- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Pretrained language models have shown promise in analysing nucleotide sequences, yet a versatile model excelling across diverse tasks with a single pretrained weight set remains
elusive. Here we introduce RNAErnie, an RNA-focused pretrained model built upon the transformer architecture, employing two simple yet effective strategies. First, RNAErnie enhances
pretraining by incorporating RNA motifs as biological priors and introducing motif-level random masking in addition to masked language modelling at base/subsequence levels. It also tokenizes
RNA types (for example, miRNA, lnRNA) as stop words, appending them to sequences during pretraining. Second, subject to out-of-distribution tasks with RNA sequences not seen during the
pretraining phase, RNAErnie proposes a type-guided fine-tuning strategy that first predicts possible RNA types using an RNA sequence and then appends the predicted type to the tail of
sequence to refine feature embedding in a post hoc way. Our extensive evaluation across seven datasets and five tasks demonstrates the superiority of RNAErnie in both supervised and
unsupervised learning. It surpasses baselines with up to 1.8% higher accuracy in classification, 2.2% greater accuracy in interaction prediction and 3.3% improved F1 score in structure
prediction, showcasing its robustness and adaptability with a unified pretrained foundation. SIMILAR CONTENT BEING VIEWED BY OTHERS PROTEINGLUE MULTI-TASK BENCHMARK SUITE FOR SELF-SUPERVISED
PROTEIN MODELING Article Open access 26 September 2022 FINE-TUNING PROTEIN LANGUAGE MODELS BOOSTS PREDICTIONS ACROSS DIVERSE TASKS Article Open access 28 August 2024 XTRIMOPGLM: UNIFIED
100-BILLION-PARAMETER PRETRAINED TRANSFORMER FOR DECIPHERING THE LANGUAGE OF PROTEINS Article 03 April 2025 MAIN RNA is a critical molecule in the central dogma of molecular biology, which
describes the flow of genetic information from DNA to RNA to protein. RNA molecules play a crucial role in various cellular processes, including gene expression, regulation and catalysis.
Given the importance of RNA in biological systems, there is a growing demand for efficient and accurate methods to analyse RNA sequences. The analysis of RNA sequences has traditionally been
performed using experimental techniques such as RNA sequencing and microarrays1,2. However, these methods are often expensive and time-consuming and require large amounts of input RNA. In
recent years, there has been increasing interest in using computational methods based on machine learning models to analyse RNA sequences. Pretrained language models, on the other hand, have
shown great success in various natural language processing tasks, including text classification3, question answering4 and language translation5. Advancements in the field of natural
language processing have led to the successful adoption of pretrained language models like BERT6 to model and analyse nucleotides (nts) and ribonucleotides from trillions of DNA/RNA
sequences. For example, preMLI7 employs rna2vec to produce RNA word vector representations. The RNA sequence features are then mined independently, and the two feature vectors are
concatenated as the input for the prediction task. DNABERT8 has been proposed to extract features from DNA sequences via the pretrained language model BERT-alike, and its derivatives9,10
with task-agnostic extensions have been studied to solve DNA analytical tasks in an ad hoc manner11. Moreover, based on T5 (ref. 12), Rm-LR13 integrates two large-scale RNA language
pretrained models to learn local key features and collect discriminative sequential information. A bilinear attention network is then used to integrate the learned features. However, there
is still some work focusing on generic models that performs well on varying downstream tasks derived from one set of pretrained weights. RNA-FM14 trains a foundation model for the community
to fit all the ncRNA sequences, although it only uses naive token masking as a pretraining strategy, which may lose high-density information hidden in continuous RNA subsequences. This
problem is further compounded by the fact that RNA is a more complex molecule than DNA15, due to the presence of additional modifications and higher-order structures, and existing pretrained
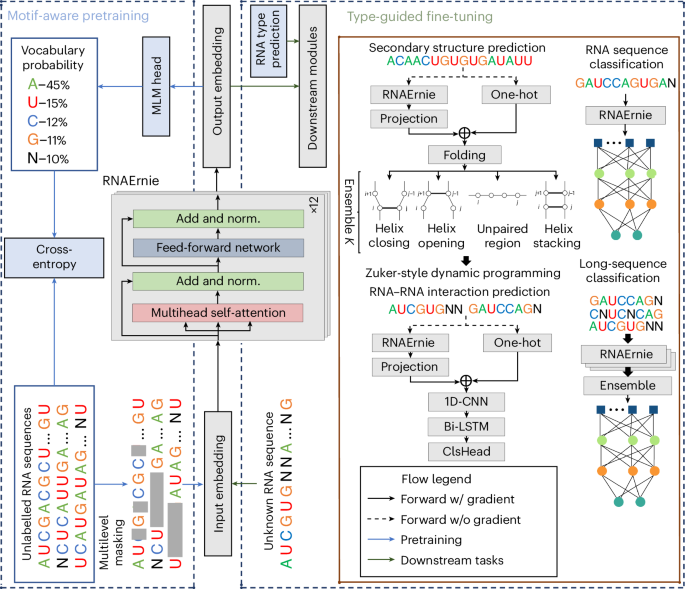
models are not optimized for RNA analysis. In response to this challenge, we have developed a pretrained RNA language model: RNAErnie. As shown in Fig. 1, this model is built upon the
Enhanced Representation through Knowledge Integration (ERNIE) framework and incorporates multilayer and multihead transformer blocks, each having a hidden state dimension of 768. Pretraining
is conducted using an extensive corpus consisting of approximately 23 million RNA sequences meticulously curated from RNAcentral16. The proposed motif-aware pretraining strategy involves
base-level masking, subsequence-level masking and motif-level random masking, which effectively captures both subsequence and motif-level knowledge17,18,19, enriching the representation of
RNA sequences as illustrated in Fig. 2a. Additionally, RNAErnie tokenizes coarse-grained RNA types as special vocabularies and appends the tokens of coarse-grained RNA types at the end of
every RNA sequence during pretraining. By doing so, the model gains the potential to discern the distinct characteristics of various RNA types, facilitating domain adaption to various
downstream tasks. Specifically, a type-guided fine-tuning strategy is employed, incorporating the predicted RNA types as ‘auxiliary information’ within a stacking architecture, as shown in
Fig. 2b. Upon receiving an RNA sequence as input, the model first employs a pretrained RNAErnie block to generate output embeddings. Subsequently, it predicts the potential coarse-grained
RNA types based on these embeddings. The sequence and the predicted RNA types are then fed into a downstream network, which consists of RNAErnie blocks and task-specific heads. This approach
enables the model to accommodate a diverse range of RNA types and enhances its utility in a broad spectrum of RNA analytical tasks. More specifically, to adapt the distribution shifts
between pretraining datasets and target domains, RNAErnie leverages domain adaptation20 that composites the pretrained backbone with downstream modules in three neural architectures: frozen
backbone with trainable head (FBTH), trainable backbone with trainable head (TBTH) and stacking for type-guided fine-tuning (STACK). In this way, the proposed method can either end-to-end
optimize the backbone and task-specific heads or fine-tune task-specific heads with embeddings extracted from the frozen backbone, subject to the downstream applications. The conducted
experiments highlight the immense potential of RNAErnie in advancing RNA analysis. The model demonstrates strong performance across diverse downstream tasks, showcasing its versatility and
effectiveness as a generic solution. Additionally, the innovative strategies employed in RNAErnie show promise in enhancing the performance of other pretrained models in RNA analysis. These
findings position RNAErnie as a valuable asset, empowering researchers with a powerful tool to unravel the complexities of RNA-related investigations. RESULTS In this section, we present the
experiment results for RNAErnie evaluation on both unsupervised learning (RNA grouping) and supervised learning (RNA sequence classification, RNA–RNA interaction prediction and RNA
secondary structure prediction) tasks. For additional experiment settings and results (such as long-sequence classification, SARS-CoV-2 variant evolutionary path visualization and so on),
please refer to Supplementary Information Section C. UNSUPERVISED CLUSTERING OF RNAERNIE-EXTRACTED FEATURES Various types of RNA exhibit distinct functions and structures, and it is expected
that these characteristics are captured within the embeddings generated by our proposed model (RNAErnie) using raw RNA sequences. To examine the patterns within the known RNA repertoire, we
utilize the suggested encoder to establish scatter plots of RNA sequences. Dimension reduction using PHATE21 is then employed to map the embeddings onto a two-dimensional plane. We evaluate
the impact of the learning process by considering both pretrained and randomly initialized RNAErnie embeddings, as well as 3mer statistical embeddings22 for visualization. Figure 3a shows
the results, where the pretrained RNAErnie embedding space effectively organizes RNA types into distinct clusters based on their structural and functional properties. We also use a random
model for comparing encoding effects, establishing a baseline for comparison with other encoding methods. This comparison allows us to evaluate the effectiveness of each method in enhancing
the encoding process. The random model exhibits a less-defined clustering structure, and the 3mer embeddings lack distinguishable features. This indicates that RNAErnie captures structural
and functional information beyond the primary structure of RNA, enabling grouping based on similar properties. To investigate the diversity of non-coding RNAs (ncRNAs), we categorize them
using sequence ontology at various levels. Figure 3b illustrates selected classes of ncRNA, such as ribosomal RNA (rRNA), long ncRNA (lncRNA) and small ncRNA (sncRNA). Figure 3c shows the
high-level ontology relationships between ncRNA, transcript, messenger RNA (mRNA) and intron RNA. Figure 3d represents the low-level ontology of small regulatory ncRNA. RNAErnie effectively
discriminates between classes at different ontology levels, while the 3mer statistical embeddings struggle to separate them. This suggests that RNAErnie captures structural or functional
similarities rather than relying solely on the length of ncRNAs. Note that the random approach seems to outperform RNAErnie in differentiating between classes across various ontology levels.
This finding suggests that RNAErnie might be less effective in capturing the ontology patterns of low-level, small regulatory ncRNA classes. We believe that this limitation in identifying
low-level ontology patterns may stem from several factors, including the complexity and heterogeneity of classes at this level or potential biases in our training dataset. Further research
and detailed analysis are needed to identify the specific causes behind RNAErnie’s reduced efficacy in discerning patterns in low-level ontology. In total, these findings demonstrate that
RNAErnie constructs scatter plots by capturing the structural and functional characteristics of ncRNAs, going beyond nucleic acid statistics alone. SUPERVISED DOMAIN ADAPTATION ON DOWNSTREAM
TASKS In this section, we demonstrate the effectiveness of RNAErnie in three essential supervised learning tasks: RNA sequence classification, RNA–RNA interaction and RNA secondary
structure prediction. To reveal the effectiveness of the designs in RNAErnie, we conducted a series of ablation studies using variant models derived from RNAErnie. These models vary in
complexity, beginning with Ernie-base, which lacks RNA-specific pretraining and includes standard fine-tuning. RNAErnie−− employs base-level masking during pretraining, and RNAErnie− adds
subsequence-level masking to the mix. The complete RNAErnie model further integrates motif-level masking and is fine-tuned using either TBTH or FBTH architectures. Extending this, RNAErnie+
represents the apogee of complexity within this family, including all three levels of masking and a STACK architecture for pretraining. Lastly, the RNAErnie without chunk model is tailored
for long RNA sequences by truncating and discarding segments to contend with computational constraints, aimed at the efficient classification of long non-coding and protein-encoding
transcripts. In addition, we also bring pretrained models from existing literature, including RNABERT23, RNA-MSM24 and RNA-FM14 for comparison. RNA SEQUENCE CLASSIFICATION We evaluate the
performance of our proposed sequence-classification models on the benchmark nRC25. This dataset consists of ncRNA sequences selected from the Rfam database release 12 (ref. 26). nRC is
composed of a balanced collection of sequences, with 20% non-redundant samples for each of the 13 classes. It has 6,320 training sequences and 2,600 testing sequences labelled with 13
classes. Table 1 presents the sequence-classification results for RNAErnie on the nRC dataset. The table includes several baseline methods as well as different variants of the RNAErnie
models. The baseline values are all taken from cited literature except the pretrained models: RNABERT, RNA-MSM and RNA-FM. Analysing the performance of the models, we observe that the
baseline methods achieve varying levels of accuracy. Notably, ncRDense demonstrates decent performance, achieving high accuracy, recall, precision, F1 score and Matthews correlation
coefficient (MCC) values. Turning our attention to the RNAErnie variants, we can see that they consistently outperform most of the baseline models across all evaluation metrics. Although
ncRDense can beat the first two (that is, Ernie-base and RNAErnie−−), RNAErnie−, RNAErnie and RNAErnie+ show better performance in all five dimensions. In the hierarchy of the RNAErnie model
family, performance metrics improve incrementally with complexity of design. The foundational model, Ernie-base, establishes a baseline that is modestly surpassed by RNAErnie−− through the
introduction of base-level masking in pretraining. Furthermore, RNAErnie− incorporates subsequence-level masking and delivers notably enhanced accuracy, recall, precision, F1 score and MCC
values, endorsing the value of a more comprehensive masking strategy. The full RNAErnie model integrates base, subsequence and motif-level masking, achieving superior performance over its
predecessors across all metrics and illustrating the cumulative benefits of multilevel masking. The apex model, RNAErnie+, which employs an exhaustive masking regimen in conjunction with a
two-stage fine-tuning architecture, outperforms all variants in our experiments. RNA–RNA INTERACTION We evaluate the performance of our model on one of the most representative benchmark
datasets, DeepMirTar27,28, which is used for predicting the interaction between microRNAs (miRNAs) and mRNAs. This dataset consists of 13,860 positive pairs and 13,860 negative pairs. The
miRNA sequences in DeepMirTar are all shorter than 26 nts, and the mRNA sequences are shorter than 53 nts. Because most of the target sites are believed to be located at the 3′ untranslated
region, DeepMirTar only considers them. Furthermore, two seeds were taken into consideration: the non-canonical seed, which pairs at position 2-7 or 3-8, permitting G-U couplings and up to
one bulged or mismatched nt; and the canonical seed, which is the precise W-C pairing of 2-7 or 3-8 nts of the miRNA. Given that RNA types (miRNA, mRNA) are fixed here, we do not test
RNAErnie+ version which uses a two-stage pipeline here. Table 2 presents the performance comparison between the proposed RNAErnie models and baseline methods from existing literature, such
as Miranda29, RNAhybrid30, PITA31, TargetScan v.7.0 (ref. 32), TarPmiR33 and DeepMirTar27. The baseline values are all taken from cited literature except the pretrained models: RNABERT,
RNA-MSM and RNA-FM. These are evaluated on the RNA–RNA interaction prediction task using the DeepMirTar dataset. DeepMirTar emerges as a strong baseline, exhibiting high scores across all
metrics. The Ernie-base model and the RNAErnie variations are then assessed, with the RNAErnie model demonstrating superior performance and particularly excelling in accuracy, precision, F1
score and area under the curve (AUC). This variation achieves an impressive accuracy score of 0.9872, a competitive precision score of 0.9901, an F1 score of 0.9873 and the highest AUC score
of 0.9976, indicating excellent overall performance and discriminative power. Overall, the results suggest that the RNAErnie model, particularly the RNAErnie variation, outperforms the
existing methods and the Ernie-base model in the RNA–RNA interaction prediction task. These findings highlight the potential of the RNAErnie model in accurately predicting RNA–RNA
interactions. RNA SECONDARY STRUCTURE PREDICTION This section presents a comprehensive comparison between our pretrained RNAErnie model and several baseline models, including the
state-of-the-art UFold model34, in the context of RNA secondary structure prediction tasks. The experiments are conducted using commonly used benchmarks employed in state-of-the-art models.
These benchmarks include: * RNAStralign35: This dataset comprises 37,149 RNA structures from eight RNA families, with lengths ranging from approximately 100 to 3,000 base pairs (bp). *
ArchiveII36: This dataset consists of 3,975 RNA structures from ten RNA families, with lengths ranging from approximately 100 to 2,000 bp. * bpRNA-1m37: This dataset contains 13,419 RNA
structures from 2,588 RNA families, with sequence similarity removed using an 80% sequence-identity cut-off. The lengths of the sequences range from approximately 100 to 500 bp. The dataset
is randomly split into three subsets: TR0 (10,814 structures) for training, TV0 (1,300 structures) for validation and TS0 (1,305 structures) for testing. We train our model on the entire
RNAStralign dataset, as well as the TR0 subset and other augmented mutated datasets, following the approach used in UFold. Subsequently, we evaluate performance on the ArchiveII600 dataset,
which is a subset of ArchiveII with lengths less than 600 bp, and the TS0 dataset. Table 3 presents a comparative analysis of the performance of various methods on the RNA secondary
structure prediction task using the ArchiveII and TS0 datasets. The table presents the results of several baseline methods, including RNAstructure, RNAsoft, RNAfold, MXfold2, Mfold,
LinearFold, Eternafold, E2Efold, Contrafold and Contextfold. Each method is assessed based on its precision, recall and F1 score for both the ArchiveII600 and TS0 datasets. The baseline
values are all taken from cited literature except the pretrained models: RNABERT, RNA-MSM and RNA-FM. Among the RNAErnie variations, RNAErnie+ achieves the highest scores in precision,
recall and F1 score, indicating its superior performance in RNA secondary structure prediction. Notably, RNAErnie+ achieves a remarkable precision score of 0.886, a high recall score of
0.870 and an impressive F1 score of 0.875 on the ArchiveII600 dataset. These results highlight the effectiveness of RNAErnie+ in accurately predicting RNA secondary structures. DISCUSSION
Our method, RNAErnie, outperforms existing advanced techniques across seven RNA sequence datasets encompassing over 17,000 major RNA motifs, 20 RNA classes/types and 50,000 RNA sequences.
Evaluation using 30 mainstream RNA sequence technologies confirms the generalization and robustness of RNAErnie. We employed accuracy, precision, recall, F1 score, MCC and AUC as evaluation
metrics to ensure a fair comparison of RNA sequence-analysis methods. Currently, little research exists on applying transformer architectures with enhanced external knowledge to RNA sequence
data analysis. Our from-scratch RNAErnie framework integrates RNA sequence embedding and a self-supervised learning strategy, resulting in superior performance, interpretability and
generalization potential for downstream RNA tasks. Additionally, RNAErnie is adaptable to other tasks through modification of the output and supervision signals. RNAErnie is publicly
available and serves as an effective tool for understanding type-guided RNA analysis and advanced applications. The RNAErnie model, despite its innovations in RNA sequence analysis,
confronts several challenges. First, the model is constrained by the size of the RNA sequences it can analyse, as sequences longer than 512 nts are dropped, potentially omitting vital
structural and functional information. The chunking method developed to handle longer sequences might result in the further loss of information about long-range interactions. Second, the
focus of this study is narrow, centred only on the RNA domain and not extending to tasks like RNA-protein prediction or binding-site identification. Additionally, the model encounters
difficulties in considering three-dimensional structural motifs of RNAs, such as loops and junctions, which are essential for understanding RNA functions. More importantly, the existing post
hoc architectural design has potential limitations, including heightened inference overhead. An alternative approach involves designing a specialized loss function that incorporates RNA
type information and pretraining the model in an end-to-end fashion. We have experimented with this concept and engaged in preliminary pretraining. Our findings indicate that although this
method proves beneficial for discriminative tasks such as sequence classification, it unfortunately leads to suboptimal token representations with performance degradation in reconstruction
of structures. Detailed information is provided Supplementary Information Section C.6. Our future work will go deeper into this issue and explore solutions. METHODS This section provides a
comprehensive overview of the design features associated with each component of RNAErnie. We will explore the specific characteristics of each element and discuss their collaborative
functionality in enabling the accomplishment of diverse downstream tasks. OVERALL DESIGN In this work, we present RNAErnie, an approach for large-scale pretraining of RNA sequences based on
the ERNIE framework38, which incorporates multilayer and multihead transformer blocks39. RNAERNIE TRANSFORMER The basic block of the RNAErnie transformer shares the same architectural
configuration as ERNIE38, employing a 12-layer transformer and a hidden state dimension of _D_h = 768. Consider an input RNA sequence denoted as X = (_x_1, _x_2, ⋯ , _x__L_), where each
element _x__i_ ∈ {‘A’, ‘U’, ‘C’, ‘G’} and _L_ represents the length of the sequence. An RNAErnie block first tokenizes RNA bases in the sequence and subsequently feeds them into the
transformer. This process enables us to extract token embeddings \({{{\bf{h}}}}=({h}_{1},{h}_{2},\cdots \,,{h}_{L})\in {{\mathbb{R}}}^{L\times {D}_{{\mathrm{h}}}}\), where _D_h represents
the dimension of the hidden representations for the tokens. Given the embeddings for every token in the RNA sequence, the RNAErnie basic block transforms the series of token embeddings into
a lower-dimensional vector (that is, 768 dimensions) using trainable parameters38 and then outputs the embedding of the RNA sequence. The total number of trainable parameters in RNAErnie is
approximately 105 million. PRETRAINING DATASETS Basically, like many other pretraining based approaches, the RNAErnie approach is structured into two main phases: pretraining and
fine-tuning. In the pretraining phase, which is agnostic to any specific task, RNAErnie is meticulously trained on a vast corpus of 23 million ncRNA sequences obtained from the RNAcentral
database16. This self-supervised autoregressive training phase allows RNAErnie to capture sequential distributions and patterns within the RNA sequences, thereby acquiring a comprehensive
understanding of their structural and functional information. In the subsequent task-specific fine-tuning phase, the pretrained RNAErnie model is either fine-tuned with downstream modules or
used to generate sequence embeddings (features) that complement a lightweight prediction layer. Regarding the tokenization of RNA bases, the sequences are tokenized to represent ‘A’, ‘T/U’,
‘C’ and ‘G’, with the initial token of each sequence reserved for the special classification embedding ([CLS]). Additionally, an indication embedding ([IND]) is appended to each RNA
sequence, followed by indication classes (for example, ‘miRNA’, ‘mRNA’, ‘lnRNA’) derived from the RNAcentral database, as depicted in Extended Data Fig. 1. The inclusion of the indication
embedding encourages the model to cluster similar RNA sequences in a latent space, facilitating retrieval-based learning40. MOTIF-AWARE PRETRAINING STRATEGIES To integrate both subsequence
and motif-level knowledge into the representation of RNA sequences, we introduce a motif-ware multilevel masking strategy to pretrain the RNAErnie basic block, as opposed to directly
incorporating motif embedding. In addition, the RNAErnie approach follows the standard routine of pretraining with all three levels of masking tasks, learning to predict the masked tokens
and also capture contextualized representations of the input RNA sequence. Specifically, the procedure of RNAErnie pretraining with motif-aware multilevel masking strategies is as follows.
BASE-LEVEL MASKING In the initial stage of the learning process, we employ base-level masking as a crucial component. Specifically, we randomly mask 15% of the nucleobases within an RNA
sequence. Among the masked positions, 10% are preserved without any alterations, and the remaining 10% are replaced with other nucleobases. The model takes the remaining nucleobases as input
and is tasked with predicting the masked positions. This stage primarily focuses on acquiring fundamental token representations; capturing intricate higher-level biological insights proves
to be a challenging endeavour. SUBSEQUENCE-LEVEL MASKING Next, we incorporate the masking of random subsequences, which are short and contiguous segments of nucleobases within an RNA
sequence. Previous studies, such as refs. 41 and 42, have demonstrated the efficacy of contiguous token masking in enhancing pretrained models for span-selection tasks. Additionally, it is
important to consider that the functionality of nucleobases often manifests within the context of sequential arrangements. By predicting these subsequences as a whole, we encourage the model
to capture a deeper understanding of the biological information inherent in the relationships between consecutive nucleobases. In our research, we specifically mask subsequences with
lengths ranging from 4 to 8 bp. MOTIF-LEVEL MASKING In the final stage of pretraining, we employ motif-level masking as part of our approach. RNA motifs, characterized as recurrent
structural elements with a high concentration of information, have been extensively observed in atomic-resolution RNA structures17. These motifs are widely recognized for their crucial
involvement in various biological activities, such as the formation of RNA tertiary structures19, interaction with dsRNA-binding proteins (RBPs) and participation in complex formation with
proteins18. To incorporate these motifs into our model as so-called biological priors, we gather them from multiple sources: * ATtRACT43: This resource provides comprehensive information on
370 RBPs and 1,583 RBP consensus binding motifs. The data is extracted and carefully curated from experimentally validated sources such as CISBP-RNA, SpliceAid-F and RBPDB databases. *
SpliceAid44: We gather information from SpliceAid, which encompasses 2,220 target sites associated with 62 human splicing proteins. Additionally, it includes expression data from 320 tissues
per cell. * We also extract the most frequently occurring contiguous nucleobase sequences, ranging from 4 to 8 bp, by scanning the entirety of the RNAcentral database. By incorporating
motifs from these diverse sources, we aim to capture a comprehensive representation of RNA structural elements for our analysis. TYPE-GUIDED FINE-TUNING STRATEGY Given the RNAErnie basic
block pretrained with motif-aware multilevel masking strategies, we need to combine the basic blocks of the RNAErnie transformer with task-specific heads—for example, a fully connected layer
for RNA classification—into a neural network for the downstream task and further train the neural network subject to labelled datasets for the downstream application in a supervised
learning manner. Here, we introduce our proposed type-guided fine-tuning strategy in two parts: neural architectures for tasks and domain-adaptation strategies. NEURAL ARCHITECTURES FOR
FINE-TUNING To adapt various downstream tasks, the RNAErnie approach follows the surgical fine-tuning strategies20 and offers three sets of neural network architectures as follows. FBTH In
the FBTH architecture, given RNA sequences and their labels for a downstream task, the RNAErnie approach simply extracts embeddings of RNA sequences from a pretrained RNAErnie basic block
and then leverages the embeddings as inputs to train a separate task-specific head subject to the downstream tasks. In this way, the parameters in the RNAErnie backbone are frozen, while the
head is trainable. According to ref. 20, this architecture would work well when the downstream tasks are out-of-distribution of pretraining datasets. TBTH In the TBTH architecture, the
RNAErnie approach directly combines the RNAErnie basic block and the task-specific head to construct an end-to-end neural network for downstream tasks and then trains the neural network
using the labelled datasets in a supervised learning manner. In this way, the parameters in both the RNAErnie backbone and the head are trainable. According to ref. 20, this architecture
would work well when the downstream tasks and pretraining datasets are in the same distribution. STACK In the STACK architecture, the RNAErnie approach first leverages an RNAErnie basic
block to predict the top-_K_ most possible coarse-grained RNA types (that is, the _K_ coarse-grained RNA types with the highest probabilities) using the input RNA sequence. Then it stacks an
additional layer of _K_ downstream modules with shared parameters for fine-tuning, where every downstream module refers to a TBTH/FBTH network and is fed with the RNA sequence and a
predicted RNA type for the downstream task. The _K_ downstream modules output _K_ prediction results, and the RNAErnie approach outputs the ensemble of _K_ results as the final outcome. More
specifically, in the STACK architecture, the RNAErnie basic block first predicts the indication of an RNA sequence following the [IND] marker by estimating the probability of the masked
indication token, denoted as _p_(_x_IND∣X; _θ_). From these predictions, the RNAErnie approach selects the top-_K_ indications, denoted as \({I}_{k}\in {{{\mathcal{I}}}}\) for _k_ = 1, ⋯ ,
_K_, along with their corresponding probabilities _σ_1, …, _σ__K_. Each selected indication is then appended to the end of the RNA sequence, resulting in _K_ parallel inputs to the
downstream module. Then the downstream module takes the _K_ parallel inputs simultaneously, enabling ensemble learning through soft majority voting. Specifically, the RNAErnie approach
calculates the weighted sum for soft majority voting as follows: $$\bar{q}=\mathop{\sum }\limits_{k=1}^{K}{\sigma }_{k}{q}_{k},$$ (1) where _q__k_ could be either scalar, vector or matrix
outputs from the downstream module for various downstream tasks (for example, logit vectors for classification tasks or pair-wise feature maps for structural analysis), while \(\bar{q}\)
refers to the weight sum. Note that although we consider the stacking architecture part of our key contributions, FBTH and TBTH sometimes deliver better performance. DOMAIN ADAPTATION TO
DOWNSTREAM TASKS Upon completion of the pretraining phase, the RNAErnie basic block is prepared for type-guided fine-tuning, enabling its application to various downstream tasks. It is
important to emphasize that RNAErnie has the potential to accommodate a diverse array of tasks, extending beyond the examples provided below, through appropriate FBTH, TBTH and STACK
architectures. RNA SEQUENCE CLASSIFICATION RNA sequence classification is a pivotal task that assigns RNA sequences to specific categories. In other words, it maps an RNA sequence X of
length _L_ to scalar labels, which refer to different categories. RNA sequence classification is crucial for understanding their functions and their roles in various biological processes.
Accurate classification of RNA sequences enables researchers to identify ontology and predict functions, which facilitates the development of new therapies and treatments for RNA-related
diseases. Our work leverages STACK with TBTH to classify RNA sequences. It stacks _K_ classification modules: the RNAErnie basic block combined with a trainable MLP as a prediction head.
However, the computational complexity of transformers, which exhibit a quadratic time complexity of \({{{\mathcal{O}}}}({n}^{2}d)\), where _n_ denotes the sequence length, posed challenges
when processing excessively long RNA sequences. To discern lncRNA amidst protein-coding transcripts, we employed a chunk strategy. This strategy entails the division of lengthy RNA sequences
into more manageable segments, which are independently fed into the RNAErnie approach. Subsequently, we aggregate the segment-level logits to obtain the sequence-level logit and employ an
MLP for classification purposes. RNA–RNA INTERACTION PREDICTION RNA–RNA interaction prediction refers to the estimation of interactions between two RNA sequences, such as miRNA and mRNA,
circular RNA and lncRNA. This task maps two RNA sequences, X_a_ of length _L_1 and X_b_ of length _L_2, to binary labels 0/1, where 0 indicates no interaction between the two RNA sequences
and 1 indicates interaction. Accurate prediction of RNA–RNA interactions can provide valuable insights into RNA-mediated regulatory mechanisms and enhance our understanding of biological
processes, including gene expression, splicing and translation45. Our work employs a TBTH architecture, which combines the RNAErnie basic block with a hybrid neural network inspired by ref.
46. This hybrid neural network acts as the interaction prediction head, sequentially incorporating several components: a convolutional neural network, a bidirectional long short-term memory
network and a MLP. Because the types of interacting RNA are fixed, it is unnecessary to employ the STACK architecture for the purpose of RNA–RNA interaction analysis. RNA SECONDARY STRUCTURE
PREDICTION RNA secondary structure prediction determines the probable arrangement of bp within an RNA sequence, which can fold back onto itself and form specific pairings. It maps an RNA
sequence X of length _L_ to a 0/1 matrix with shape _L_ × _L_, where element _i_, _j_ means whether nt _i_ forms bp with nt _j_. The secondary structure of RNA plays a critical role in
understanding its interactions with other molecules and its functional importance. This prediction technique is a valuable tool in molecular biology, aiding in the identification of
potential targets for drug design and enhancing our understanding of gene expression and regulation mechanisms. Our work utilizes the STACK architecture with FBTH to fold RNA sequences. We
combined the RNAErnie basic block with a folding neural network inspired by the methodology described in ref. 47. It computes four distinct folding scores—helix stacking, unpaired region,
helix opening and helix closing—for each pair of nt bases. Subsequently, we utilize a Zuker-style dynamic programming approach48 to predict the most favourable secondary structure. This is
achieved by maximizing the cumulative scores of adjacent loops, following a systematic and rigorous computational procedure. To facilitate the training of our deep neural network, we adopt
the max-margin framework. Within this framework, the network minimizes the structured hinge loss function while incorporating thermodynamic regularization. HYPERPARAMETERS AND CONFIGURATIONS
During the pretraining phase, our model underwent approximately 2,580,000 steps of training, with a batch size set to 50 and a maximum sequence length for ERNIE limited to 512. We utilized
the AdamW optimizer, which was regulated by a learning-rate schedule involving anneal warm-up and decay. The initial learning rate was set at 1 × 10−4, with a minimum learning rate of 5 ×
10−5. The learning-rate scheduler was designed to warm up during the first 5% of the steps and then decay in the final 5% of the steps. In terms of masking strategies, we maintained a
proportion of 1:1:1 across the three different masking levels, with the training algorithm randomly selecting one strategy for each training session. The pretraining was conducted on four
Nvidia Tesla V100 32 GB graphics processing units, taking around 250 hours to reach convergence. Here, in additional to the hyperparameters for pretraining, we introduce the configurations
of variant pretrained models derived from RNAErnie and used in experiments: * Ernie-base: this model represents the vanilla ERNIE architecture without any pretraining on RNA sequence
datasets. It underwent standard fine-tuning. * RNAErnie−−: in this model, only base-level masking was employed during the pretraining phase of the RNAErnie family. It was then fine-tuned
using the standard approach. * RNAErnie−: the RNAErnie family model with both base and subsequence-level masking during pretraining, followed by standard fine-tuning. * RNAErnie: this model
encompasses the complete set of masking strategies, including base, subsequence and motif-level masking during pretraining. It was fine-tuned using the TBTH or FBTH architecture. *
RNAErnie+: the most comprehensive model in the RNAErnie family, incorporating all three levels of masking during pretraining and the STACK architecture. * RNAErnie without chunk: this model
truncates RNA sequences and discards any remaining segments when classifying long RNA sequences, specifically lncRNA (for example, in lnRC_H and lnRC_M datasets) alongside protein-encoding
transcripts. DATA AVAILABILITY The datasets used for pretraining and fine-tuning are all derived from previous studies. Here we include the official links. Note that the lncRNA_H and
lncRNA_M datasets are used for long-sequence classification in the Supplementary Information. RNAcentral16: https://ftp.ebi.ac.uk/pub/databases/RNAcentral/releases/21.0/; ATtRACT43:
https://attract.cnic.es/download; SpliceAid44: http://193.206.120.249/cgi-bin/SpliceAid.pl?sites=Download; nRC25: http://tblab.pa.icar.cnr.it/public/nRC/paper_dataset/; lncRNA_H49:
https://www.gencodegenes.org/human/release_25.html; lncRNA_M49: https://www.gencodegenes.org/mouse/; DeepMirTar27: https://github.com/tjgu/miTAR/tree/master/scripts_data_models; ArchiveII36:
https://rna.urmc.rochester.edu/publications.html; RNAStrAlign35: https://github.com/mxfold/mxfold2/releases/tag/v0.1.0; bpRNA37: https://bprna.cgrb.oregonstate.edu/download.php#bpRNA.
Source data are provided with this paper. CODE AVAILABILITY We built RNAErnie using Python and the PaddlePaddle deep learning framework. The code repository of RNAErnie, readme files and
tutorials are all available at ref. 50. A docker image with configured environments and dependent libraries is available for download at ref. 51. To compare pretrained RNA language
baselines, see the code repository at ref. 52. REFERENCES * Kukurba, K. & Montgomery, S. RNA sequencing and analysis. _Cold Spring Harb. Protoc._ 2015, pdb–top084970 (2015). Article
Google Scholar * Conesa, A. et al. A survey of best practices for RNA-seq data analysis. _Genome Biol._ 17, 1–19 (2016). Google Scholar * Dharmadhikari, S., Ingle, M. & Kulkarni, P.
Empirical studies on machine learning based text classification algorithms. _Adv. Comput._ 2, 161 (2011). Google Scholar * Zheng, S., Li, Y., Chen, S., Xu, J. & Yang, Y. Predicting
drug-protein interaction using quasi-visual question answering system. _Nat. Mach. Intell._ 2, 134–140 (2020). Article Google Scholar * Min, B. et al. Recent advances in natural language
processing via large pre-trained language models: a survey. _ACM Comput. Surv._ 56, 1–40 (2021). * Kenton, J. & Toutanova, L. BERT: pre-training of deep bidirectional transformers for
language understanding. In _Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies_ (eds Burstein, J. et al.)
4171–4186 (Association for Computational Linguistics, 2019). * Yu, X., Jiang, L., Jin, S., Zeng, X. & Liu, X. preMLI: a pre-trained method to uncover microRNA-lncRNA potential
interactions. _Brief. Bioinform._ 23, bbab470 (2022). Article Google Scholar * Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. DNABERT: pre-trained bidirectional encoder representations from
transformers model for DNA-language in genome. _Bioinformatics_ 37, 2112–2120 (2021). Article Google Scholar * Leksono, M. & Purwarianti, A. Sequential labelling and DNABERT For splice
site prediction in Homo Sapiens DNA. Preprint at https://arXiv.org/quant-ph/2212.07638 (2022). * Zhou, Z. et al. DNABERT-2: efficient foundation model and benchmark for multi-species
genome. In _Twelfth International Conference on Learning Representations_ (2024). * Altenburg, T., Giese, S., Wang, S., Muth, T. & Renard, B. Ad hoc learning of peptide fragmentation
from mass spectra enables an interpretable detection of phosphorylated and cross-linked peptides. _Nat. Mach. Intell._ 4, 378–388 (2022). Article Google Scholar * Raffel, C. et al.
Exploring the limits of transfer learning with a unified text-to-text transformer. _J. Mach. Learn. Res._ 21, 5485–5551 (2020). MathSciNet Google Scholar * Liang, S. et al. Rm-LR: a
long-range-based deep learning model for predicting multiple types of RNA modifications. _Comput. Biol. Med._ 164, 107238 (2023). Article Google Scholar * Chen, J. et al. Interpretable RNA
foundation model from unannotated data for highly accurate RNA structure and function predictions. Preprint at _bioRxiv_ https://doi.org/10.1101/2022.08.06.503062 (2022). * Holbrook, S. RNA
structure: the long and the short of it. _Curr. Opin. Struct. Biol._ 15, 302–308 (2005). Article Google Scholar * Sweeney, B. et al. RNAcentral 2021: secondary structure integration,
improved sequence search and new member databases. _Nucleic Acids Res._ 49, D212–D220 (2021). * Leontis, N., Lescoute, A. & Westhof, E. The building blocks and motifs of RNA
architecture. _Curr. Opin. Struct. Biol._ 16, 279–287 (2006). Article Google Scholar * Fierro-Monti, I. & Mathews, M. Proteins binding to duplexed RNA: one motif, multiple functions.
_Trends Biochem. Sci._ 25, 241–246 (2000). Article Google Scholar * Butcher, S. & Pyle, A. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and
networks. _Acc. Chem. Res._ 44, 1302–1311 (2011). Article Google Scholar * Lee, Y. et al. Surgical fine-tuning improves adaptation to distribution shifts. In _Eleventh International
Conference on Learning Representations_ (2023). * Moon, K. et al. Visualizing structure and transitions in high-dimensional biological data. _Nat. Biotechnol._ 37, 1482–1492 (2019). Article
Google Scholar * Kirk, J. et al. Functional classification of long non-coding RNAs by k-mer content. _Nat. Genet._ 50, 1474–1482 (2018). Article Google Scholar * Akiyama, M. &
Sakakibara, Y. Informative RNA base embedding for RNA structural alignment and clustering by deep representation learning. _NAR Genom. Bioinform._ 4, lqac012 (2022). Article Google Scholar
* Zhang, Y. et al. Multiple sequence alignment-based RNA language model and its application to structural inference. _Nucleic Acids Res._ 52, e3–e3 (2024). Article Google Scholar *
Fiannaca, A., La Rosa, M., La Paglia, L., Rizzo, R. & Urso, A. nRC: non-coding RNA classifier based on structural features. _BioData Min._ 10, 1–18 (2017). Article Google Scholar *
Nawrocki, E. et al. Rfam 12.0: updates to the RNA families database. _Nucleic Acids Res._ 43, D130–D137 (2015). Article Google Scholar * Wen, M., Cong, P., Zhang, Z., Lu, H. & Li, T.
DeepMirTar: a deep-learning approach for predicting human miRNA targets. _Bioinformatics_ 34, 3781–3787 (2018). Article Google Scholar * Pla, A., Zhong, X. & Rayner, S. miRAW: a deep
learning-based approach to predict microRNA targets by analyzing whole microRNA transcripts. _PLoS Comput. Biol._ 14, e1006185 (2018). Article Google Scholar * Enright, A. et al. MicroRNA
targets in Drosophila. _Genome Biol._ 4, 1–27 (2003). Article Google Scholar * Krüger, J. & Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. _Nucleic Acids
Res._ 34, W451–W454 (2006). Article Google Scholar * Pita, T., Feliciano, J. & Leitão, J. Identification of Burkholderia cenocepacia non-coding RNAs expressed during Caenorhabditis
elegans infection. _Appl. Microbiol. Biotechnol._ 107, 3653–3671 (2023). * Agarwal, V., Bell, G., Nam, J. & Bartel, D. Predicting effective microRNA target sites in mammalian mRNAs.
_eLife_ 4, e05005 (2015). Article Google Scholar * Ding, J., Li, X. & Hu, H. TarPmiR: a new approach for microRNA target site prediction. _Bioinformatics_ 32, 2768–2775 (2016). Article
Google Scholar * Fu, L. et al. UFold: fast and accurate RNA secondary structure prediction with deep learning. _Nucleic Acids Res._ 50, e14–e14 (2022). Article Google Scholar * Tan, Z.,
Fu, Y., Sharma, G. & Mathews, D. TurboFold II: RNA structural alignment and secondary structure prediction informed by multiple homologs. _Nucleic Acids Res._ 45, 11570–11581 (2017).
Article Google Scholar * Sloma, M. & Mathews, D. Exact calculation of loop formation probability identifies folding motifs in RNA secondary structures. _RNA_ 22, 1808–1818 (2016).
Article Google Scholar * Danaee, P. et al. bpRNA: large-scale automated annotation and analysis of RNA secondary structure. _Nucleic Acids Res._ 46, 5381–5394 (2018). Article Google
Scholar * Sun, Y. et al. Ernie 2.0: a continual pre-training framework for language understanding. In _Proc. AAAI Conference on Artificial Intelligence 34_ (eds Wooldridge, M., Dy, J. &
Natarajan, S.) 8968–8975 (AAAI, 2020). * Vaswani, A. et al. Attention is all you need. In _Proc. Advances in Information Processing Systems 30_ (eds Guyon, I. et al.) 5999–6009 (NeurIPS,
2017). * Karpicke, J. D., Lehman, M. & Aue, W. R. Retrieval-based learning: an episodic context account. In _Psychology of Learning and Motivation_ Vol. 61, 237–284 (Academic Press,
2014). * Joshi, M. et al. SpanBERT: improving pre-training by representing and predicting spans. _Trans. Assoc. Comput. Linguist._ 8, 64–77 (2020). Article Google Scholar * Wu, R. et al.
High-resolution de novo structure prediction from primary sequence. Preprint at _bioRxiv_ https://doi.org/10.1101/2022.07.21.500999 (2022). * Giudice, G., Sánchez-Cabo, F., Torroja, C. &
Lara-Pezzi, E. ATtRACT—a database of RNA-binding proteins and associated motifs. _Database_ 2016, baw035 (2016). * Piva, F., Giulietti, M., Burini, A. & Principato, G. SpliceAid 2: a
database of human splicing factors expression data and RNA target motifs. _Hum. Mutat._ 33, 81–85 (2012). Article Google Scholar * Fang, Y., Pan, X. & Shen, H. Recent deep learning
methodology development for RNA-RNA interaction prediction. _Symmetry_ 14, 1302 (2022). Article Google Scholar * Gu, T., Zhao, X., Barbazuk, W. & Lee, J. miTAR: a hybrid deep
learning-based approach for predicting miRNA targets. _BMC Bioinform._ 22, 1–16 (2021). Article Google Scholar * Sato, K., Akiyama, M. & Sakakibara, Y. RNA secondary structure
prediction using deep learning with thermodynamic integration. _Nat. Commun._ 12, 1–9 (2021). Article Google Scholar * Zuker, M. & Stiegler, P. Optimal computer folding of large RNA
sequences using thermodynamics and auxiliary information. _Nucleic Acids Res._ 9, 133–148 (1981). Article Google Scholar * Frankish, A. et al. GENCODE 2021. _Nucleic Acids Res._ 49,
D916–D923 (2021). Article Google Scholar * Ning, W. CatIIIIIIII/RNAErnie: v.1.0. _Zenodo_ https://doi.org/10.5281/zenodo.10847621 (2024). * Ning, W. RNAErnie docker. _Zenodo_
https://doi.org/10.5281/zenodo.10847856 (2024). * Ning, W. CatIIIIIIII/RNAErnie_baselines: v.1.0.0. _Zenodo_ https://doi.org/10.5281/zenodo.10851577 (2024). * Panwar, B., Arora, A. &
Raghava, G. Prediction and classification of ncRNAs using structural information. _BMC Genomics_ 15, 1–13 (2014). Article Google Scholar * Wang, L. et al. ncRFP: a novel end-to-end method
for non-coding RNAs family prediction based on deep learning. _IEEE/ACM Trans. Comput. Biol. Bioinform._ 18, 784–789 (2020). Article Google Scholar * Deng, C. et al. RNAGCN: RNA tertiary
structure assessment with a graph convolutional network. _Chin. Phys. B_ 31, 118702 (2022). Article Google Scholar * Chantsalnyam, T., Lim, D., Tayara, H. & Chong, K. ncRDeep:
non-coding RNA classification with convolutional neural network. _Comput. Biol. Chem._ 88, 107364 (2020). Article Google Scholar * Chantsalnyam, T., Siraj, A., Tayara, H. & Chong, K.
ncRDense: a novel computational approach for classification of non-coding RNA family by deep learning. _Genomics_ 113, 3030–3038 (2021). Article Google Scholar * Reuter, J. & Mathews,
D. RNAstructure: software for RNA secondary structure prediction and analysis. _BMC Bioinform._ 11, 1–9 (2010). Article Google Scholar * Andronescu, M., Aguirre-Hernandez, R., Condon, A.
& Hoos, H. RNAsoft: a suite of RNA secondary structure prediction and design software tools. _Nucleic Acids Res._ 31, 3416–3422 (2003). Article Google Scholar * Lorenz, R. et al.
ViennaRNA package 2.0. _Algorithms Mol. Biol._ 6, 1–14 (2011). Article Google Scholar * Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. _Nucleic Acids
Res._ 31, 3406–3415 (2003). Article Google Scholar * Huang, L. et al. LinearFold: linear-time approximate RNA folding by 5′-to-3′ dynamic programming and beam search. _Bioinformatics_ 35,
i295–i304 (2019). Article Google Scholar * Wayment-Steele, H. K. et al. RNA secondary structure packages evaluated and improved by high-throughput experiments. _Nat. Methods_ 19, 1234–1242
(2022). Article Google Scholar * Chen, X., Li, Y., Umarov, R., Gao, X. & Song, L. RNA secondary structure prediction by learning unrolled algorithms. In _International Conference on
Learning Representations_ (2020). * Do, C., Woods, D. & Batzoglou, S. CONTRAfold: RNA secondary structure prediction without physics-based models. _Bioinformatics_ 22, e90–e98 (2006).
Article Google Scholar * Zakov, S., Goldberg, Y., Elhadad, M. & Ziv-Ukelson, M. Rich parameterization improves RNA structure prediction. _J. Comput. Biol._ 18, 1525–1542 (2011).
Article MathSciNet Google Scholar Download references ACKNOWLEDGEMENTS This work is kindly supported by the National Science and Technology Major Project under grant no. 2021ZD0110303
(N.W., J.B., X.L. and H.X.) and the National Science Foundation of China under grant no. 62141220 (Y.L. and L.K.). AUTHOR INFORMATION Author notes * These authors contributed equally: Ning
Wang, Jiang Bian, Haoyi Xiong. AUTHORS AND AFFILIATIONS * Big Data Lab, Baidu Inc., Beijing, China Ning Wang, Jiang Bian, Yuchen Li, Xuhong Li & Haoyi Xiong * Department of Computer
Science, City University of Hong Kong, Hong Kong, China Ning Wang * Department of Computer Science and Engineering, Shanghai Jiao Tong University, Shanghai, China Yuchen Li & Linghe Kong
* Department of Computer Science, Nottingham Trent University, Nottingham, UK Shahid Mumtaz * Department of Applied Informatics, Silesian University of Technology, Gliwice, Poland Shahid
Mumtaz Authors * Ning Wang View author publications You can also search for this author inPubMed Google Scholar * Jiang Bian View author publications You can also search for this author
inPubMed Google Scholar * Yuchen Li View author publications You can also search for this author inPubMed Google Scholar * Xuhong Li View author publications You can also search for this
author inPubMed Google Scholar * Shahid Mumtaz View author publications You can also search for this author inPubMed Google Scholar * Linghe Kong View author publications You can also search
for this author inPubMed Google Scholar * Haoyi Xiong View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS All authors made contributions to
this paper. N.W. and J.B. conducted experiments and wrote part of the paper. Y.L., X.L. and S.M. were involved in the discussion and wrote part of the paper. L.K. oversaw the research
progress, was involved in the discussion and wrote part of the paper. H.X. oversaw the research progress, designed the study and experiments, was involved in the discussion and wrote the
paper. H.X. is the senior author, and L.K. is the co-senior contributor. CORRESPONDING AUTHORS Correspondence to Linghe Kong or Haoyi Xiong. ETHICS DECLARATIONS COMPETING INTERESTS The
authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Machine Intelligence_ thanks Xiangfu Zhong and the other, anonymous, reviewer(s) for their contribution to
the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
EXTENDED DATA EXTENDED DATA FIG. 1 THE FIGURE ILLUSTRATES THE USE OF A SPECIAL ‘[IND]’ TOKEN FOLLOWED BY THE RNACENTRAL INSTANCE TYPE AS AN INDICATOR. During the pre-training phase, the
instance type is masked out and RNAErnie attempts to predict it. In downstream tasks, a two-stage pipeline is employed, which aggregates the top-K predicted indicators to improve
performance. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Related work, Supplementary Figs. 1 and 2, and results and analysis. SOURCE DATA SOURCE DATA FIG. 1 Scatter coordinates and
labels. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Wang, N., Bian, J., Li, Y. _et al._ Multi-purpose RNA language modelling with motif-aware pretraining and type-guided fine-tuning. _Nat Mach Intell_ 6, 548–557 (2024).
https://doi.org/10.1038/s42256-024-00836-4 Download citation * Received: 19 August 2023 * Accepted: 10 April 2024 * Published: 13 May 2024 * Issue Date: May 2024 * DOI:
https://doi.org/10.1038/s42256-024-00836-4 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative