- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Sterile inflammation resulting in an altered immune response is a key determinant of renal injury in diabetic nephropathy (DN). In this investigation, we evaluated the hypothesis
that hyperglycemic conditions augment the pro-inflammatory immune response in the kidney by promoting podocyte-specific expression of the stress response protein regulated in development and
DNA damage response 1 (REDD1). In support of the hypothesis, streptozotocin (STZ)-induced diabetes increased REDD1 protein abundance in the kidney concomitant with renal immune cell
infiltration. In diabetic mice, administration of the SGLT2 inhibitor dapagliflozin was followed by reductions in blood glucose concentration, renal REDD1 protein abundance, and immune cell
infiltration. In contrast with diabetic REDD1+/+ mice, diabetic REDD1−/− mice did not exhibit albuminuria, increased pro-inflammatory factors, or renal macrophage infiltration. In cultured
human podocytes, exposure to hyperglycemic conditions promoted REDD1-dependent activation of NF-κB signaling. REDD1 deletion in podocytes attenuated both the increase in chemokine expression
and macrophage chemotaxis under hyperglycemic conditions. Notably, podocyte-specific REDD1 deletion prevented the pro-inflammatory immune cell infiltration in the kidneys of diabetic mice.
Furthermore, exposure of podocytes to hyperglycemic conditions promoted REDD1-dependent pyroptotic cell death, evidenced by an NLRP3-mediated increase in caspase-1 activity and LDH release.
REDD1 expression in podocytes was also required for an increase in pyroptosis markers in the glomeruli of diabetic mice. The data support that podocyte-specific REDD1 is necessary for
chronic NF-κB activation in the context of diabetes and raises the prospect that therapies targeting podocyte-specific REDD1 may be helpful in DN. SIMILAR CONTENT BEING VIEWED BY OTHERS
CASPASE-11/4 AND GASDERMIN D-MEDIATED PYROPTOSIS CONTRIBUTES TO PODOCYTE INJURY IN MOUSE DIABETIC NEPHROPATHY Article 23 September 2020 CERS6 LINKS CERAMIDE METABOLISM TO INNATE IMMUNE
RESPONSES IN DIABETIC KIDNEY DISEASE Article Open access 11 February 2025 ABCA1 DEFICIENCY CONTRIBUTES TO PODOCYTE PYROPTOSIS PRIMING VIA THE APE1/IRF1 AXIS IN DIABETIC KIDNEY DISEASE
Article Open access 14 June 2023 INTRODUCTION Diabetic nephropathy (DN) is one of the major complications in people with type 1 or type 2 diabetes and the most common cause of end-stage
kidney disease [1]. As per the 2022 United States Renal Data System (USRDS) annual report, the prevalence of diabetes has increased to 35.6% in patients with chronic kidney complications
[2]. Despite the improved prognosis of diabetes with the advent of sodium glucose cotransporter 2 (SGLT2) inhibitors and glucagon-like peptide-1 receptor agonists, a great number of patients
with diabetes still develop renal failure and the risk of death remains high [3]; so much so that diabetes and chronic kidney disease are among the top 10 causes of mortality in the US [4].
This is in part due to a lack of understanding of the specific molecular events that contribute to diabetes-induced renal pathology. Although the etiology of DN is complicated and
multifactorial, it is widely accepted that inflammation plays a critical role in renal dysfunction. DN is often viewed as a chronic inflammatory disease and the role of innate and adaptive
immune responses are recognized as important etiological components of DN pathogenesis [5]. The transcription factor nuclear factor κ-light-chain enhancer of activated B cells (NF-κB) plays
a major role in mediating inflammation and immune function, with enhanced activity seen in the kidneys of diabetic patients and in preclinical models of diabetes [6, 7]. A complex network of
extracellular perturbagens and signaling pathways are regulated by the NF-κB family of pleiotropic transcription factors [RelA (p65), RelB, c-Rel, p50, and p52] that act to promote the
expression of a variety of pro-inflammatory cytokines, such as TNFα, IL-6, and IL-1β, as well as chemokines like CCL2 and RANTES. Inflammatory cytokines not only regulate immune responses,
but are also cardinal effectors of renal injury. Increased circulating and local renal expression of pro-inflammatory cytokines and chemokines have been reported in diabetic patients and are
associated with albuminuria and clinical markers of renal injury [8, 9]. Sterile inflammation in DN is often attributed to activation of the NOD-like receptor (NLR) family pyrin domain
containing 3 (NLRP3) inflammasome complex in response to metabolic stimuli associated with diabetes [10, 11]. The NLRP3 inflammasome is unique among inflammasome complexes because it is
sensitive to a wide range of stimuli. When activated, the NLRP3 inflammasome complex acts to not only process the maturation of interleukin (IL)-1β that contributes to an inflammatory
response, but also causes pyroptosis, a pro-inflammatory form of lytic cell death [12]. Activation of the NLRP3 inflammasome has been closely linked with diabetic kidney disease in humans
and in preclinical experimental models [10, 11, 13, 14]. NLRP3 inflammasome activation has been reported in a variety of renal cells including podocytes [14]. In fact, recent evidence
supports immune cell-like functions of podocytes, as they produce chemokines that recruit immune cells and cytokines that drive their differentiation [11, 14, 15]. Shazad et al. demonstrated
that suppression of NLRP3 specifically in podocytes is sufficient to attenuate renal injury and dysfunction in diabetic mice [11]. However, the specific signaling events that promote NLRP3
inflammasome activation and consequently the canonical and non-canonical signaling that is triggered in the context of DN remain to be fully defined. The stress response protein REDD1
(Regulated in Development and DNA Damage 1; also known as DDIT4 or RTP801) is upregulated in the kidneys of diabetic patients, as well as in preclinical rodent models of diabetes [16, 17].
Our laboratory recently demonstrated that whole-body REDD1 deletion is sufficient to prevent albuminuria, renal injury, and podocyte loss in diabetic mice [16]. The significance of this
observation is supported by a strong positive correlation between clinical indicators of renal disease and kidney REDD1 protein content in diabetic patients [17]. However, the mechanism
responsible for reno-protection in diabetic REDD1 knockout mice remains to be fully established. Notably, REDD1 is required for the development of diabetes-associated inflammation [18].
REDD1 has been shown to promote and sustain NF-κB activation, thereby upregulating pro-inflammatory and immune responses in the context of disease [19,20,21,22,23,24]. To date, a role for
REDD1 in diabetes-induced immune responses in the kidney has not been fully elucidated. Studies herein investigated the role of REDD1-dependent signaling in the development of renal
inflammation. MATERIAL AND METHODS ANIMAL EXPERIMENTS All procedures adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and ARRIVE guidelines, and
were approved by the Penn State College of Medicine Institutional Animal Care and Use Committee. Mice were maintained as littermate cages (4 mice/cage) on a 12:12 h reverse light dark cycle
with ad libitum access to food and water. At 6 weeks of age, littermate cages were randomly divided into treatment groups. Diabetes was induced in male mice by administering 50 mg/kg
streptozotocin (STZ; Sigma Aldrich, St. Louis, MO, US) intraperitoneally (ip) over 5 consecutive days, and phenotype was confirmed by fasting blood glucose concentrations >250 mg/dL.
Non-diabetic mice received sodium citrate buffer (0.01 M, pH 6) as a vehicle (Veh) control. Hyperglycemia was controlled via daily administration of 1 mg/kg Dapagliflozin (DG in 0.1% DMSO,
ip; Selleck Chemicals, Houston, TX, USA) for 2 weeks beginning 14 weeks post-diabetes induction [25]. Male B6;129 REDD1+/+ and REDD1−/− mice [26] were made diabetic as described.
Podocyte-specific REDD1 knockout (PodKO) mice were generated by crossing hemizygous B6.Cg-Tg(NPHS2-cre)295Lbh/J (Stock #008205; The Jackson Laboratory, Bar Harbor, ME, USA) with REDD1fl/fl
mice [27] and administered STZ or Veh as described above. At 16 weeks of diabetes, all treatment indicators were removed from cages by a technician and after 4 h of fasting, one mouse from
each group was euthanized under isoflurane anesthesia in a random order. Urine was collected from the bladder, and kidneys were analyzed as described below. Urine albumin and creatinine
levels were measured as previously described [16]. IMMUNOHISTOLOGY Renal sections (6 µm) were cut from 10% formalin-fixed, paraffin-embedded (FFPE) kidneys and processed for
immunohistochemistry (IHC) or immunofluorescence (IF) staining as described previously [16]. Antibodies are listed in Table S1. For IHC, sections were incubated with ImmPRESS HRP-conjugated
secondary antibody and detected using 3,3ʹ-diaminobenzidine (Vector Laboratories, Newark, CA, USA). Tissue sections were counterstained with hematoxylin, mounted and micrographs were
captured using an AmScope T720Q compound microscope (AmScope, Irvine, CA, USA). For IF labeling, tissue sections were incubated with appropriate secondary antibodies (Table S1) and
counterstained with 1.6 μmol/L Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA, USA). Immunocytochemistry analysis was done as previously described [28]. Cells were fixed with 4% PFA,
permeabilized in 0.1% triton X-100, blocked with 5% BSA and then incubated with appropriate antibodies (Table S1). Cell nuclei were counterstained with 1 μM DAPI (Invitrogen, Carlsbad, CA,
USA). All slides were mounted with Fluoromount aqueous mounting media (Sigma-Aldrich) and imaged with a Leica SP8 confocal laser microscope (Leica, Deerfield, IL, USA) using frame-stack
sequential scanning. FLOW CYTOMETRY For flow cytometric analysis, cell suspensions of kidney tissue were made using Liberase digestion as previously described [29]. Briefly, kidneys were
washed in cold PBS, minced finely and then shaken for 30 min at 37 °C in serum free RPMI medium containing 100 µg/ml Liberase (Roche, Basel, Switzerland) enzyme. Digestion was stopped by
adding RPMI medium containing 10% fetal bovine serum, centrifuged, and resuspended in PBS. Cell debris was separated using Debris Removal Solution (Miltenyi Biotec, Bergisch Gladbach,
Germany). Erythrocytes were lysed using RBC Lysis Buffer (eBioscience, San Diego, CA, USA) and washed with PBS. Samples were resuspended in Cell Staining Buffer (Biolegend, San Diego, CA,
USA) and incubated in Fc Block (BD Biosciences, Franklin Lakes, NJ, USA) for 15 min prior to labeling with antibodies (Table S1) for 45 min. After washing, fixed cells were analyzed by flow
cytometry using a BD LSRFortessa (BD Biosciences) instrument in Penn State College of Medicine’s Flow Cytometry Core (RRID:SCR_021134). CELL CULTURE Conditionally immortalized human
podocytes (CIHP-1) were cultured as previously described [16]. CIHP-1 cells were cultured in RPMI 1640 media at 33 °C in 5% CO2, then differentiated for 10 days at 37 °C in 5% CO2 before
treatments. Human leukemia monocytic THP-1 cells (ATCC TIB-202) were differentiated into macrophages using 50 ng/mL phorbol 12-myristate-13-acetate (Cayman Chemicals, Ann Arbor, MI, USA) for
48 h. CRISPR/Cas9 genome editing was used to generate a stable CIHP-1 REDD1 knockout (REDD1 KO) cell line [16]. Plasmids including a pCMV5 vector (EV), HA-tagged pCMV-HA-REDD1, or
pBabe-GFP-IκB alpha-mut (IκBα super repressor; Addgene plasmid # 15264) were transiently transfected using Jet PRIME (Polyplus transfection, New York, NY, USA). To model hyperglycemia, cells
were exposed to culture medium containing 30 mM glucose (HG) versus 5 mM glucose supplemented with 25 mM mannitol as an osmotic control (OC). Lactate dehydrogenase (LDH) released into cell
culture supernatant was quantified after 48 h HG by LDH Cytotoxicity Assay Kit (Cayman Chemicals) following manufacturer’s instructions. To quantify NF-κB activity, differentiated CIHP-1
cells were co-transfected with the Renilla luciferase (Promega, Madison, WI, USA) and NF-κB-TATA-luciferase [30] plasmids. After 24 h, cells were exposed to hyperglycemic conditions, and
luciferase activity was measured. PROTEIN ANALYSIS Nuclear or total proteins were extracted from cells or renal cortical tissue. Western blot analysis was carried out as previously described
[19] with the appropriate antibodies (Table S1). Uncropped western blots are presented in the supplemental information. IL-1β protein was determined in culture medium or kidney homogenates
by ELISA (DuoSet ELISA, R&D systems, Minneapolis, MN, USA). CCL2 recombinant protein (R&D systems, Minneapolis, MN, USA) was subjected to western blotting and CCL2 protein in cell
(105 cells) and tissue lysates were quantified (Fig. S1B,D & E). Nuclear NF-κB activity was quantified in renal tissue using a NF-κB p65 DNA-binding ELISA (TransAM NF-κB p65; Active
Motif, Carlsbad, CA, USA). The FAM-FLICA Caspase-1 Kit (Bio-Rad Laboratories, Hercules, CA, USA) was used to detect active caspase-1 in podocyte cultures. Slides were counterstained with
Hoechst 33342 (Thermo fisher Scientific), mounted, and imaged using a Leica SP8 confocal microscope (Leica Microsystems). CHROMATIN IMMUNOPRECIPITATION NF-κB p65 binding to the promoter
region of the _CCL2_ gene was determined by performing chromatin-immunoprecipitation (ChIP) using a Simple ChIP Plus Enzymatic ChIP Kit (Cell Signaling, Danvers, MA, USA) and quantitative
PCR (ChIP-qPCR). Chromatin was crosslinked with proteins from CIHP-1 podocytes and the protein-chromatin complex was disrupted by ultrasonication. The soluble chromatin was subjected to an
overnight incubation at 4 °C with either anti-p65 antibody or IgG (negative control), and protein G magnetic beads (Cell Signaling) were used for IP. RT-PCR analysis of the recovered DNA was
performed with CCL2 primers (Table S2) encompassing the region of the human CCL2 promoter ( − 209 to −9; RefSeq accession NM_002982) [31]. Fold enrichment adjusted to the IgG controls was
used to tabulate the results [32]. PCR ANALYSIS Total RNA was extracted, reverse transcribed, and subjected to quantitative real-time PCR (QuantStudio 12 K Flex Real-Time PCR System, Thermo
Fisher Scientific, RRID:SCR_021098) with primers listed in Table S2. Mean cycle threshold values were determined. Change in mRNA expression relative to GAPDH mRNA was calculated. TRANSWELL
MIGRATION ASSAY Migration of THP-1 cells across Transwell inserts was measured as described [33]. Differentiated CIHP-1 were seeded into the lower chamber of the Transwell system (Corning,
Kennebunk, ME, USA) and exposed to hyperglycemic conditions or osmotic control. Activated THP-1 cells were transferred into the top chamber of the inserts and allowed to migrate for 24 h.
Cells attached to the bottom surface were fixed, stained, and imaged using a Nikon Eclipse TS100 inverted microscope (Nikon Instruments, Melville, NY, USA). Ten fields of view were imaged
per sample, the number of migrated cells was quantified using ImageJ software, and counts were manually verified. STATISTICAL ANALYSIS Based on A priori power analysis of urinary albumin:
creatinine ratio (ACR) in diabetic vs non-diabetic wild type mice from prior works [16, 34], an N of 4 was determined to yield statistically significant results (Effect size d = 2, α = 5%).
Data are expressed as mean ± SD. Statistical analysis of data with more than two groups were analyzed by one-way or two-way ANOVA, with Tukey’s test for multiple comparisons used for
pairwise analysis. The relationships between urine albumin to creatinine ratio (ACR) and blood glucose levels were tested by Spearman’s correlation analysis. Significance was defined as p
< 0.05 for all analyses. Sample size for each experimental group and exact p-values for significantly different groups are listed in Table S3. Model assumptions were checked using the
Shapiro-Wilk normality test and by visual inspection of residual plots. RESULTS DIABETES-INDUCED HYPERGLYCEMIA PROMOTED RENAL REDD1 CONTENT AND IMMUNE CELL INFILTRATION IN THE KIDNEY As
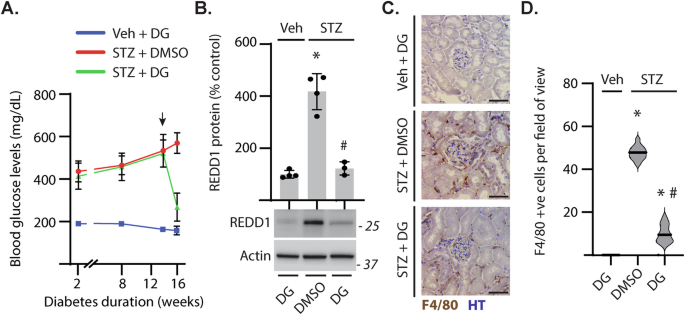
compared to non-diabetic mice, fasting blood glucose levels were elevated in STZ-induced diabetic mice. Treatment with DG was followed by reduced hyperglycemia (Fig. 1A). STZ-diabetes
increased REDD1 protein content in renal cortical homogenates (Fig. 1B) and enhanced renal immune cell infiltration (Fig. 1C, D). When diabetic mice were treated with DG, REDD1 protein
content and immune cell infiltration in the kidneys were reduced (Fig. 1B–D). Together the data supports a role for diabetes-induced hyperglycemia in promoting renal REDD1 protein abundance
and activation of the immune response in the kidneys. REDD1 ABLATION ATTENUATED THE DIABETES-INDUCED RENAL PRO-INFLAMMATORY RESPONSE To evaluate the role of REDD1 in renal inflammation,
wild-type and REDD1-deficient mice were administered STZ. REDD1 protein in the kidneys of STZ-diabetic REDD1+/+ mice was increased compared to non-diabetic controls (Fig. 2A). Elevated blood
glucose levels were observed in both genotypes with STZ administration; however, a positive correlation between blood glucose concentrations with urine ACR was only observed in REDD1+/+ but
not REDD1−/− mice (Fig. 2B; Pearson r: REDD1+/+ = 0.77 _vs_ REDD1−/− = 0.4) [16]. Global REDD1 deletion attenuated STZ-diabetes induced renal mRNA expression of proinflammatory genes
including _Ccl5, Vegfa_, and _Icam-1_ (Fig S1A). Monocyte chemoattractant protein-1 (MCP-1/CCL2) is a ligand of C-C motif chemokine receptor 2 (CCR-2) and a predictive biomarker for the
development of DN [35]. Diabetes increased _Ccl2_ mRNA expression (Fig. 2C) and protein levels (Fig. 2D, S1B) in the kidneys of REDD1+/+ mice. By contrast, CCL2 was not increased in the
kidneys of diabetic REDD1−/− mice. Similarly, diabetes also increased renal interleukin 1β mRNA (Fig. 2E) and protein (Fig. 2F) in a REDD1-dependent manner. The data support that REDD1 is
necessary for enhanced renal inflammatory cytokine and chemokine expression in DN. REDD1 DELETION ATTENUATED RENAL IMMUNE CELL INFILTRATION IN DIABETIC MICE Due to the critical role of
macrophage infiltration in DN pathogenesis [36], renal immune cell infiltrates were evaluated by labeling for F4/80 (Fig. 3A). The number of F4/80+ cells in the renal cortex of diabetic
REDD1+/+ mice was increased as compared to non-diabetic controls (Fig. 3B). Diabetes-induced immune cell infiltration was absent in REDD1−/− mice. Infiltrating immune cells were
characterized by flow cytometry (Fig. 3C, S2). STZ-diabetes increased infiltrating CD45+ cells within the kidney (Fig. 3D), and REDD1 deletion prevented this effect. Within the population of
CD45+ cells, an increase in CD11b + F4/80+ macrophages was observed in the kidneys of diabetic REDD1+/+ mice, but not in diabetic REDD1−/− mice (Fig. 3E). Macrophages were further
characterized based on polarization as CD86+ (M1) or CD206+ (M2) cells. REDD1 ablation prevented the diabetes-induced increase in CD86 + M1 macrophages (Fig. 3F). No significant changes were
observed in CD206+ macrophage populations (Fig. 3G). These data support that REDD1 expression is necessary for elevated pro-inflammatory innate immune responses in the kidneys of diabetic
mice. REDD1 EXPRESSION WAS REQUIRED FOR NF-ΚB ACTIVATION IN PODOCYTES NF-κB signaling involves the degradation of the inhibitor of κB (IκB) to facilitate the phosphorylation and nuclear
translocation of NF-κB. Degradation of IκBα (Fig. S3A) and enhanced nuclear localization of NF-κB and increased NF-κB activity were observed in the kidneys of diabetic REDD1+/+ mice (Fig.
4A). As compared to REDD1+/+ mice, diabetes-induced NF-κB activation was reduced in the kidneys of REDD1−/− mice. Prior reports suggest a role for podocytes in mediating inflammatory
responses in diabetes [11, 37]. REDD1 partially colocalized with the podocyte marker nephrin in the kidneys of diabetic mice (Fig. 4B). To investigate the role of REDD1 in podocytes, CIHP-1
cells were exposed to hyperglycemic culture conditions. Hyperglycemic conditions promoted both the degradation of IκBα (Fig. S3B) and phosphorylation of the p65 NF-κB subunit at S536 (Fig.
4C), NF-κB nuclear localization (Fig. 4D), and NF-κB luciferase reporter activity (Fig. 4E) in podocytes. CRISPR-Cas9-mediated REDD1 deletion prevented these effects in CIHP-1 cells (Fig.
4C–E). To assess specificity of NF-κB signaling in podocytes, CIHP cells expressing either EV or an IκBα mutant plasmid were exposed to hyperglycemic conditions. High glucose-induced
expression of mRNAs encoding the NF-κB target genes _IL1B_ and _CCL2_ was attenuated with suppressed activation of NF-κB (Fig. S3C). Notably, expression of these genes were upregulated in
wild-type cells, but not in REDD1-deficient cells, upon exposure to hyperglycemic conditions (Fig. 4F). REDD1 deletion also attenuated mRNA expression of pro-inflammatory genes including
_TNFA_, _VEGFA_, and _ICAM-1_ in podocytes exposed to hyperglycemic conditions (Fig. S1C). Hyperglycemic conditions also upregulated IL-1β protein released into media in a manner that was
dependent on REDD1 (Fig. 4G). ChIP PCR analysis indicated increased binding of p65 to the _CCL2_ promoter in wild-type cells exposed to hyperglycemic conditions, but not in REDD1-deficient
cells exposed to hyperglycemic conditions (Fig. 4H). We also observed a REDD1-dependent increase in CCL2 protein in cell lysates upon exposure to hyperglycemic conditions (Fig. 4I, S1D). To
confirm the role of REDD1 in NF-κB activation in podocytes, REDD1 was rescued in REDD1 knockout podocytes by expression of an HA-tagged REDD1. HA-REDD1 enhanced NF-κB p65 phosphorylation at
S536 in REDD1 knockout podocytes and restored the increase in NF-κB activity upon exposure to hyperglycemic conditions (Fig. 4J). These data support that REDD1 is both necessary and
sufficient to increase NF-κB activation in podocytes under hyperglycemic conditions. PODOCYTE-SPECIFIC DELETION OF REDD1 PROTECTED AGAINST MACROPHAGE INFILTRATION IN DN To evaluate the role
of podocyte-specific REDD1 in macrophage infiltration, wild-type and REDD1-deficient CIHP-1 cells were exposed to hyperglycemic conditions. A Transwell migration assay was then used to
assess macrophage chemotaxis by podocytes (Fig. 5A). Increased transmigration of THP-1 macrophages was observed when co-cultured with wild-type CIHP-1 cells exposed to hyperglycemic
conditions (Fig. 5B). However, a similar increase in chemotaxis was not observed when macrophages were co-cultured with REDD1-deficient CIHP-1 cells exposed to hyperglycemic conditions.
Thus, REDD1 deletion in podocytes attenuated both the increase in chemokine expression and macrophage chemotaxis under hyperglycemic conditions. To evaluate the role of podocyte-specific
REDD1 expression in the kidney, targeted REDD1 deletion in podocytes was carried out by NPHS2-cre directed excision of exons 2 and 3 from the REDD1 gene (Fig. 5C) [34]. To evaluate a role
for podocyte-specific REDD1 expression in diabetes-induced renal inflammatory responses, REDD1fl/fl and REDD1 PodKO mice were administered STZ. After 16 weeks of STZ-induced diabetes,
increased urine ACR was observed in REDD1 fl/fl mice but not in REDD1 PodKO mice (Fig. 5D). Urine ACR positively correlated with elevated fasting blood glucose concentrations in REDD1fl/fl
mice (Fig. S4A, Pearson r = 0.812). As compared to REDD1fl/fl mice, the linear correlation between urine ACR and blood glucose was reduced in REDD1 PodKO mice (Pearson r = 0.646). REDD1
protein was increased throughout the kidneys of diabetic REDD1fl/fl mice and colocalized with the podocyte marker nephrin within the glomerulus (Fig. 5E). REDD1 protein expression was absent
within glomeruli and attenuated in tubules of diabetic REDD1 PodKO mouse kidneys, as compared to diabetic REDD1fl/fl mice. Moreover, increased CCL2 protein levels (Fig. 5F, S1E) and F4/80+
immune cell infiltration (Fig. 5G) were observed in the kidneys of diabetic REDD1fl/fl mice, but not diabetic REDD1 PodKO mice. Flow cytometry analysis (Fig. S4B) revealed a large proportion
of the infiltrating CD45+ immune cells in diabetic REDD1fl/fl mice to be macrophages (CD11b + F4/80 + ; Fig. 5H) that were further characterized as pro-inflammatory CD86 + M1 macrophages
(Fig. 5I). As compared to the kidney of REDD1fl/fl mice, fewer F4/80+ cells were observed in diabetic REDD1 PodKO mice and the M1 macrophage population was reduced. The data support that
REDD1 expression specifically in podocytes promotes renal infiltration of pro-inflammatory macrophages in the context of diabetes. PODOCYTE-SPECIFIC REDD1 DELETION ATTENUATED ACTIVATION OF
THE NLRP3 INFLAMMASOME AND PYROPTOSIS IN DN Under metabolic stress, cells undergo pyroptosis, which is a pro-inflammatory form of cell death characterized by increased caspase-1 activity,
Gasdermin D (GSDMD) cleavage, and lactate dehydrogenase (LDH) release [38]. In podocyte cultures, exposure to high glucose concentrations increased _NLRP3_ mRNA expression (Fig. S3C), and
suppression of NF-κB with the IκBα super repressor mutant plasmid attenuated this effect. To investigate the role of REDD1 in NLRP3 activation and pyroptosis, differentiated wild-type and
REDD1-deficient podocytes were exposed to hyperglycemic conditions. _NLRP3_ mRNA expression (Fig. 6A) and NLRP3 protein abundance (Fig. 6B) were increased in wild-type podocytes exposed to
hyperglycemic conditions, but not in podocytes deficient for REDD1. In podocytes exposed to hyperglycemic conditions, there was increased caspase-1 activity (Fig. 6C), GSDMD cleavage (Fig.
6D), and LDH release (Fig. 6E) in a manner that was dependent on REDD1. To investigate if REDD1 expression in podocytes was necessary for diabetes-induced pyroptosis, kidneys and glomerular
isolates from STZ-diabetic and non-diabetic REDD1fl/fl and REDD1 PodKO mice were assessed. As compared to non-diabetic mice, _Nlrp3_ mRNA expression (Fig. 6F) and NLRP3 protein content (Fig.
6G) were increased in glomeruli isolated from diabetic REDD1fl/fl mice. We also observed an increase in GSDMD protein in glomeruli of diabetic REDD1fl/fl mice compared to non-diabetic mice
(Fig. 6G). However, increases in NLRP3 and GSDMD were both blunted in glomeruli isolated from diabetic REDD1 PodKO mice. Immunofluorescence microscopy also showed an increased colocalization
of NLRP3 and GSDMD with the podocyte marker nephrin in diabetic REDD1fl/fl mice, but not in diabetic REDD1 PodKO mice (Fig. 6H). In support of a role for podocyte-specific REDD1 expression
in podocyte loss with STZ-diabetes, the reduction in nephrin and WT-1 staining in diabetic REDD1fl/fl mice was reduced in diabetic REDD1 PodKO mice (Fig. 6H). We also observed that
podocyte-specific REDD1 deletion attenuated the increase in IL-1β levels in kidney homogenates from diabetic mice (Fig. 6I). Together, the data are consistent with a role for
diabetes-induced REDD1 expression in mediating NLRP3-associated pyroptotic cell death in podocytes. DISCUSSION Studies from the last two decades support a critical role for inflammation in
the etiology of DN [39]. Herein, we investigated the role of REDD1 in diabetes-induced renal inflammation. Diabetes increased renal NF-κB activation and enhanced pro-inflammatory cytokine
expression, with augmented renal infiltration of M1 pro-inflammatory macrophages in a manner that was dependent on REDD1. REDD1 was necessary and sufficient to promote NF-κB activation in
podocytes exposed to hyperglycemic conditions. Importantly, the deletion of REDD1 specifically in podocytes attenuated macrophage infiltration in the kidneys of diabetic mice. Overall, the
studies support a model wherein REDD1 expression in podocytes promotes NF-κB- and NLRP3-mediated inflammatory responses in the kidney including podocyte pyroptosis and the recruitment and
polarization of macrophages in DN (Fig. 7). Hyperglycemia is a determining factor in the development and progression of DN [40, 41]. Herein, hypo-insulinemia was induced in mice by
administration of STZ, resulting in secondary hyperglycemia. While STZ is a valuable tool for modeling diabetic complications in genetically manipulated mice, it is important to note that
strain-dependent variability in renal responses to STZ have been reported. With STZ administration, the C57BL/6 and B6;129 strains used in this study develop mild pathological changes in the
kidney with mild-to-moderate albuminuria, as compared to other inbred mouse lines (e.g., DBA/2, KK-H1J) [42,43,44]. Importantly, the STZ-diabetic C57BL/6 mouse strain continues to be a
helpful experimental paradigm to investigate diabetes-associated renal inflammation [45,46,47]. REDD1 levels are elevated in the kidneys of diabetic patients and in preclinical murine models
of type 1 and type 2 diabetes [16, 17]. Upregulation of REDD1 occurs in multiple cell types exposed to diabetogenic conditions [16, 17, 27, 48, 49]. Normalization of blood glucose
concentrations by SGLT2 inhibition was followed by attenuated REDD1 protein abundance in the kidney of diabetic mice concomitant with a reduction in immune cell infiltration. The observation
builds on prior studies demonstrating increased REDD1 in renal cell cultures exposed to hyperglycemic conditions [16, 17, 49]. A growing body of research demonstrates that REDD1 controls
critical cellular and metabolic functions [50], and is vital in the pathogenesis of metabolic disorders including diabetic retinopathy [19, 20, 27, 48] and nephropathy [16, 17, 49]. In the
past decade, increasing evidence supports a pro-inflammatory role for REDD1 [19, 20, 22, 23, 49]. Chronic low-grade inflammation and activation of the innate immune response are integral to
the pathogenesis of diabetes and its complications [51]. Inflammatory mediators like IL-1β, IL6 and CCL2 are upregulated in the kidneys of diabetic patients and act as pathogenic mediators
in DN [9, 52]. Our data agree with these works and advance the understanding of mechanisms whereby REDD1 drives immune signaling in the context of DN. Specifically, REDD1 was necessary for
activation of NF-κB, increased expression of cytokines and chemokines, and immune cell infiltration in the kidneys of diabetic mice. Lee et al. previously reported that REDD1 plays a role in
the recruitment of immune cells into adipose tissue in murine model an obesity [22]. The data here support that REDD1 has a similar role in the recruitment of M1 pro-inflammatory
macrophages into the kidney in the context of diabetes. Podocyte dysfunction and loss is an early event in DN pathogenesis and predicts diabetic kidney injury [53]. Damaged podocytes produce
inflammatory cytokines and chemokines that drive immune cell recruitment and glomerular inflammation [11, 49]. Activation of inflammatory pathways in non-hematopoietic kidney resident cells
including podocytes promote inflammatory processes that aggravate renal injury in DN [14]. Indeed, podocyte-specific suppression of the NLRP3 inflammasome prevents diabetes-induced
proteinuria [11]. Herein, REDD1 was required for NF-κB activation and the production of inflammatory cytokines and chemokines by podocytes. This advances findings from Wang et al. showing
that REDD1 knockdown attenuates expression of TNFα, IL6, and IL-1β in podocyte cultures exposed to hyperglycemic conditions [49]. Additionally, in vitro transmigration assays demonstrated
that hyperglycemia-induced REDD1 in podocytes was required for macrophage chemotaxis to the site of inflammatory injury. Importantly, in mice with podocyte-specific REDD1 deletion, diabetes
failed to increase immune cell infiltration and renal recruitment of M1 pro-inflammatory macrophages. Notably, the attenuated inflammatory response within glomeruli observed with
podocyte-specific REDD1 deletion correlated with preserved glomerular architecture and filtration function, as well as reduced podocyte loss [34]. Independent investigations have
demonstrated that canonical and non-canonical activation of the inflammasome is a characteristic event in diabetic complications [38]. In the context of diabetic kidney disease, studies have
shown that excessive cell pyroptosis mediated by caspase-1-associated canonical [54] cleavage of GSDMD (as well as caspase-11/4 non-canonical GSDMD cleavage [10]) promotes podocyte damage
and renal immune cell infiltration. Moreover, in preclinical models of DN, podocyte-specific activation of the NLRP3 inflammasome is both necessary and sufficient to promote glomerular
dysfunction and kidney damage [11]. In recent years, investigations delineating the regulation of NLRP3 inflammasome activation have implicated a role for REDD1 in both priming and
activation of the inflammasome complex [21, 23, 55]. Prior work from our laboratory demonstrated a role for REDD1 in NF-κB-dependent NLRP3 inflammasome activity in the context of diabetic
retinopathy [21]. The findings presented herein extend these prior studies and demonstrate that REDD1 expression in podocytes is required for NF-κB-dependent NLRP3 inflammasome activation
and subsequent induction of pyroptosis in experimental models of diabetes. A major limitation of the current standards of care for DN is that they predominantly focus on controlling blood
glucose levels and fail to address the specific underlying cause of DN. The studies here delineate specific molecular events that contribute to renal inflammation caused by diabetes.
Podocytes perform immune-surveillance functions and initiate immune responses that make the glomerular filtration barrier vulnerable to inflammatory disorders like DN [11]. The
proof-of-concept studies here are consistent with a mechanism of action whereby REDD1 drives renal injury by promoting NF-κB activation in podocytes, thereby enhancing the renal
pro-inflammatory immune response to diabetes. Given that REDD1 expression is also upregulated in acute kidney injury (AKI) [56, 57], interventions targeting REDD1 in the context of
nephropathies including AKI and DN could improve current treatment paradigms. Novel podocyte-centric therapies like PS-001 [58], which recently entered clinical development, also offer great
promise in developing therapeutics that can suppress REDD1 specifically in podocytes to combat proteinuric kidney disease. DATA AVAILABILITY All primary data including original western
blots and immunofluorescent images supporting the findings of this study are presented within the manuscript or in the supplemental information. REFERENCES * Koye DN, Shaw JE, Reid CM,
Atkins RC, Reutens AT, Magliano DJ. Incidence of chronic kidney disease among people with diabetes: a systematic review of observational studies. Diabet Med J Br Diabet Assoc.
2017;34:887–901. Article CAS Google Scholar * United States Renal Data System. 2022 USRDS Annual Data Report: Epidemiology of kidney disease in the United States. National Institutes of
Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD %8, 2022. https://adr.usrds.org/2022. * Yamazaki T, Mimura I, Tanaka T, Nangaku M. Treatment of
Diabetic Kidney Disease: Current and Future. dmj. 2021;45:11–26. PubMed PubMed Central Google Scholar * Xu J, Murphy SL, Kochanek KD, Arias E. Mortality in the United States, 2021.
2022.https://stacks.cdc.gov/view/cdc/122516. * Chen J, Liu Q, He J, Li Y. Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front Immunol.
2022;13:958790. Article PubMed PubMed Central CAS Google Scholar * Mezzano S, Aros C, Droguett A, Burgos ME, Ardiles L, Flores C, et al. NF-κB activation and overexpression of regulated
genes in human diabetic nephropathy. Nephrol Dial Transplant. 2004;19:2505–12. Article PubMed CAS Google Scholar * Foresto-Neto O, Albino AH, Arias SCA, Faustino VD, Zambom FFF,
Cenedeze MA et al. NF-κB System Is Chronically Activated and Promotes Glomerular Injury in Experimental Type 1 Diabetic Kidney Disease. Front Physiol. 2020; 11.
https://www.frontiersin.org/articles/10.3389/fphys.2020.00084. * Nguyen D, Ping F, Mu W, Hill P, Atkins RC, Chadban SJ. Macrophage accumulation in human progressive diabetic nephropathy.
Nephrol Carlton Vic. 2006;11:226–31. Article Google Scholar * Araújo LS, Torquato BGS, da Silva CA, dos Reis Monteiro MLG, dos Santos Martins ALM, da Silva MV, et al. Renal expression of
cytokines and chemokines in diabetic nephropathy. BMC Nephrol. 2020;21:308. Article PubMed PubMed Central Google Scholar * Cheng Q, Pan J, Zhou Z-L, Yin F, Xie H-Y, Chen P-P, et al.
Caspase-11/4 and gasdermin D-mediated pyroptosis contributes to podocyte injury in mouse diabetic nephropathy. Acta Pharmacol Sin. 2021;42:954–63. Article PubMed CAS Google Scholar *
Shahzad K, Fatima S, Khawaja H, Elwakiel A, Gadi I, Ambreen S, et al. Podocyte-specific Nlrp3 inflammasome activation promotes diabetic kidney disease. Kidney Int. 2022;102:766–79. Article
PubMed CAS Google Scholar * Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89. Article PubMed
PubMed Central CAS Google Scholar * Shahzad K, Bock F, Al-Dabet MM, Gadi I, Kohli S, Nazir S, et al. Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. J Am Soc Nephrol JASN.
2016;27:2270–5. Article PubMed CAS Google Scholar * Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates
diabetic nephropathy. Kidney Int. 2015;87:74–84. Article PubMed CAS Google Scholar * Chung CH, Fan J, Lee EY, Kang JS, Lee SJ, Pyagay PE, et al. Effects of Tumor Necrosis Factor-α on
Podocyte Expression of Monocyte Chemoattractant Protein-1 and in Diabetic Nephropathy. Nephron Extra. 2015;5:1–18. Article PubMed PubMed Central Google Scholar * Sunilkumar S, Yerlikaya
EI, Toro AL, Miller WP, Chen H, Hu K, et al. REDD1 Ablation Attenuates the Development of Renal Complications in Diabetic Mice. Diabetes. 2022;71:2412–25. Article PubMed PubMed Central
CAS Google Scholar * Mu L, Chen N, Chen Y, Yang Z, Zhou H, Song S, et al. Blocking REDD1/TXNIP Complex Ameliorates HG-Induced Renal Tubular Epithelial Cell Apoptosis and EMT through
Repressing Oxidative Stress. Int J Endocrinol. 2022;2022:6073911. Article PubMed PubMed Central Google Scholar * Sunilkumar S, Dennis MD. REDD1 Is a Promising Therapeutic Target to
Combat the Development of Diabetes Complications: A Report on Research Supported by Pathway to Stop Diabetes. Diabetes. 2024;73:1553–62. * Sunilkumar S, Toro AL, McCurry CM, VanCleave AM,
Stevens SA, Miller WP, et al. Stress response protein REDD1 promotes diabetes-induced retinal inflammation by sustaining canonical NF-κB signaling. J Biol Chem. 2022; 298.
https://doi.org/10.1016/j.jbc.2022.102638. * Sunilkumar S, VanCleave AM, McCurry CM, Toro AL, Stevens SA, Kimball SR, et al. REDD1-dependent GSK3β dephosphorylation promotes NF-κB activation
and macrophage infiltration in the retina of diabetic mice. J Biol Chem. 2023;299:104991. Article PubMed PubMed Central CAS Google Scholar * McCurry CM, Sunilkumar S, Toro A, VanCleave
A, Dennis MD. NLRP3 inflammasome priming in the retina of diabetic mice requires REDD1-dependent activation of GSK3. Invest Ophthalmol Vis Sci. 2023;64:496. Google Scholar * Lee D-K, Kim
T, Byeon J, Park M, Kim S, Kim J, et al. REDD1 promotes obesity-induced metabolic dysfunction via atypical NF-κB activation. Nat Commun. 2022;13:6303. Article PubMed PubMed Central CAS
Google Scholar * Pastor F, Dumas K, Barthelemy MA, Regazzetti C, Druelle N, Peraldi P, et al. Implication of REDD1 in the activation of inflammatory pathways. Sci Rep. 2017;7:7023. Article
PubMed PubMed Central Google Scholar * Stevens SA, Sunilkumar S, Subrahmanian SM, Toro AL, Cavus O, Omorogbe EV, et al. REDD1 Deletion Suppresses NF-κB Signaling in Cardiomyocytes and
Prevents Deficits in Cardiac Function in Diabetic Mice. Int J Mol Sci. 2024;25:6461. Article PubMed PubMed Central CAS Google Scholar * Han S, Hagan DL, Taylor JR, Xin L, Meng W, Biller
SA, et al. Dapagliflozin, a Selective SGLT2 Inhibitor, Improves Glucose Homeostasis in Normal and Diabetic Rats. Diabetes. 2008;57:1723. Article PubMed CAS Google Scholar * Brafman A,
Mett I, Shafir M, Gottlieb H, Damari G, Gozlan-Kelner S, et al. Inhibition of Oxygen-Induced Retinopathy in RTP801-Deficient Mice. Invest Ophthalmol Vis Sci. 2004;45:3796–805. Article
PubMed Google Scholar * Miller WP, Toro AL, Sunilkumar S, Stevens SA, VanCleave AM, Williamson DL, et al. Müller Glial Expression of REDD1 Is Required for Retinal Neurodegeneration and
Visual Dysfunction in Diabetic Mice. Diabetes. 2022;71:1051–62. Article PubMed PubMed Central CAS Google Scholar * Sunilkumar S, Ford SM. Elevated glucose concentration in culture media
decreases membrane trafficking of SGLT2 in LLC-PK1 cells via a cAMP/PKA-dependent pathway. Am J Physiol-Cell Physiol. 2019;316:C913–C924. Article PubMed CAS Google Scholar * Laurence J,
Elhadad S, Gostynska S, Yu Z, Terry H, Varshney R, et al. HIV protease inhibitor ritonavir induces renal fibrosis and dysfunction: role of platelet-derived TGF-β1 and intervention via
anti-oxidant pathways. AIDS Lond Engl. 2020;34:989–1000. Article CAS Google Scholar * Mohanty S, Han T, Choi YB, Lavorgna A, Zhang J, Harhaj EW. The E3/E4 ubiquitin conjugation factor
UBE4B interacts with and ubiquitinates the HTLV-1 Tax oncoprotein to promote NF-κB activation. PLoS Pathog. 2020;16:e1008504. Article PubMed PubMed Central CAS Google Scholar * Deng X,
Xu M, Yuan C, Yin L, Chen X, Zhou X, et al. Transcriptional regulation of increased CCL2 expression in pulmonary fibrosis involves nuclear factor-κB and activator protein-1. Int J Biochem
Cell Biol. 2013;45:1366–76. Article PubMed CAS Google Scholar * Tariq M, Saze H, Probst AV, Lichota J, Habu Y, Paszkowski J. Erasure of CpG methylation in Arabidopsis alters patterns of
histone H3 methylation in heterochromatin. Proc Natl Acad Sci USA. 2003;100:8823–7. Article PubMed PubMed Central CAS Google Scholar * AnandBabu K, Sen P, Angayarkanni N. Oxidized LDL,
homocysteine, homocysteine thiolactone and advanced glycation end products act as pro-oxidant metabolites inducing cytokine release, macrophage infiltration and pro-angiogenic effect in
ARPE-19 cells. PloS One. 2019;14:e0216899. Article PubMed PubMed Central CAS Google Scholar * Sunilkumar S, Yerlikaya EI, Toro AL, Chen H, Zhou Y, Gill DL, et al. Podocyte-Specific
Expression of the Stress Response Protein REDD1 is Necessary for Diabetes-induced Podocytopenia. Diabetes. 2024;db240533. https://doi.org/10.2337/db24-0533. * Scurt FG, Menne J, Brandt S,
Bernhardt A, Mertens PR, Haller H, et al. Monocyte chemoattractant protein-1 predicts the development of diabetic nephropathy. Diabetes Metab Res Rev. 2022;38:e3497. Article PubMed CAS
Google Scholar * You H, Gao T, Cooper TK, Brian Reeves W, Awad AS. Macrophages directly mediate diabetic renal injury. Am J Physiol-Ren Physiol. 2013;305:F1719–F1727. Article CAS Google
Scholar * Banas MC, Banas B, Hudkins KL, Wietecha TA, Iyoda M, Bock E, et al. TLR4 Links Podocytes with the Innate Immune System to Mediate Glomerular Injury. J Am Soc Nephrol JASN.
2008;19:704–13. Article PubMed CAS Google Scholar * Wan J, Liu D, Pan S, Zhou S, Liu Z. NLRP3-mediated pyroptosis in diabetic nephropathy. Front Pharmacol. 2022;13:998574. Article
PubMed PubMed Central CAS Google Scholar * Rayego-Mateos S, Morgado-Pascual JL, Opazo-Ríos L, Guerrero-Hue M, García-Caballero C, Vázquez-Carballo C, et al. Pathogenic Pathways and
Therapeutic Approaches Targeting Inflammation in Diabetic Nephropathy. Int J Mol Sci. 2020;21:3798. Article PubMed PubMed Central CAS Google Scholar * Schrijvers BF, De Vriese AS,
Flyvbjerg A. From Hyperglycemia to Diabetic Kidney Disease: The Role of Metabolic, Hemodynamic, Intracellular Factors and Growth Factors/Cytokines. Endocr Rev. 2004;25:971–1010. Article
PubMed CAS Google Scholar * Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J
Med. 2016;375:323–34. Article PubMed CAS Google Scholar * Breyer MD, Brosius FCI, Coffman TM, Harris RC, Heilig CW, Levi M, et al. Mouse Models of Diabetic Nephropathy. J Am Soc Nephrol.
2005;16:27. Article PubMed Google Scholar * Qi Z, Fujita H, Jin J, Davis LS, Wang Y, Fogo AB, et al. Characterization of Susceptibility of Inbred Mouse Strains to Diabetic Nephropathy.
Diabetes. 2005;54:2628–37. Article PubMed CAS Google Scholar * Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. Am
J Physiol-Ren Physiol. 2006;290:F214–F222. Article CAS Google Scholar * Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes
the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. Article PubMed CAS Google Scholar * Matsushita Y, Ogawa D, Wada J, Yamamoto N, Shikata
K, Sato C, et al. Activation of Peroxisome Proliferator–Activated Receptor δ Inhibits Streptozotocin-Induced Diabetic Nephropathy Through Anti-Inflammatory Mechanisms in Mice. Diabetes.
2011;60:960–8. Article PubMed PubMed Central CAS Google Scholar * Huang X, Shi Y, Chen H, Le R, Gong X, Xu K, et al. Isoliquiritigenin prevents hyperglycemia-induced renal injuries by
inhibiting inflammation and oxidative stress via SIRT1-dependent mechanism. Cell Death Dis. 2020;11:1–14. Article Google Scholar * Miller WP, Yang C, Mihailescu ML, Moore JA, Dai W, Barber
AJ, et al. Deletion of the Akt/mTORC1 Repressor REDD1 Prevents Visual Dysfunction in a Rodent Model of Type 1 Diabetes. Diabetes. 2018;67:110–9. Article PubMed CAS Google Scholar * Wang
X, Yang J, Wang W, Li Y, Yang Y. Decreasing REDD1 expression protects against high glucose-induced apoptosis, oxidative stress and inflammatory injury in podocytes through regulation of the
AKT/GSK-3β/Nrf2 pathway. Immunopharmacol Immunotoxicol. 2023;45:527–38. * Kim J-Y, Kwon Y-G, Kim Y-M. The stress-responsive protein REDD1 and its pathophysiological functions. Exp Mol Med.
2023;55:1933–44. Article PubMed PubMed Central CAS Google Scholar * Navarro-González JF, Mora-Fernández C. The Role of Inflammatory Cytokines in Diabetic Nephropathy. J Am Soc Nephrol.
2008;19:433. Article PubMed Google Scholar * Wada T, Furuichi K, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, et al. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial
lesions of human diabetic nephropathy. Kidney Int. 2000;58:1492–9. Article PubMed CAS Google Scholar * Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary
albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia. 1999;42:1341–4. Article PubMed CAS Google Scholar * Chen J, Yang Y, Lv Z, Shu A, Du Q, Wang W,
et al. Study on the inhibitive effect of Catalpol on diabetic nephropathy. Life Sci. 2020;257:118120. Article PubMed CAS Google Scholar * Yang Z, Wu M, Chen N, Zhou H, Yuan C, Zhou Z,
et al. REDD1 Deficiency Alleviates Autophagic Flux Impairment Via Inhibiting NLRP3 Inflammasome-Mediated Notch1 Signaling Pathway in Diabetic Kidney Disease. 2022.
https://doi.org/10.2139/ssrn.4061018. * Huang D, Chen D, Hu T, Liang H. GATA2 promotes oxidative stress to aggravate renal ischemia-reperfusion injury by up-regulating Redd1. Mol Immunol.
2023;153:75–84. Article PubMed CAS Google Scholar * Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell
Types and Novel Cell States Revealed in Fibrosis. J Am Soc Nephrol JASN. 2019;30:23–32. Article PubMed CAS Google Scholar * Illingworth S, Gurung K, Kuzmuk V, Wood H, Asfahani R, Gennari
A, et al. #1118 Direct delivery of novel gene therapy to podocytes enables pathway to clinical translation for the treatment of glomerular diseases. Nephrol Dial Transplant.
2024;39:gfae069-0032–1118. Article Google Scholar Download references ACKNOWLEDGEMENTS We thank Elena Feinstein (Quark Pharmaceuticals) for permission to use the REDD1 knockout mice and
Dr. Moin Saleem (University of Bristol) for providing the CIHP cell line. We are grateful to Ellen Mullady and Gretchen Snavely of the Department of Comparative Medicine at Penn State
College of Medicine for their preparation of histology sections. FUNDING This research was supported by grants from the American Diabetes Association (Grant#11-23-PDF-84), Children’s Miracle
Network Trainee Research Award, the Judy S. Finkelstein Memorial Student Research Award, and the Penn State College of Medicine’s Comprehensive Health Studies Program (to S.S.); as well as
National Institutes of Health grant R01 EY032879 and an Innovative Award 1-INO-2024-1538-A-N from the Juvenile Diabetes Research Association (to M.D.D.). S.M.S. was supported by a
postdoctoral fellowship M2024006F from the BrightFocus Foundation. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Cellular and Molecular Physiology, Penn State College of
Medicine, Hershey, PA, USA Siddharth Sunilkumar, Sandeep M. Subrahmanian, Esma I. Yerlikaya, Allyson L. Toro, Scot R. Kimball & Michael D. Dennis * Department of Microbiology and
Immunology, Penn State College of Medicine, Hershey, PA, USA Edward W. Harhaj Authors * Siddharth Sunilkumar View author publications You can also search for this author inPubMed Google
Scholar * Sandeep M. Subrahmanian View author publications You can also search for this author inPubMed Google Scholar * Esma I. Yerlikaya View author publications You can also search for
this author inPubMed Google Scholar * Allyson L. Toro View author publications You can also search for this author inPubMed Google Scholar * Edward W. Harhaj View author publications You can
also search for this author inPubMed Google Scholar * Scot R. Kimball View author publications You can also search for this author inPubMed Google Scholar * Michael D. Dennis View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS S.S., S.R.K, and M.D.D. conceptualization; S.S., and M.D.D. data curation; S.S., and M.D.D. formal
analysis; S.S. and M.D.D. funding acquisition; S.S., S.M.S., E.I.Y, and A.L.T., investigation; S.S., S.M.S., and M.D.D. visualization; S.S., S.M.S., and M.D.D. methodology; S.S., and M.D.D.
writing; S.S., S.M.S., E.I.Y., A.L.T., S.R.K., E.W.H., and M.D.D. reviewing and editing; S.R.K., E.W.H. and M.D.D. resources; S.R.K. and M.D.D. supervision. M.D.D. is guarantor of this work
and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and accuracy of the data analysis. CORRESPONDING AUTHOR Correspondence to
Michael D. Dennis. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICAL APPROVAL All procedures adhered to the National Institutes of Health Guide for
the Care and Use of Laboratory Animals and were approved by the Penn State College of Medicine Institutional Animal Care and Use Committee (PROTO202302452). ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by Massimiliano Agostini SUPPLEMENTARY
INFORMATION SUPPLEMENTAL MATERIALS RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Sunilkumar, S., Subrahmanian, S.M., Yerlikaya, E.I. _et al._ REDD1 expression in podocytes facilitates renal inflammation and pyroptosis in
streptozotocin-induced diabetic nephropathy. _Cell Death Dis_ 16, 79 (2025). https://doi.org/10.1038/s41419-025-07396-4 Download citation * Received: 23 August 2024 * Revised: 10 January
2025 * Accepted: 27 January 2025 * Published: 07 February 2025 * DOI: https://doi.org/10.1038/s41419-025-07396-4 SHARE THIS ARTICLE Anyone you share the following link with will be able to
read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative