- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Rett syndrome is a severe neurodevelopmental disorder that arises from mutations in the X-linked _MECP2_ gene. It is almost exclusively seen in girls due to the predominant
occurrence of the mutations on the paternal X-chromosome, and also the early postnatal lethal effect of the disease causing mutations in hemizygous boys. We identified a boy with features of
classic Rett syndrome who is mosaic for the truncating _MECP2_ mutation R270X. Chromosome analysis showed normal karyotype. These results indicate that a _MECP2_ mutation associated with
Rett syndrome in females could lead to a similar phenotype in males as a result of somatic mosaicism. SIMILAR CONTENT BEING VIEWED BY OTHERS NONRECURRENT 17P DUPLICATIONS IN TWO PATIENTS
WITH DEVELOPMENTAL AND NEUROLOGICAL ABNORMALITIES Article Open access 26 March 2025 RARE MOSAIC VARIANT OF _GJA1_ IN A PATIENT WITH A NEURODEVELOPMENTAL DISORDER Article Open access 15
January 2024 GITELMAN SYNDROME CAUSED BY A NOVEL HEMIALLELIC MISSENSE MUTATION IN _SLC12A3_ REVEALED BY 16Q12.2Q21 MICRODELETION Article Open access 27 May 2020 INTRODUCTION Classical Rett
syndrome (RTT-MIM 312750) was first defined as a neurodevelopmental disorder that is primarily seen in girls.1,2 Following the hypothesis of X-linked dominant mutations that are lethal in
hemizygous males,2 the RTT gene was localised to Xq28 by exclusion mapping studies in rare familial cases.3 Subsequently, _MECP2_ (methyl-CpG-binding protein 2) was identified as the gene
responsible for this disorder by a systematic search for mutations in genes from the Xq28 region in RTT patients.4 Normal development until 6–18 months of age, followed by gradual loss of
speech and purposeful hand use, microcephaly, seizures, ataxia, and stereotypic hand movements are among the cardinal features of Rett syndrome.5 Femaleness is also included in the
above-mentioned diagnostic criteria. A wide range of _MECP2_ mutations has been identified in Rett syndrome patients.6 Interestingly, mutations of the same gene have been documented in male
patients affected by severe encephalopathy,3,6 and X-linked mental retardation.7 One boy affected with a nonfatal neurodevelopmental disorder with similarities to Rett syndrome was found to
be mosaic for a _MECP2_ gene mutation.8 In addition, an Angelman syndrome-like phenotype has recently been documented in girls with _MECP2_ mutations.9 We and others have observed boys with
clinical features of Rett syndrome and a normal male karyotype.10,11,12 Several cases of 47,XXY males with a clinical diagnosis of Rett syndrome have been reported, although to our knowledge
only one has been reported so far as having a _MECP2_ mutation.13 In order to determine whether _MECP2_ is involved in the development of the disease phenotype in males, we performed
mutation screening in a patient not reported before, and diagnosed as Rett syndrome. MATERIALS AND METHODS CASE REPORT BF was the product of a non-eventful pregnancy born to a 40-year-old
father and 29-year-old mother. He has two brothers (15 and 17 years old), and one sister (18 years old), who are all healthy. His birth weight was 3500 g. Although he appeared to be a
hypotonic child, he had normal psychomotor development through the first 6 months. Head control was achieved during the first 4 months. He sat with support at age 5 months, and without
support at 12 months. He rolled over, transferred toys from one hand to the other, and babbled nonspecifically at around 6–8 months. He was also able to bring the milk bottle to his mouth
alone. Loss of acquired purposeful hand skills began around 11 months, and stereotypic hand movements such as patting, squeezing became apparent at 15 months. He never crawled or walked. He
has never spoken. None of the exclusion criteria (evidence of intrauterine growth retardation, organomegaly, retinopathy, microcephaly at birth etc.) was ever present. We first saw him at 2
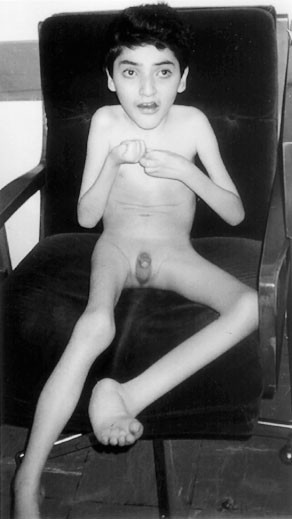
years of age, and a tentative diagnosis of Rett syndrome was made. On examination at 12 years of age (Figure 1, Table 1), he was microcephalic with a head circumference of 47 cm (below
second percentile), and brachycephalic with titubation of the head. His height was 110 cm and weight 18 kg, which are both below the third percentile. Sialorrhea and bruxism were present.
Stereotypic hand movements, tremors, and apraxia were noted. Deep tendon reflexes were increased bilaterally, and the clonus and Babinski signs were positive. His parents describe BF as a
happy child who likes listening to music. No breath holding or abnormal skin pigmentation was observed. He had a thoracic scoliosis and poor lower-limb musculature. His hands and feet were
small, and cold. Examination of the genitalia revealed presence of hypospadias and cryptorchidism. He experienced a total of three febrile seizures at the ages of five, 11, and 15 months,
which was treated with phenobarbital for a short period of time. Currently he is not on this medication. Electroencephalography showed excess of slow-wave activity and paroxysmal sharp theta
wave activity prominent on wake recordings of frontal regions. A magnetic resonance scan at age 12 was normal. Based on these findings, RTT currently at stage IV was considered. Therefore,
we obtained genomic DNA isolated from venous blood and hair root samples. Informed consent was obtained from each subject. GENOTYPING As a first step of the _MECP2_ gene screening procedure,
we looked for eight recurrent mutations (R106W, P152R, T158M, R168X, R255X, R270X, R288X, and R306C) by PCR, and restriction enzyme digestion,4,6 which was further confirmed by sequencing.
Two different sets of primers were used for the detection of the R270X mutation. The first set is 5′-GGC AGG AAG CGA AAA GCT GAG-3′ and 5′-TGA GTG GTG GTG ATG GTG GTG GG-3′ which yields a
366 bp fragment. The second set of primers which amplify a 380 bp fragment was used to confirm the results: 5′AAC CAC CTA AGA AGC CAA A-3′ and 5′-CTG CAC AGA TCG GAT AGA AGA C-3′. All
reactions were performed in a total volume of 25 μl, with 10× buffer, 1.5 U Taq polymerase, 1.5 mM MgCl2, 200 μM dNTP, and 10 pmol of each primer. PCR conditions were as follows: 95°C for 3
min, 30 cycles of 30 s at 94°C, 58°C and 30 s at 72°C. Ten minutes of 72°C was added at the extension step. RESULTS We identified the truncating mutation 808C→T (R270X) in BF's DNA
sample (Figure 2A). The same mutation was present also in two girls diagnosed with RTT.14 Interestingly, the mutant allele in the affected boy was present along with the wild-type allele
though at a reduced dosage when compared with the samples of the two girls. Whereas the mutant (T) and the wild-type (C) nucleotide peaks were equal in the two girls, the T allele peak was
smaller in the boy. In order to quantitate the dosage of the alleles, genomic DNA samples of the boy obtained from three independent blood samplings done at different times, and from one of
the girls was re-amplified with the same primers used in the sequencing reactions, and then subjected to restriction endonuclease treatment since the mutation abolishes an _Nla_IV
restriction site. Densitometric scanning of the restriction fragments revealed that the T : C allele ratio is approximately 56 : 44 in the girl and approximately 36 : 64 in the boy (Figure
2B). Furthermore, hair root samples of BF was obtained for DNA isolation. Three independent samples were subjected to PCR amplification and restriction endonuclease treatment as indicated
above. The results of these experiments also revealed the presence of somatic mosaicism with the T : C allele ration of 41 : 59. Since the restriction digestion method described above relies
on the absence of digestion of the mutated allele, but does not include a control for complete digestion of the DNA, we amplified a new 380 bp fragment, which contains four constant _Nla_IV
sites in addition to the sequences encompassing the R270X mutation. Digestion of the PCR products obtained from BF's DNA sample, and the positive control sample revealed the presence
of a mutation specific 177 bp fragment in addition to the constant fragments of 91, 86, 80, 74, and 49 bp (Figure 2C). The 177 bp fragment was absent in the normal control sample. This
result confirmed the presence of the R270X mutation in BF and the positive control sample. Chromosome analysis on blood revealed a normal 46,XY karyotype. These results suggest that the boy
is a somatic mosaic for the R270X mutation. DISCUSSION So far, _MECP2_ mutations have been shown to be associated with Rett syndrome in females, predominantly as _de novo_ mutations on the
paternal X-chromosome;15 neonatal encephalopathy6 or X-linked non-specific mental retardation7 in males which is transmitted through the maternal X-chromosome; X-linked non-specific mental
retardation in females;7 and finally Rett syndrome in XY males when the mutation is present in cells alongside a normal cell line,8 or in XXY males. Somatic mosaicism for the R270X mutation
in BF could be the result of an early post-zygotic mutation or chimerism. We think chimerism – which is defined as the coexistence of different cell populations of more than one zygote in
one body – is less likely since whole body chimeras result from fertilisation of one or more egg nuclei by two sperms, and is often associated with 46,XY/46,XX karyotype.16 BF's
karyotype analysis did not show a chromosomal abnormality. Analysis of highly polymorphic markers used in DNA profiling for individual identification would provide the definitive answer for
the post-zygotic mutation _vs_ chimerism issue since one would expect to see the inheritance of two alleles of a locus from one parent in addition to the allele from the other parent if
enough loci are studied in a chimeric individual. Based on the function of the _MECP2_ gene abnormal epigenetic regulation has been proposed as the mechanism underlying the pathogenesis of
RTT.4 DNA methylation-dependent silencing which is a known mechanism for maintenance of X-chromosome inactivation (XCI) and gametic imprinting are good candidates to study for abnormalities
in individuals with RTT.6 In this model, escape from XCI causing biallelic overexpression of X-linked genes and/or misexpression of imprinted genes is expected. However, with the observation
of a male RTT patient who is mosaic for a _MECP2_ mutation, disturbances in the expression of genes subject to XCI as a causative mechanism for RTT becomes unlikely since males are not
subject to X-inactivation yet they display the RTT phenotype. Somatic mosaicism has been documented in various genetic diseases including X-linked disorders in both males and females. These
include ornithine transcarbamylase deficiency,17 Duchenne muscular dystrophy,18 and hemophilia A.19 Although it is not clear whether the single-gene mutation occurred as a postzygotic event
or at the half-chromatid stage, before fertilisation,20 somatic mosaicism should be considered when an X-linked dominant disease is observed in a male. REFERENCES * Rett A . Uber ein
eigenartiges hirnatrophisches Syndrom bei Hyperammonemie im Kindesalter _Wien Med Wochenschr_ 1966 116: 723–738 CAS PubMed Google Scholar * Hagberg B, Aicardi J, Dias K, Ramos O . A
progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases _Ann Neurol_ 1983 14: 471–479 Article CAS Google
Scholar * Schanen NC, Francke U . A severely affected male born into a Rett syndrome kindred supports X-linked inheritance and allows extension of the exclusion map _Am J Hum Genet_ 1998
63: 267–269 Article CAS Google Scholar * Amir RE, Van den Veyver IB, Wan M _et al_. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2 _Nat
Genet_ 1999 23: 185–188 Article CAS Google Scholar * Trevathan E, Naidu S . The clinical recognition and differential diagnosis of Rett syndrome _J Child Neurol_ 1988 3 Suppl: S6–S16
Article Google Scholar * Wan M, Sung Jae Lee S, Zhang X _et al_. Rett Syndrome and Beyond: Recurrent Spontaneous and Familial _MECP2_ Mutations at CpG Hotspots _Am J Hum Genet_ 1999 65:
1520–1529 Article CAS Google Scholar * Meloni I, Bruttini M, Longo I _et al_. A mutation in the Rett syndrome gene, MECP2, causes X-linked mental retardation and progressive spasticity in
males _Am J Hum Genet_ 2000 67: 982–985 Article CAS Google Scholar * Clayton-Smith J, Watson P, Ramsden S, Black GCM . Somatic mutation in MECP2 as a non-fatal neurodevelopmental
disorder in males _Lancet_ 2000 356: 830–832 Article CAS Google Scholar * Watson P, Black G, Ramsden S _et al_. Angelman syndrome phenotype associated with mutations in MECP2, a gene
encoding a methyl CpG binding protein _J Med Genet_ 2001 38: 224–228 Article CAS Google Scholar * Philippart M . The Rett syndrome in males _Brain Dev_ 1990 12: 33–36 Article CAS Google
Scholar * Eeg-Olofsson O, Al-Zuhair AGH, Teebi AS _et al_. A boy with Rett syndrome? _Brain Dev_ 1990 12: 529–532 Article CAS Google Scholar * Topçu M, Topaloglu H, Renda Y _et al_. The
Rett syndrome in males _Brain Dev_ 1991 13: 62 Article Google Scholar * Hoffbuhr K, Devaney JM, LaFleur B _et al_. MeCP2 mutations in children with and without the phenotype of Rett
syndrome _Neurology_ 2001 56: 1489–1495 Article Google Scholar * Akyerli C, Sayi A, Kocoglu RS, Cimbis M, Topcu M, Ozcelik T . Analysis of MECP2 gene mutations in Turkish Rett syndrome
patients _Eur J Hum Genet_ 2001 9 suppl. 1: P1450 Google Scholar * Trappe R, Laccone F, Cobilanschi J _et al_. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of
paternal origin _Am J Hum Genet_ 2001 68: 1093–1101 Article CAS Google Scholar * Repas-Humpe LM, Humpe A, Lynen R _et al_. A dispermic chimerism in a 2-year-old Caucasian boy _Ann
Hematol_ 1999 78: 431–434 Article CAS Google Scholar * Maddalena A, Sosnoski DM, Berry GT, Nussbaum RL . Mosaicism for an intragenic deletion in a boy with mild ornithine transcarbamylase
deficiency _New Engl J Med_ 1988 319: 999–1003 Article CAS Google Scholar * Bakker E, Veenema H, Den Dunnen JT _et al_. Germinal mosaicism increases the recurrence risk for “new”
Duchenne muscular dystropy mutations _J Med Genet_ 1989 26: 553–559 Article CAS Google Scholar * Leuer M, Oldenburg J, Levergne J-M _et al_. Somatic mosaicism in hemophilia: A fairly
common event _Am J Hum Genet_ 2001 69: 75–87 Article CAS Google Scholar * Gartler SM, Francke U . Half Chromatid Mutations: Transmission in Humans? _Am J Hum Genet_ 1975 27: 218–223 CAS
PubMed PubMed Central Google Scholar * Kerr AM, Nomura Y, Armstrong D _et al_. Guidelines for reporting different clinical features in cases with _MECP2_ mutations _Brain Dev_ 2001 23:
208–211 Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We would like to express our gratitude to the family for their help with this project. We thank Dr Uta Francke for
critical reading of the manuscript, Drs Ergül Tunçbilek and Dilek Aktaş for performing the cytogenetic analysis, Dr Göknur Haliloğlu for help in obtaining patient samples, and Emre Sayan,
Tolga Çağatay, Tülay Arayıcı for dosage analysis experiments and DNA sequencing. This work was supported by Bilkent University, and TÜBİTAK. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS *
Department of Pediatric Neurology, Hacettepe University School of Medicine, Sihhiye, Ankara, 0600, Turkey Meral Topçu & Mine Cimbiş * Department of Molecular Biology and Genetics,
Faculty of Science, Bilkent University, Ankara, 06533, Turkey Cemaliye Akyerli, Ayça Sayı, Gökçe A Törüner, Süha R Koçoğlu & Tayfun Özçelik Authors * Meral Topçu View author publications
You can also search for this author inPubMed Google Scholar * Cemaliye Akyerli View author publications You can also search for this author inPubMed Google Scholar * Ayça Sayı View author
publications You can also search for this author inPubMed Google Scholar * Gökçe A Törüner View author publications You can also search for this author inPubMed Google Scholar * Süha R
Koçoğlu View author publications You can also search for this author inPubMed Google Scholar * Mine Cimbiş View author publications You can also search for this author inPubMed Google
Scholar * Tayfun Özçelik View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Tayfun Özçelik. RIGHTS AND PERMISSIONS
Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Topçu, M., Akyerli, C., Sayı, A. _et al._ Somatic mosaicism for a _MECP2_ mutation associated with classic Rett syndrome in a
boy. _Eur J Hum Genet_ 10, 77–81 (2002). https://doi.org/10.1038/sj.ejhg.5200745 Download citation * Received: 12 July 2001 * Revised: 16 October 2001 * Accepted: 18 October 2001 *
Published: 28 February 2002 * Issue Date: 01 January 2002 * DOI: https://doi.org/10.1038/sj.ejhg.5200745 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
KEYWORDS * Rett syndrome * MECP2 * somatic mosaicism