun._ 9, 5018 (2018). Article ADS PubMed PubMed Central MATH
Google Scholar * Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. _BMC Bioinform._ 15, 356 (2014). Article MATH Google Scholar *
Schiffels, S. sequenceTools. _GitHub_ https://github.com/stschiff/sequenceTools.git (2018). * Andrews, R. M. et al. Reanalysis and revision of the cambridge reference sequence for human
mitochondrial DNA. _Nat. Genet._ 23, 147 (1999). Article CAS PubMed MATH Google Scholar * Weissensteiner, H. et al. HaploGrep 2: mitochondrial haplogroup classification in the era of
high-throughput sequencing. _Nucleic Acids Res._ 44, W58–W63 (2016). Article CAS PubMed PubMed Central Google Scholar * Vianello, D. et al. HAPLOFIND: a new method for high-throughput
mtDNA haplogroup assignment. _Hum. Mutat._ 34, 1189–1194 (2013). Article PubMed MATH Google Scholar * Weissensteiner, H. et al. Contamination detection in sequencing studies using the
mitochondrial phylogeny. _Genome Res._ 31, 309–316 (2021). * Rohrlach, A. B. et al. Using Y-chromosome capture enrichment to resolve haplogroup H2 shows new evidence for a two-Path Neolithic
expansion to Western Europe. _Sci. Rep._ 11, 15005 (2021). Article CAS PubMed PubMed Central Google Scholar * Bergström, A. et al. Insights into human genetic variation and population
history from 929 diverse genomes. _Science_ 367, eaay5012 (2020). Article PubMed PubMed Central MATH Google Scholar * Lazaridis, I. et al. Ancient human genomes suggest three ancestral
populations for present-day Europeans. _Nature_ 513, 409–413 (2014). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Patterson, N. et al. Ancient admixture in human
history. _Genetics_ 192, 1065–1093 (2012). Article PubMed PubMed Central MATH Google Scholar * Biagini, S. A. et al. People from Ibiza: an unexpected isolate in the Western
Mediterranean. _Eur. J. Hum. Genet._ 27, 941–951 (2019). Article PubMed PubMed Central MATH Google Scholar * Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation
of ancestry in unrelated individuals. _Genome Res._ 19, 1655–1664 (2009). Article CAS PubMed PubMed Central MATH Google Scholar * Lamnidis, T. C. AdmixturePlotter. _GitHub_
https://github.com/TCLamnidis/AdmixturePlotter (2019). * Schiffels, S. Xerxes CLI software. _GitHub_ https://github.com/poseidon-framework/poseidon-framework.github.io/blob/master/xerxes.md
(2022). * Radolf, J. et al. _Treponema pallidum_, the syphilis spirochete: making a living as a stealth pathogen. _Nat. Rev. Microbiol._ 14, 744–759 (2016). Article CAS PubMed PubMed
Central Google Scholar * Weinstock, G. M. et al. The genome of _Treponema pallidum_: new light on the agent of syphilis. _FEMS Microbiol. Rev._ 22, 323–332 (1998). Article CAS PubMed
MATH Google Scholar * Mikalová, L. et al. Genome analysis of _Treponema pallidum_ subsp. _pallidum_ and subsp. _pertenue_ strains: most of the genetic differences are localized in six
regions. _PLoS ONE_ 5, e15713 (2010). Article ADS PubMed PubMed Central Google Scholar * Wright, E. S. & Vetsigian, K. H. Quality filtering of Illumina index reads mitigates sample
cross-talk. _BMC Genom._ 17, 876 (2016). Article MATH Google Scholar * Quinland, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features.
_Bioinformatics_ 26, 841–842 (2010). Article Google Scholar * Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in
the genome of _Drosophila melanogaster_ strain w1118; iso-2; iso-3. _Fly_ 6, 80–92 (2012). Article CAS PubMed PubMed Central MATH Google Scholar * Bos, K. I. et al. Pre-Columbian
mycobacterial genomes reveal seals as a source of New World human tuberculosis. _Nature_ 514, 494–497 (2014). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Baele, G.
& Lemey, P. in _Bayesian Phylogenetics, Methods, Algorithms, and Applications_ (eds Chen, M. H. et al.) 59–94 (Chapman and Hall/CRC press, 2014). * Yang, Z. Maximum likelihood
phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. _J. Mol. Evol._ 39, 306–314 (1994). Article ADS CAS PubMed MATH Google Scholar *
Drummond, A. J., Rambaut, A., Shapiro, B. & Pybus, O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. _Mol. Biol. Evol._ 22, 1185–1192 (2005).
Article CAS PubMed Google Scholar * Drummond, A. J., Ho, S. Y. W., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. _PLoS Biol._ 4, e88 (2006). Article
PubMed PubMed Central Google Scholar * Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. _Syst.
Biol._ 67, 901–904 (2018). Article CAS PubMed PubMed Central MATH Google Scholar * Heled, J. & Bouckaert, R. R. Looking for trees in the forest: summary tree from posterior
samples. _BMC Evol. Biol._ 13, 221 (2013). Article PubMed PubMed Central MATH Google Scholar * Suchard, A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST
1.10. _Virus Evol._ 4, vey016 (2018). Article PubMed PubMed Central MATH Google Scholar Download references ACKNOWLEDGEMENTS The authors thank the laboratory processing teams of the Max
Planck Institute for Evolutionary Anthropology at both the Jena satellite laboratory and the Leipzig Core Unit for their assistance in data generation; M. Meyer and S. Nagel for support in
the UDG treatment of the JUC013 extract; J. Ilger for assistance with isotope analysis; D. Kühnert and S. Duchêne for valuable discussions regarding molecular dating; the Department of
Physical Anthropology of the University of Chile for allowing us access to the Chonos collection; the Consejo de Monumentos Nacionales de Chile for the authorization to export the samples
analysed (document no. 063 dated 24 January 2003); H. Poinar and the McMaster Ancient DNA Centre for housing the Chilean archaeological tissues for a period after the passing of S. R.
Saunders; S. T. Díaz de la Cruz for assistance with osteopathological assessment of the MXV collection; J. Gretzinger and T. Lamnidis for helpful discussions on human DNA analyses. We
acknowledge the Council of Archaeology, the Direction of Archaeological Salvage/Direction of Physical Anthropology from the National Institute of Anthropology and History (INAH; Mexico City,
Mexico) for the permit granted to analyse the individuals from the Manuel González no. 95 and Avenida Vallejo no. 1864 archaeological salvage projects (project: Estudio molecular de
posibles casos de treponematosis y otras patologías antes del periodo de contacto: el caso de la Cuenca de México en los periodos Clásico y Postclásico; official notice no.
401.1S.3-2019/1201; official export permit no. 401-3-32 AA-76) and critically review and approve our results before publication (partial report approval for this section of the registered
project: 401.1S.3-2023/713). We acknowledge the authorization of the Archaeological Council of INAH for the performed analysis on the tepanecan (RAZ007) individual (official dispatch no.
401-1S.3-2021/1414 and 401.1S.3-2024/72; export: 401-3-440 AA-01-2022). Special thanks to D. E. Volcanes, from the Physical Anthropology Direction of INAH for the unconditional support for
handling the human remains. We thank the Consejo de Comunidades de Pueblos Indígenas de la Provincia de Córdoba, Argentina, for endorsing and supporting the execution of this research. The
Argentina samples analysed in this work were exported with authorization from the Agencia Córdoba Cultura and the Instituto Nacional de Antropología y Pensamiento Latinoamericano (INAPL) no.
DI-2021-87-APN-INAPL#MC dated 9 December 2021, and no. DI-2022-38-APN-INAPL#MC dated 19 May 2022. We also acknowledge the Peruvian Ministry of Culture for granting permission to carry out
genomic research and export ancient Peruvian samples (Project permit RD000013-2017/DGM/VMPCIC/MC and export permit RV167-2017-VMPCIC-MC). This study was funded by the Max Planck Society,
European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 805268 CoDisEASe to K.I.B., Social Sciences and Humanities
Research Council of Canada doctoral dissertation grant no. 752-2001-1058 to T.v.H., Social Sciences and Humanities Research Council of Canada Postdoctoral Fellowship grant no. 756-2023-0246
to C.L.K., Programa de Becas de Reciprocidad para la Formación de Postgrado de Académicos, Científicos y Profesionales de Canadá from the International Council for Canadian Studies
(ICCS/CIEC) y Agencia de Cooperación Internacional de la República de Chile (AGCI) to T.v.H., German Academic Exchange Service (DAAD) to D.A.R. under Short Term Grant no. 57588366, Kone
Foundation, the Faculty of Medicine and the Faculty of Biological and Environmental Sciences at the University of Helsinki for grants to E.K.G., Finnish Cultural Foundation, Magnus Ehrnrooth
Foundation, Sigrid Juselius Foundation, Life and Health Medical Foundation, Finnish Society of Sciences and Letters, Kone Foundation and Academy of Finland to A.S., and CONICET, ANPCyT and
SeCyT (UNC) to M.F. and R.N. FUNDING Open access funding provided by Max Planck Society. AUTHOR INFORMATION Author notes * Casey L. Kirkpatrick Present address: Department of Archaeology,
Simon Fraser University, Burnaby, British Columbia, Canada * Elizabeth A. Nelson Present address: Department of Anthropology, Dedman College of Humanities and Sciences, Southern Methodist
University, Dallas, TX, USA * These authors contributed equally: Rodrigo Barquera, T. Lesley Sitter, Casey L. Kirkpatrick, Darío A. Ramirez AUTHORS AND AFFILIATIONS * Max Planck Institute
for Evolutionary Anthropology, Leipzig, Germany Rodrigo Barquera, T. Lesley Sitter, Casey L. Kirkpatrick, Arthur Kocher, Maria A. Spyrou, Anthi Tiliakou, Elizabeth A. Nelson, Karen L.
Giffin, Raffaela A. Bianco, Adam B. Rohrlach, Alexander Herbig, Johannes Krause & Kirsten I. Bos * Western University, London, Ontario, Canada Casey L. Kirkpatrick * Instituto de
Antropología de Córdoba, Consejo Nacional de Investigaciones Científicas y Técnicas, Universidad Nacional de Córdoba, Museo de Antropologías, Córdoba, Argentina Darío A. Ramirez, Mariana
Fabra & Rodrigo Nores * Departamento de Antropología, Facultad de Filosofía y Humanidades, Universidad Nacional de Córdoba, Córdoba, Argentina Darío A. Ramirez, Mariana Fabra &
Rodrigo Nores * Institute for Archaeological Sciences, Eberhard Karls Universität Tübingen, Tübingen, Germany Maria A. Spyrou * Dirección de Antropología Física, Instituto Nacional de
Antropología e Historia, Mexico City, Mexico Lourdes R. Couoh, Jorge A. Talavera-González & Fabiola A. Ballesteros Solís * Museo Nacional de Historia Natural, Santiago, Chile Mario
Castro * Department of Morphology, Faculty of Medicine, Clínica Alemana–Universidad del Desarrollo, Santiago, Chile Mario Castro * McMaster University, Department of Anthropology, Hamilton,
Ontario, Canada Tanya von Hunnius & Shelley R. Saunders * Department of Forensic Medicine, University of Helsinki, Helsinki, Finland Evelyn K. Guevara & Antti Sajantila * Scottish
Universities Environmental Research Centre, East Kilbride, UK W. Derek Hamilton * Department of Coevolution of Land Use and Urbanisation, Max Planck Institute for Geoanthropology, Jena,
Germany Patrick Roberts * Laboratory Unit, Max Planck Institute of Geoanthropology, Jena, Germany Erin Scott * Consejo Nacional de Investigaciones Científicas y Técnicas, Museo de Ciencias
Naturales y Antropológicas Juan C. Moyano, Mendoza, Argentina Gabriela V. Da Peña * Departamento de Arqueología, Facultad de Filosofía y Letras, Universidad Nacional de Cuyo, Mendoza,
Argentina Gabriela V. Da Peña * Bioarchaeology Research Group, Durham University, Durham, UK Aryel Pacheco * Independent researcher, Santiago, Chile Mónica Rodriguez * Department of
Anthropology, University of Chile, Santiago, Chile Eugenio Aspillaga * School of Computer and Mathematical Sciences, University of Adelaide, Adelaide, South Australia, Australia Adam B.
Rohrlach * Adelaide Data Science Centre, University of Adelaide, Adelaide, South Australia, Australia Adam B. Rohrlach * Dirección de Salvamento Arqueológico, Instituto Nacional de
Antropología e Historia, Mexico City, Mexico María de los Ángeles García Martínez * Forensic Medicine Unit, Finnish Institute of Health and Welfare, Helsinki, Finland Antti Sajantila Authors
* Rodrigo Barquera View author publications You can also search for this author inPubMed Google Scholar * T. Lesley Sitter View author publications You can also search for this author
inPubMed Google Scholar * Casey L. Kirkpatrick View author publications You can also search for this author inPubMed Google Scholar * Darío A. Ramirez View author publications You can also
search for this author inPubMed Google Scholar * Arthur Kocher View author publications You can also search for this author inPubMed Google Scholar * Maria A. Spyrou View author publications
You can also search for this author inPubMed Google Scholar * Lourdes R. Couoh View author publications You can also search for this author inPubMed Google Scholar * Jorge A.
Talavera-González View author publications You can also search for this author inPubMed Google Scholar * Mario Castro View author publications You can also search for this author inPubMed
Google Scholar * Tanya von Hunnius View author publications You can also search for this author inPubMed Google Scholar * Evelyn K. Guevara View author publications You can also search for
this author inPubMed Google Scholar * W. Derek Hamilton View author publications You can also search for this author inPubMed Google Scholar * Patrick Roberts View author publications You
can also search for this author inPubMed Google Scholar * Erin Scott View author publications You can also search for this author inPubMed Google Scholar * Mariana Fabra View author
publications You can also search for this author inPubMed Google Scholar * Gabriela V. Da Peña View author publications You can also search for this author inPubMed Google Scholar * Aryel
Pacheco View author publications You can also search for this author inPubMed Google Scholar * Mónica Rodriguez View author publications You can also search for this author inPubMed Google
Scholar * Eugenio Aspillaga View author publications You can also search for this author inPubMed Google Scholar * Anthi Tiliakou View author publications You can also search for this author
inPubMed Google Scholar * Elizabeth A. Nelson View author publications You can also search for this author inPubMed Google Scholar * Karen L. Giffin View author publications You can also
search for this author inPubMed Google Scholar * Raffaela A. Bianco View author publications You can also search for this author inPubMed Google Scholar * Adam B. Rohrlach View author
publications You can also search for this author inPubMed Google Scholar * María de los Ángeles García Martínez View author publications You can also search for this author inPubMed Google
Scholar * Fabiola A. Ballesteros Solís View author publications You can also search for this author inPubMed Google Scholar * Antti Sajantila View author publications You can also search for
this author inPubMed Google Scholar * Shelley R. Saunders View author publications You can also search for this author inPubMed Google Scholar * Rodrigo Nores View author publications You
can also search for this author inPubMed Google Scholar * Alexander Herbig View author publications You can also search for this author inPubMed Google Scholar * Johannes Krause View author
publications You can also search for this author inPubMed Google Scholar * Kirsten I. Bos View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
K.I.B., J.K., R.B., S.R.S., M.C., R.N., A.S., L.R.C. and J.A.T.-G. conceived of the investigation. M.C., E.K.G., A.P., M.R., E.A., J.A.T.-G., M.d.l.A.G.M., M.F., L.R.C. and F.A.B.S. provided
archaeological tissues for analysis. C.L.K., T.v.H., A.T., E.A.N., A.P., M.R., G.V.D.P., L.R.C. and J.A.T.-G. performed osteological analyses. C.L.K., D.A.R., E.A.N., R.A.B., P.R., E.S.,
R.N. and R.B. performed laboratory processing. T.L.S., A.K., M.A.S., R.B., D.A.R., R.N., C.L.K., W.D.H., R.A.B., A.B.R., A.H., K.L.G. and K.I.B. performed analyses. R.B. independently
drafted a report on MXV001 for a doctoral dissertation. K.I.B. wrote the current manuscript with contributions from all coauthors. CORRESPONDING AUTHORS Correspondence to Johannes Krause or
Kirsten I. Bos. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature_ thanks Maarten Blaauw, Sébastien
Calvignac-Spencer, Martin Sikora and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA FIGURES AND TABLES EXTENDED DATA FIG. 1 AUTHENTICATION OF ANCIENT TREPONEMAL DNA
IN SHOTGUN AND CAPTURE LIBRARIES VIA DAMAGEPROFILER AS IMPLEMENTED IN EAGER2 VIA MAPPING TO THE HUMAN REFERENCE GENOME (LEFT) AND _T. PALLIDUM PALLIDUM_ NICHOLS (RIGHT). Damage signals are
predictably low in the partial-UDG treated GAP009 and MXV001 libraries. EXTENDED DATA FIG. 2 COMPUTATIONAL SCREENING PIPELINE FOR TREPONEMAL DATA PROCESSING OF MODERN AND ANCIENT DATASETS.
Image Created in BioRender. Sitter, L. (2024) https://BioRender.com/d39q601. EXTENDED DATA FIG. 3 COVERAGE OF PUTATIVE VIRULENCE-ASSOCIATED GENES ACROSS THE NICHOLS REFERENCE GENOME
(NC_021490.2). TEN = _T. pallidum endemicum_, TPE = _T. pallidum pertenue_, and TPA = _T. pallidum pallidum_. Genes indicated with an asterisk are reported to be potentially implicated in
phenotypic differences between TPE and TPA (supplementary section 11). For additional information on gene coordinates and function, see Table S24. EXTENDED DATA FIG. 4 BAYESIAN SKYLINE PLOT
SHOWING INFERRED EFFECTIVE POPULATION SIZE (_N__E_) CHANGES OF _T. PALLIDUM_ THROUGH TIME. The mean estimate and 95% HPD interval are shown. The red dashed line indicates the year 1492 CE.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION This document includes Supplementary Methods, Supplementary Figs. 1–21, Supplementary Tables 3 and 11–14, additional references, and a
list of references cited in the Supplementary Tables. REPORTING SUMMARY SUPPLEMENTARY TABLES Supplementary Tables 1, 2, 4–10 and 15–29 SUPPLEMENTARY DATA Molecular dating files (.xml) for
use within BEAST. BEAST1_TDR.xml – “BEAST1 Time Dependent Rate”. BESAT2_UCLN.xml – “BEAST2 Relaxed Clock”. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative
Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the
original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in
the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended
use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Barquera, R., Sitter, T.L., Kirkpatrick, C.L. _et al._ Ancient genomes reveal a
deep history of _Treponema pallidum_ in the Americas. _Nature_ 640, 186–193 (2025). https://doi.org/10.1038/s41586-024-08515-5 Download citation * Received: 23 June 2023 * Accepted: 11
December 2024 * Published: 18 December 2024 * Issue Date: 03 April 2025 * DOI: https://doi.org/10.1038/s41586-024-08515-5 SHARE THIS ARTICLE Anyone you share the following link with will be
able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative