- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Download PDF Article Open access Published: 06 February 2017 New insights into the electrochemical behavior of acid orange 7: Convergent paired electrochemical synthesis of new aminonaphthol
derivatives Shima Momeni1 & Davood Nematollahi1 Scientific Reports volume 7, Article number: 41963 (2017) Cite this article
5907 Accesses
Metrics details
Subjects ElectrochemistryOrganic chemistryReaction mechanisms AbstractElectrochemical behavior of acid orange 7 has been exhaustively studied in aqueous solutions with different pH values, using cyclic voltammetry and constant current coulometry. This study
has provided new insights into the mechanistic details, pH dependence and intermediate structure of both electrochemical oxidation and reduction of acid orange 7. Surprisingly, the results
indicate that a same redox couple (1-iminonaphthalen-2(1H)-one/1-aminonaphthalen-2-ol) is formed from both oxidation and reduction of acid orange 7. Also, an additional purpose of this work
is electrochemical synthesis of three new derivatives of 1-amino-4-(phenylsulfonyl)naphthalen-2-ol (3a–3c) under constant current electrolysis via electrochemical oxidation (and reduction)
of acid orange 7 in the presence of arylsulfinic acids as nucleophiles. The results indicate that the electrogenerated 1-iminonaphthalen-2(1 H)-one participates in Michael addition reaction
with arylsulfinic acids to form the 1-amino-3-(phenylsulfonyl)naphthalen-2-ol derivatives. The synthesis was carried out in an undivided cell equipped with carbon rods as an anode and
cathode.
Similar content being viewed by others Green electrochemical method for the synthesis of nitro and azo derivatives based on mefenamic acid Article Open access 20 January 2022 Anew type of convergent paired electrochemical synthesis of sulfonamides under green and catalyst-free conditions Article Open access 16 October 2023 Electrochemical generation of
phenothiazin-5-ium. A sustainable strategy for the synthesis of new bis(phenylsulfonyl)-10H-phenothiazine derivatives Article Open access 21 February 2024 Introduction
2-Naphthol orange (acid orange 7), C16H11N2NaO4S, is a mono-azo water-soluble dye that extensively used for dyeing paper, leather and textiles1,2. The structure of acid orange 7 involves a
hydroxyl group in the ortho-position to the azo group. This resulted an azo-hydrazone tautomerism, and the formation of two tautomers, which each show an acid−base
equilibrium3,4,5,6,7,8,9,10,11,12. Despite the number of articles dealing with acid−base properties of the acid orange 7, this topic is not yet well known, and only one pKa, (pKa = 11.4) is
reported6. On the other hand, azo dyes have been widely used for developing and testing theories of color and constitution, tautomerism, indicator action, and acid-base equilibria5.
Therefore, detailed mechanistic information is important in understanding of the stability and in identifying of the intermediates structure resulting from the oxidative or reductive
decomposition of dye. Consequently, detailed mechanistic information is particularly attractive from the point of view of environmental pollution because of residual dye and the commercial
applications13,14. Additionally, green/sustainable synthesis is much more important than conventional synthetic methods. The concept and significance of green sustainable chemistry (GSC),
has been recognized throughout the world, and nowadays new processes cannot be developed without consideration of GSC. In recent years, much attention has been paid to electroorganic
synthesis as a typical environmentally friendly process15,16,17,18,19,20,21,22,23. This method contains the simultaneous incidence of both oxidation (at the anode) and reduction (at the
cathode). In conventional electroorganic synthesis, the synthesis of the desired products is done either by the anodic or by the cathodic reaction and so; the reaction product at the counter
electrode is undesirable. The simultaneous use of both oxidation and reduction reactions to synthesis of a product is the dream of an organic electrochemist and is a wonderful
strategy24,25,26,27,28. At ideal conditions, a 200% current efficiency is achievable for paired electrosynthesis when both anodic and cathodic reactions to provide the similar product
(convergent strategy)15. Furthermore, sulfone compounds and naphthalene derivatives are found in antibiotic drugs such as nafacillin and 4,4-diaminodiphenylsulfone (dapsone) and antifungal
drugs such as naftifine, tolnaftate and terbinafine29,30,31. These compounds have an effective inhibitory effect against the bacteria30,31 and antimicrobials effect against wide range of
human pathogens29. Based on these advances, we anticipate that naphthalene derivatives containing sulfone groups reveals such properties.
The results discussed above prompted us to investigate the electrochemical oxidation and reduction of acid orange 7 in aqueous solutions with different pH values to achieve the following
goals: (i) new insights into the electrochemical oxidation and reduction of acid orange 7, (ii) definitive detection of intermediates formed during the oxidative and reductive degradation of
acid orange 7, and (iii) convergent paired electrochemical synthesis of new 1-amino-2-naphthol derivatives by constant current electrolysis of acid orange 7 in the presence of arylsulfinic
acids as nucleophiles.
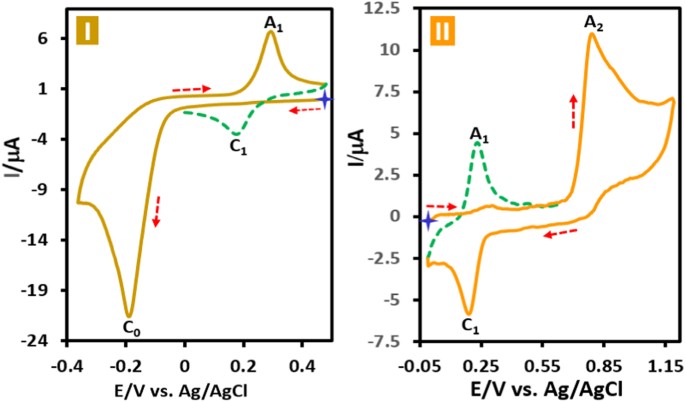
Results and DiscussionElectrochemical Study of Acid Orange 7Cyclic voltammograms of acid orange 7 (AO7) in aqueous phosphate buffer solution (c = 0.2 M, pH = 2.0) in two different potential regions (+0.5 to −0.4 and 0.0 to +1.2 V vs. Ag/AgCl) is
shown in Fig. 1. When the electrode potential was scanned from +0.5 V versus Ag/AgCl to a sufficiently negative voltage (−0.4 V versus Ag/AgCl), the cyclic voltammogram exhibits a large
cathodic peak (C0) at −0.19 with an anodic peak (A1) at +0.30 V versus Ag/AgCl at 50 mV s−1 (Fig. 1I). Under these conditions, in the second cycle a new cathodic peak (C1), which is the
counterpart of anodic peak (A1) appears with an Ep value of +0.18 V versus Ag/AgCl. Moreover, when the electrode potential was scanned from 0.0 V vs. Ag/AgCl to the positive potentials (+1.2
V vs. Ag/AgCl), the voltammogram shows an anodic peak (A2) at +0.80 V vs. Ag/AgCl with a cathodic peak (C1) (Fig. 1II). As in the previous experiment, a quasi-reversible couple, A1/C1,
appears in the second cycle of the voltammogram. A very important point in this study is the presence of the same redox couple in both cyclic voltammograms. This confirms the generation of
the same intermediates from both oxidation and reduction of AO7.
Figure 1Cyclic voltammogram of AO7 (1.0 mM) (first and second cycles) in two different direction of scanning of potential, (I) in negative-going scan and (II) in positive-going scan in aqueous
phosphate buffer (c = 0.2 M, pH = 2.0). Scan rate: 50 mV s−1; T = 25 ± 1 °C.
Full size imageWhen the potential scan rate increases from 250 to 8000 mV s−1, the cyclic voltammograms of AO7 in anodic region (Fig. S1) show the following changes: (1) the appearance of peak C2, which is
the counterpart of peak A2. (2) The increase of the anodic and cathodic peak current ratios (IpA2/IpA1) and (IpC2/IpCA1) and (3) the decrease of peak current function for anodic peak A2
(IpA2/v1/2). Increase in the potential scan rate causes a decrease in the CV time-scale and therefore a decrease in the progress of the following chemical reaction. These data confirm the
occurrence of a following chemical reaction and generation of a quasi-reversible system after oxidation of AO732. In addition, the effect of potential scan rate, v, (250 to 8000 mV s−1) on
the cyclic voltammetric response of AO7 in cathodic region which confirms the Irreversibility of the reduction process corresponding to peak C0 (Fig. S2).
The oxidative and reductive controlled-potential coulometry of AO7 was performed by applying potentials +0.90 and −0.20 V vs. Ag/AgCl, respectively. The solutions after coulometry are shown
in Supporting Information (Fig. S3). The monitoring of the electrolysis progress was carried out by cyclic voltammetry (Fig. 2). This Figure show that, in both experiments, proportional to
the advancement of coulometry, parallel to the decrease in the current of peaks A2 (Fig. 2I) and C0 (Fig. 2II), the peaks A1 and C1, increases. In these conditions, the number of transferred
electrons in oxidative controlled potential coulometry was obtained 2.9 electrons pre each AO7 molecule. On the other hand, the number of transferred electrons in reductive controlled
potential coulometry was obtained 4.1 electrons pre each AO7 molecule. An important point on the oxidation behaviour of AO7 can be seen in Fig. 2Ia. This Figure (and also Fig. 1II)
represents a typical behaviour of an ECE pathway in the kinetic region32. However, the comparison of IpA2 at the start of coulometry with IpC1 at the end of coulometry shows that, IpC1 at
the end of coulometry is equal to IpA2 at the start of coulometry . It should be noted that, in an ECE mechanism, the peak current ratio of the starting compound to the product (Ist/Ipr) in
the kinetic region is {(n1 + n2)/n2}3/2. Where, n1 and n2 are the number of electrons involved in the oxidation of starting compound and product, respectively32. This discrepancy implying
that oxidation pathway of AO7, is not an ECE and confirms the reaction pathway presented in Fig. 3, for oxidative behaviour of AO7. Contrary to Fig. 2I,II shows that, IpC0 at the start of
coulometry, , is more than 2.5 times than that of IpA1 at the end of coulometry which is near to theoretical value of 2.8 when n1 = n233.
Figure 2Cyclic voltammograms of AO7 (0.1 mmol) in aqueous phosphate buffer (c = 0.2 M, pH = 2.0) at a glassy carbon electrode in a divided cell, during controlled potential electrolysis at: (I) Eapp
= +0.9 V and (II) Eapp = −0.2 V vs. Ag/AgCl, (a) at the beginning of electrolysis and (b) at the end of electrolysis. Scan rate: 50 mV s−1. T = 25 ± 1.
Full size imageFigure 3Oxidation and reduction pathways of AO7 in acidic media.
Full size imageDiagnostic criteria of cyclic voltammetry accompanied by previously published data on oxidation of AO734,35,36,37,38,39,40,41,42,43 allow us to propose the mechanism presented in Fig. 3 for
the electrochemical oxidation and reduction of AO7. In oxidation pathway, generation of AO7ox is followed by the addition of H2O and formation of AO7OH. At the final step, AO7OH degraded
into 4-nitrosobenzenesulfonate (4NB) and 1-iminonaphthalen-2(1 H)-one (INO). It should be noted that, according to the Fig. 3, the number of transferred electron in oxidative pathway is two
electrons, while, the obtained number of transferred electron (from controlled potential electrolysis) was 2.9 electrons. This discrepancy can be related to partially oxidation of 4NB19.
In the reduction pathway, the first two-electron reduction converts AO7 to AO7R. In the next step, AO7R via an irreversible two-electron degradation process converts to
4-aminobenzenesulfonate (4AB) and 1-aminonaphthalen-2-ol (ANO).
According to the above data, the cathodic peak C0 corresponds to reduction of AO7 to its reduced form (AO7R). The anodic peak A2 corresponds to the two-electron oxidation of AO7 to its
oxidized form (AO7ox). Obviously, the cathodic peak C2 corresponds to the reduction of AO7ox into AO7 and the redox couple A1/C1, are related to the oxidation of ANO to INO and reduction of
INO to ANO within a quasi-reversible two-electron, two-proton process.
Adsorption StudyFig. S4 shows the normalized oxidative cyclic voltammograms of AO7 (the data of Fig. S4, obtained from Fig. S1). The normalization was performed by dividing the current by the square root of
the potential scan rate (I/v1/2). According to the proposed pathway for the electrochemical oxidation of AO7, the increasing of normalized A2 peak current (IpA2/v1/2), was unexpected. One
possibility for such inconsistency is adsorption of AO7 on the electrode surface. To confirm this finding, the plot of log IPA2 vs. log v at pH values 2.4, 7.2 and 10.3 is shown in Fig S5.
It was reported that when the slope log IPA2 vs. log v is 0.5, the electrochemical reaction is a diffusion controlled process, while when the slope increases to 1, the electrochemical
reaction occurs via an adsorption-controlled process32. It is clear that, in all pHs, the slope is more than 0.5 and increases with increasing pH from 0.58 to 0.74. These values are greater
than 0.5 for the diffusion controlled process and are less than one, which is theoretical value for the adsorption-controlled electrode process. Therefore, it is clear that the
electrochemical oxidation of AO7 at glassy carbon electrode in aqueous media is adsorption/diffusion process. These results show that the interaction between anionic forms of AO7 and the
electrode surface is stronger than neutral form.
The Effect of pHThe electrochemical behavior of AO7 has been studied in different pH values. The oxidative and reductive cyclic voltammograms of AO7 in aqueous solution with various pHs are shown in Figs S6
and S7, respectively. As seen in Fig. S6, with increasing pH, EpA2 shifts to negative values. This confirms the participation of proton(s) in the oxidation of AO7. The variation of EpA2
with pH is given by:
where m is the number of protons involved in the reaction, is the anodic peak potential at pH = 0.0 and R, T, and F have their usual meaning. The EpA2–pH diagram comprise three linear
segments with different equations and slopes at pH values 7.4 and 11.4 (Fig. 4I). This diagram indicates that in the aqueous solutions, AO7 is in different reduced and oxidized forms, that
their relative amounts are dependent on the pH and electrode potential. At pHs lower than 7.4, the EpA2 value shifts by −29 mV/pH indicating that the redox reaction is
two-electron/one-proton process involving the oxidation protonated AO7 (HAO7) to protonated AO7ox (Fig. 5, Eq. 1)44,45,46. Moreover, at pH range 7.4–11.4, the EpA2 value shifts by −61 mV/pH.
In this range of pH, the redox reaction is two-electron/two-proton process involving the oxidation of HAO7 to AO7ox (Fig. 5, Eq. 2).
Figure 4The potential-pH diagram for oxidation and reduction of AO7.
Full size imageFigure 5Oxidation and reduction pathways of AO7 at different pH values and acid/base equilibriums of HAO7ox/AO7ox, AO7/AO7− and HANO/INO.
Full size imageFinally, at pHs >11.4, the EpA2 value is independent of pH, showing that the redox reaction involves a two-electron process without participation of any proton including the oxidation of AO7
anion (AO7−) to AO7ox (Fig. 5, Eq. 3). The important point of Fig. 5 is the absence of neutral AO7, which can be related to the high tendency of AO7 to keep the proton on the nitrogen atoms
due to the intramolecular hydrogen bonding (Fig. S8). In addition, the calculated pKa for HAO7ox/AO7ox and AO7/AO7− equilibria which is also shown in Fig. 5, Eqs 4 and 5, are: 7.4 and 11.4,
respectively.
The reductive cyclic voltammograms of AO7 in aqueous solution with different pHs are shown in Fig. S7. As seen, with increasing pH, peak C0 shifts to negative potentials, indicating the
participation of proton(s) in the electrode process. The EPC0–pH diagram is shown in Fig. 4II. It has two linear segments at pH 7.7. At pH values lower than 7.7, EpC0 value shifts by −90
mV/pH indicating the redox reaction is two-electron/three-proton process involving the reduction of HAO7 to H2AO7R (Fig. 5, Eq. 6). However, at pHs >7.7, the situation is a little
complicated, as the EpC0 value shifts by −40 mV/pH. It probably including unrecognizable reductions HAO7 to HAO7R (two-electron/two-proton process) and HAO7 to AO7R (two-electron/one-proton
process) (Fig. 5, Eqs. 7 and 8).
The cyclic voltammograms regarding A1/C1 peaks at different pHs and its E1/2–pH diagram are shown in Fig. S9. The E1/2 values were calculated as the average of the anodic and cathodic peak
potentials (EpA1 + EpC1)/2. The E1/2–pH diagram displays a simple linear dependence of the E1/2 on pH, with the slope of −85 mV/pH, indicating that the redox reaction is
two-electron/three-proton process involving the oxidation of HANO to INO in the forward scan and reduction of INO to HANO in the reverse scan (Fig. 5, Eq. 9). The extrapolation of this line
to pH = 0.0 provides the E1/2 = +0.60 V versus Ag/AgCl for redox couple A1/C1.
Electrochemical Study of AO7 in the Presence of Arylsulfinic acidsBecause of two reasons: a) confirmation of the proposed mechanism in Fig. 3 and b) electrochemical synthesis of some new organic compounds, in this part, the electrochemical behavior of AO7
in the presence of 4-toluenesulfinic acid (1a) was studied and compared with that of AO7 in the absence of 1a (Fig. 6). The cyclic voltammogram of AO7 in aqueous phosphate buffer (c = 0.2 M,
pH = 2.0), in the presence of 1a is shown in Fig. 6b. Comparison of the voltammogram with that of AO7 in the absence of 1a (Fig. 6a), shows two important differences: (a) the appearance of
a new redox couple (peaks A3 and C3) at E1/2 = 0.39 V vs Ag/AgCl. (b) The disappearance of the cathodic peak C1 in the reverse scan. Under these conditions, the peak current ratio,
IpC1/IpA1, depends on the both potential scan rate and 1a concentration, so that, IpC1/IpA1 increases with increasing scan rate and decreasing 1a concentration. In Fig. 6, curve c is the
voltammogram of 1a in the same conditions and in the absence of AO7 that does not show any peak in the working potential range. In addition, cyclic voltammogram d, is belong to the isolated
product from the electrolysis of AO7 in the presence of 1a. In this part, the same results were obtained for the other two arylsulfinic acids (1b and 1c). These results and the spectroscopic
data of the isolated electrolysis product all point out to compound 3a (final product), which would have been formed according to the pathway shown in Fig. 7.
Figure 6: (a) Cyclicvoltammogram of AO7 (1.0 mM) in the absence, (b) in the presence of 4-toluenesulfinic acid (1a) (1.0 mM), (c) 4-toluenesulfinic acid (1.0 mM) and (d) isolated product (3a) (0.05 mM), at a
glassy carbon electrode, in aqueous phosphate buffer (c = 0.2 M, pH = 2.0).
Scan rate: 100 mV s−1, T = 25 ± 1 °C.
Full size imageFigure 7Proposed mechanism for the electrochemical oxidation of AO7 in the presence of arylsulfinic acids (1a–1c).
Full size imageThe first step in the synthesis of 3a is the generation of INO. This compound is directly generated from oxidative cleavage of AO7 (Fig. 3), and/or from the two-electron oxidation of ANO
(Fig. 7). In the next step, INO would serve as a Michael acceptor in a reaction with 1a to form the final product 3a. The oxidation of 3a did not occur during the electrolysis due to the
insolubility of 3a in electrolysis solution (aqueous phosphate buffer). Based on Fig. 7, the anodic and cathodic peaks A3 and C3 pertain to the oxidation of 3a to 4a and vice versa.
According to the proposed mechanism in Fig. 3, we have designed a paired electrochemical strategy for the synthesis of the sulfone derivatives 3a–3c. The paired electrochemical synthesis of
3a–3c has been successfully performed in a one-pot process at the current density of 0.32 mA/cm2, in an undivided cell equipped with carbon rods as cathode and anode. The electrolysis was
terminated when the cathodic peak that corresponds to the reduction of AO7 (C0) disappears. This peak disappears after consumption of 4.0 F/mol electricity. Under these conditions, the
monitoring of the electrolysis progress was carried out by cyclic voltammetry and shown in Fig. 8. This Figure shows that, proportional to the advancement of coulometry, IpC0, IpA1 and IpA2
decreases while IpA3 and IpC3 increases. The variation of IpC0 vs. charge consumed is also shown in Fig. 8 (inset). This curve show that IpC0 decrease exponentially with advancing coulometry
(IpC0 = 10.15 e−0.03Q). The total amount of charge passed for terminating the reaction was determined from the extrapolation to the X-axis. The calculated charge passed confirms consumption
of about 4e− per molecule of AO7.
Figure 8: Cyclic voltammograms of AO7 (0.25 mmol) in the presence of 4-toluenesulfinic acid (1a) (0.25 mmol) in aqueous phosphate buffer (c = 0.2 M, pH= 2.0), at a glassy carbon electrode during constant current coulometry, after consumption of (a) 0, (b) 20, (c) 35, (d) 50, (e) 65, (f) 80 and (g) 95 C.
Current density: 0.32 mA cm−1. Scan rate: 50 mV s−1. Inset: variation of peak current (IpC0) vs. charge consumed. T = 25 ± 1.
Full size imageIn addition, the UV–visible spectra of AO7 in the presence of 4-toluenesulfinic acid (1a) were collected during a constant current coulometry in the same conditions as before (Fig. S11).
Under these conditions, the absorption spectrum of AO7 consists of three absorption bands at 312, 410 and 488 nm. Our data show as the coulometry is carried out, the height of all three
peaks decrease and a new peak appears at 352 nm and grows in intensity.
INO is an asymmetric Michael acceptor and can be attacked by 1a via 1,4 (site A) or 1,6 (site B) Michael addition reaction to yield two types of products (3a and 3a’) (Fig. 9). However, the
recorded NMR spectrum shows a singlet peak at 8.04 ppm belongs to aromatic proton. This result confirms that the 1,4-Michael addition is more probable reaction and compound 3a is the final
product of the electrochemical oxidation of AO7 in the presence of 1a.
Figure 9The structures of possible compounds 3a and 3a’.
Full size imageIn order to increase the yield of 3a–3c, some affecting factors must be optimized. Therefore, the effects of two of the most important factors, applied current density and charge were
investigated by setting all parameters to be constant and optimizing one each time. The effect of charge passed was studied in the range of 1 to 6 F mol−1, while the other parameters are as
follows: temperature = 298 K, current density = 0.32 mA/cm2, electrode surface = 31.2 cm2, AO7 = 0.1 mmol and 1a = 0.1 mmol are kept constant. As is shown in Fig. S10a, the maximum product
yield appears in 4.1 F mol−1 charge consumed. The product yield decreases with increasing charge passed from 4.1 F mol−1 probably due to the occurrence of side reaction(s) such as
over-oxidation. Furthermore, the effect of applied current density on product yield was studied in the range 0.16–1.6 mA cm−2, while the other parameters (temperature = 298 K, charge
consumed = 40 C, electrode surface = 31.2 cm2, AO7 = 0.1 mmol, and of 1a = 0.1 mmol) are kept constant. The results show that, with increasing the current density from 0.32 mA cm−2, the
product yield decreases (Fig. S10b). The product yield decreasing in current densities greater than 0.32 mA cm−2, can be related to some side reactions such as oxidation of solvent,
nucleophile or over-oxidation of 3a and/or INO. To evaluate the usefulness of the pair strategy in the synthesis of 3a–3c, electrochemical synthesis of 3a was performed in a divided cell in
both oxidative and reductive conditions. Our data confirms that in a divided cell (in both cases) (unpaired condition), (a) the yield of 3a is lower and (b) the charge consumption is greater
than that of in undivided cell.
ConclusionsThis work provides new insights into the electrochemical behavior of AO7 in aqueous solutions in both oxidative and reductive regions and shows that both oxidation and reduction of AO7
leading to the formation of a redox couple (ANO/INO) (Fig. 3). In addition, the pH dependence of AO7 and other intermediates was studied in order to understand the predominant species,
oxidation and reduction pathways and adsorption study. For example, our data shows that, the interaction between anionic forms of AO7 and the electrode surface is stronger than neutral form.
Furthermore, in this work, the electrochemical oxidation/reduction of AO7 has been investigated in the presence of arylsulfinic acids (1a–1c) as nucleophiles, in acidic solutions. Our data
display that the intermediate (INO) is attacked by the nucleophile, 1a–1c, to give the final product 3a–3c (Fig. 7). Clean synthesis, technical feasibility (using galvanostatic method and
simple cell), use of electricity instead of oxidative or reductive reagents, one-step process, work in room temperature and pressure and use of aqueous solution instead of organic solvents,
are the advantages of this method.
Materials and MethodsApparatus and ReagentsCyclic voltammetry, controlled-potential coulometry and preparative electrolysis were performed using an Autolab model PGSTAT 30 and a Behpazho potentiostat/galvanostat. The working and
counter electrode used in macro-scale electrolysis and coulometry was an assembly of four ordinary soft carbon rods (6 mm diameter and 4 cm length). Working electrode used in the cyclic
voltammetry experiments was a glassy carbon disc (1.8 mm diameter) and a platinum rod was used as a counter electrode. The electrosynthesis were performed under constant-current condition in
an undivided cell. The glassy carbon electrode was polished using alumina slurry (from Iran Alumina Co.). More details are described in our previous paper47. Acid orange 7, arylsulfinic
acids and phosphate salts were obtained from commercial sources. These chemicals were used without further purification.
Electroorganic Synthesis of 3a–3cAn aqueous phosphate buffer solution (70 ml, c = 0.2 M, pH 2.0) containing AO7 (0.25 mmol) and arylsulfinic acid (1a–1c) (0.25 mmol) was electrolyzed in an undivided cell under constant
current conditions (current density = 0.32 mA cm−2) for 2 h 45 min. At the end of electrolysis, the cell was placed in a refrigerator overnight. The precipitated solid was collected by
filtration and washed several times with water. After recrystallization in ethyl ether, the products were characterized by IR, 1H NMR, 13C NMR and mass
spectroscopy.
1-Amino-3-tosylnaphthalen-2-ol (C17H15NO3S) (3a)mp: 163–164 °C; isolated yield 65%. 1H NMR (400 MHz, DMSO-d6): δ = 2.32 (s, 3 H, methyl), 6.22 (s, ~1 H, NH, this peak disappeared upon addition of D2O), 7.36 (m, 4 H, J = 8 Hz, aromatic),
7.71 (d, 2 H, J = 8.4, aromatic), 8.04 (s, 1 H, aromatic), 8.11 (m, 1 H, aromatic), 8.29 (m, 1 H, aromatic), 9.86 (s, ~1 H, OH, this peak disappeared upon addition of D2O); 13C NMR (100 MHz,
DMSO-d6): δ = 21.3, 119.5, 120.5, 122.7, 123.1, 123.9, 124.8, 125.0, 125.8, 126.9, 130.2, 136.7, 137.7, 140.4, 143.7; IR (KBr): 3384, 2926, 1704, 1622, 1354, 1266, 1200, 1140, 1083, 951,
755, 668, 571, 529 cm−1; MS (EI, 70 eV): m/z (relative intensity %): 313 (M+, 31), 270 (6), 158 (100), 139 (11), 130 (65), 91 (24), 77 (15),
65(18).
1-amino-3-(phenylsulfonyl)naphthalen-2-ol (C16H13NO3S) (3b)mp: 209–210 °C; isolated yield 60%. 1H NMR (400 MHz, DMSO-d6): δ = 6.27 (s, ~1 H, NH, this peak disappeared upon addition of D2O), 7.38 (dd, 2 H, J = 3.2 and 10.0 Hz, aromatic), 7.57 (m, 3
H, aromatic), 7.85 (dd, 2 H, J = 2.0 and 6.8 Hz, aromatic), 8.09 (s, 1 H, aromatic), 8.18 (dd, 1 H, J = 3.2 and 10.0 Hz, aromatic), 8.33 (dd, 1 H, J = 3.2 and 10.0 Hz, aromatic), 9.91 (s, ~1
H, OH, this peak disappeared upon addition of D2O); 13C NMR (100 MHz, DMSO-d6): δ = 118.8, 120.7, 122.7, 123.2, 123.9, 124.7, 125.1, 125.8, 126.8, 129.8, 133.1, 136.8, 138.2, 143.4; IR
(KBr): 3473, 3379, 3073, 3025, 1739, 1616, 1358, 1286, 1207, 1136, 1082, 952, 784, 740, 557 cm−1; MS (EI, 70 eV): m/z (relative intensity %): 299 (M+, 21), 257 (7), 160 (16), 159 (55), 158
(100), 130 (58), 103 (16), 77(44), 51(24), 43 (74).
1-amino-3-((4-chlorophenyl)sulfonyl)naphthalen-2-ol (C16H12ClNO3S) (3c)mp: 164–165 °C; isolated yield 62%. 1H NMR (400 MHz, DMSO-d6): δ = 6.33 (s, ~1 H, NH, this peak disappeared upon addition of D2O), 7.38 (dd, 2 H, J = 3.2 and 9.6 Hz, aromatic), 7.62 (d, 2 H,
J = 8.8 Hz, aromatic), 7.85 (d, 2 H, J = 8.8 Hz, aromatic), 8.06 (d, 1 H, J = 2.8 Hz, aromatic), 8.19 (m, 1 H, aromatic), 8.28 (m, 1 H, aromatic), 9.92 (s, ~1 H, OH, this peak disappeared
upon addition of D2O); 13C NMR (100 MHz, DMSO-d6): δ = 118.2, 120.6, 122.6, 123.1, 123.7, 124.3, 124.9, 125.0, 126.2, 128.8, 129.9, 136.7, 138.2, 142.0; IR (KBr): 3362, 3286, 3068, 1630,
1309, 1284, 1174, 1144, 1085, 906, 824, 781, 752, 648, 580 cm−1; MS (EI, 70 eV): m/z (relative intensity %): 333 (M+, 24), 174 (12), 158 (100), 130 (58), 111 (21), 103 (18), 77 (17), 75
(28), 50 (18).
Additional InformationHow to cite this article: Momeni, S. and Nematollahi, D. New insights into the electrochemical behavior of acid orange 7: Convergent paired electrochemical synthesis of new aminonaphthol
derivatives. Sci. Rep. 7, 41963; doi: 10.1038/srep41963 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References Silva, J. P. et al. Adsorption of acid orange 7 dye in aqueous solutions by spent brewery grains. Sep. Purif. Technol. 40, 309–315 (2004).
Article CAS Google Scholar
Zollinger, H. Color Chemistry: Syntheses, Properties, and Applications of Organic Dyes and Pigments. (John Wiley & Sons, 2003).
Hihara, T., Okada, Y. & Morita, Z. Azo-hydrazone tautomerism of phenylazonaphthol sulfonates and their analysis using the semiempirical molecular orbital PM5 method. Dyes Pigm. 59, 25–41
(2003).
Article CAS Google Scholar
Özen, A. S., Doruker, P. & Aviyente, V. Effect of cooperative hydrogen bonding in azo-hydrazone tautomerism of azo dyes. J. Phys. Chem. A 111, 13506–13514 (2007).
Article CAS PubMed Google Scholar
Gordon, P. F. & Gregory, P. Organic Chemistry in Colour (Springer Science & Business Media, 2012).
Oakes, J. & Gratton, P. Kinetic investigations of azo dye oxidation in aqueous media. J. Chem. Soc. Perk. T. 2, 1857–1864 (1998).
Article Google Scholar
Oakes, J., Gratton, P., Clark, R. & Wilkes, I. Kinetic investigation of the oxidation of substituted arylazonaphthol dyes by hydrogen peroxide in alkaline solution. J. Chem. Soc. Perk. T. 2,
2569–2576 (1998).
Article Google Scholar
Hihara, T., Okada, Y. & Morita, Z. Reactivity of phenylazonaphthol sulfonates, their estimation by semiempirical molecular orbital PM5 method, and the relation between their reactivity and
azo-hydrazone tautomerism. Dyes Pigm. 59, 201–222 (2003).
Article CAS Google Scholar
Allen, R. L. Colour Chemistry (Springer Science & Business Media, 2013).
Christie, R. Colour Chemistry (Royal Society of Chemistry, 2014).
Thomas, F. & Boto, K. The Chemistry of the Hydrazo, Azo and Azoxy Groups. 443–493 (Wiley-VCH, Weinheim, 1975).
Ember, E., Rothbart, S., Puchta, R. & van Eldik, R. Metal ion-catalyzed oxidative degradation of orange II by H2O2. High catalytic activity of simple manganese salts. New J. Chem. 33, 34–49
(2009).
Article CAS Google Scholar
Robinson, T., McMullan, G., Marchant, R. & Nigam, P. Remediation of dyes in textile effluent: a critical review on current treatment technologies with a proposed alternative. Bioresource
Technol. 77, 247–255 (2001).
Article CAS Google Scholar
Waring, D. R. & Hallas, G. The Chemistry and Application of Dyes (Springer Science & Business Media, 2013).
Fuchigami, T., Atobe, M. & Inagi, S. Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices (John Wiley & Sons, 2014).
Varmaghani, F., Nematollahi, D., Mallakpour, S. & Esmaili, R. Electrochemical oxidation of 4-substituted urazoles in the presence of arylsulfinic acids: an efficient method for the synthesis
of new sulfonamide derivatives. Green Chem. 14, 963–967 (2012).
Article CAS Google Scholar
Salehzadeh, H., Nematollahi, D. & Hesari, H. An efficient electrochemical method for the atom economical synthesis of some benzoxazole derivatives. Green Chem. 15, 2441–2446 (2013).
Article CAS Google Scholar
Nematollahi, D., Hosseiny Davarani, S. S. & Mirahmadpour, P. A green approach for the electroorganic synthesis of new dihydroxyphenyl-indolin-2-one derivatives. ACS Sustainable Chem. Eng. 2,
579–583 (2014).
Article CAS Google Scholar
Khazalpour, S. & Nematollahi, D. Electrochemical and chemical synthesis of different types of sulfonamide derivatives of N,N-dimethyl-1,4-benzenediamine using 4-nitroso-Nα-dimethylaniline.
Green Chem. 17, 3508–3514 (2015).
Article CAS Google Scholar
Maleki, A., Nematollahi, D., Rasouli, F. & Zeinodini-Meimand, A. Electrode instead of catalyst and enzyme. A greener protocol for the synthesis of new 2-hydroxyacetamide derivatives
containing a γ-lactone ring. Green Chem. 18, 672–675 (2016).
Article CAS Google Scholar
Nematollahi, D. & Rafiee, M. Diversity in electrochemical oxidation of dihydroxybenzoic acids in the presence of acetylacetone. A green method for synthesis of new benzofuran derivatives.
Green Chem. 7, 638–644 (2005).
Article CAS Google Scholar
Salahifar, E., Nematollahi, D., Bayat, M., Mahyari, A. & Amiri Rudbari, H. Regioselective green electrochemical approach to the synthesis of nitroacetaminophen derivatives. Org. Lett. 17,
4666–4669 (2015).
Article CAS PubMed Google Scholar
Paddon, C. A., Pritchard, G. J., Thiemann, T. & Marken, F. Paired electrosynthesis: micro-flow cell processes with and without added electrolyte. Electrochem. Commun. 4, 825–831 (2002).
Article CAS Google Scholar
Amatore, C., Lund, H. & Baizer, M. Organic Electrochemistry (Marcel Dekker, New York, 1991).
Hammerich, O. & Lund, H. Organic Electrochemistry (CRC Press, 2000).
Nematollahi, D. & Varmaghani, F. Paired electrochemical synthesis of new organosulfone derivatives. Electrochim. Acta 53, 3350–3355 (2008)
Article CAS Google Scholar
Habibi, D., Pakravan, N. & Nematollahi, D. The green and convergent paired Diels–Alder electro-synthetic reaction of 1,4-hydroquinone with 1,2-bis (bromomethyl) benzene. Electrochem. Commun.
49, 65–69 (2014).
Article CAS Google Scholar
Sharafi-Kolkeshvandi, M., Nematollahi, D. & Nikpour, F. A Green CC bond formation reaction between N,N′-diphenylbenzene-1, 4-diamine and Michael donors: A convergent paired strategy. J.
Electrochem. Soc. 163, G75–G78 (2016).
Article CAS Google Scholar
Beale, Jr. J. M. & Block, J. (eds) Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry. 12th ed. (Lippincott Williams and Wilkins, Philadelphia, 2011).
Nematollahi, D., Baniardalan, M., Khazalpour, S. & Pajohi-Alamoti, M. R. Product diversity by changing the electrode potential. Synthesis, kinetic evaluation and antibacterial activity of
arylsulfonyl-4,4′-biphenol and bis-arylsulfonyl-4, 4′-biphenol derivatives. Electrochim. Acta 191, 98–105 (2016).
CAS Google Scholar
Khazalpour, S., Nematollahi, D. & Pajohi-Alamoti, M. R. A green approach for the synthesis of bis (substituted sulfabenzamide) para-benzoquinone based on the reaction of sulfabenzamide with
electrochemically generated para-benzoquinone and its antibacterial evaluation. New J. Chem. 39, 6734–6737 (2015).
Article CAS Google Scholar
Bard, A. J. & Faulkner, L. R. Electrochemical Methods: Fundamentals and Applications (Wiley, 2000).
Nicholson, R. S. & Shain, I. Theory of stationary electrode polarography. Single scan and cyclic methods applied to reversible, irreversible, and kinetic systems. Anal. Chem. 36, 706–723
(1964).
CAS Google Scholar
Abbott, L. C., Batchelor, S. N., Smith, J. R. L. & Moore, J. N. Reductive reaction mechanisms of the azo dye orange II in aqueous solution and in cellulose: from radical intermediates to
products. J. Phys. Chem. A 113, 6091–6103 (2009).
Article CAS PubMed Google Scholar
Mu, Y., Rabaey, K., Rozendal, R. A., Yuan, Z. & Keller, J. Decolorization of azo dyes in bioelectrochemical systems. Environ. Sci. Technol. 43, 5137–5143 (2009).
Article ADS CAS PubMed Google Scholar
Zhang, S. J., Yu, H. Q. & Li, Q. R. Radiolytic degradation of acid orange 7: A mechanistic study. Chemosphere 61, 1003–1011 (2005).
Article ADS CAS PubMed Google Scholar
Lopez, C. et al. Mechanism of enzymatic degradation of the azo dye orange II determined by ex situ1H nuclear magnetic resonance and electrospray ionization-ion trap mass spectrometry. Anal.
Biochem. 335, 135–149 (2004).
Article CAS PubMed Google Scholar
Ozcan, A., Oturan, M. A., Oturan, N. & Şahin, Y. Removal of acid orange 7 from water by electrochemically generated Fenton’s reagent. J. Hazard. Mater. 163, 1213–1220 (2009).
Article CAS PubMed Google Scholar
Hammami, S., Bellakhal, N., Oturan, N., Oturan, M. A. & Dachraoui, M. Degradation of acid orange 7 by electrochemically generated OH radicals in acidic aqueous medium using a boron-doped
diamond or platinum anode: A mechanistic study. Chemosphere 73, 678–684 (2008).;
Article ADS CAS Google Scholar
Wu, J., Zhang, H. & Qiu, J. Degradation of acid orange 7 in aqueous solution by a novel electro/Fe 2+/peroxydisulfate process. J. Hazard. Mater. 215, 138–145 (2012).
Article ADS CAS PubMed Google Scholar
Zhao, H. Z., Sun, Y., Xu, L. N. & Ni, J. R. Removal of acid orange 7 in simulated wastewater using a three-dimensional electrode reactor: Removal mechanisms and dye degradation pathway.
Chemosphere 78, 46–51 (2010).
Article ADS CAS PubMed Google Scholar
Zheng, J., Gao, Z., He, H., Yang, S. & Sun, C. Efficient degradation of acid orange 7 in aqueous solution by iron ore tailing Fenton-like process. Chemosphere 150, 40–48 (2016).
Article ADS CAS PubMed Google Scholar
Cai, C., Zhang, Z. & Zhang, H. Electro-assisted heterogeneous activation of persulfate by Fe/SBA-15 for the degradation of orange II. J. Hazard. Mater. 313, 209–218 (2016).
Article ADS CAS PubMed Google Scholar
Baymaky, M. S. & Zuman, P. Equilibria of formation and dehydration of the carbinolamine intermediate in the reaction of benzaldehyde with hydrazine. Tetrahedron 63, 5450–5454 (2007).
Article CAS Google Scholar
Baymak, M. S., Celik, H., Ludvik, J., Lund H. & Zuman P. Diprotonated hydrazones and oximes as reactive intermediates in electrochemical reductions. Tetrahedron Lett. 45, 5113–5115 (2004).
Article CAS Google Scholar
Shine, H. J., Zmuda, H. & Park, K. H. Mechanism of the benzidine rearrangement. Kinetic isotope effects and transition states. Evidence for concerted rearrangement. J. Am. Chem. Soc. 103,
955–956 (1981).
Article CAS Google Scholar
Nematollahi, D., Dehdashtian, S. & Niazi, A. Electrochemical oxidation of some dihydroxybenzene derivatives in the presence of indole. J. Electroanal. Chem. 616, 79–86 (2008).
Article CAS Google Scholar
Download references
AcknowledgementsWe acknowledge the Bu-Ali Sina University Research Council and Center of Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS) for their support of
this work.
Author informationAuthors and Affiliations Faculty of Chemistry, Bu-Ali Sina University, Hamedan, Zip Code 65178-38683, Iran
Shima Momeni & Davood Nematollahi
AuthorsShima MomeniView author publications You can also search for this author inPubMed Google Scholar
Davood NematollahiView author publications You can also search for this author inPubMed Google Scholar
ContributionsS.M. and D.N. conceived and designed the study. S.M. did the experiments. S.M. and D.N. wrote the manuscript. D.N. directed the research.
Corresponding author Correspondence to Davood Nematollahi.
Ethics declarations Competing interestsThe authors declare no competing financial interests.
Supplementary informationSupporting Information (PDF 1981 kb)Rights and permissionsThis work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons
license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to
reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
Reprints and permissions
About this articleCite this article Momeni, S., Nematollahi, D. New insights into the electrochemical behavior of acid orange 7: Convergent paired electrochemical synthesis of new
aminonaphthol derivatives. Sci Rep 7, 41963 (2017). https://doi.org/10.1038/srep41963
Download citation
Received: 24 August 2016
Accepted: 29 December 2016
Published: 06 February 2017
DOI: https://doi.org/10.1038/srep41963
Share this article Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article.
Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative







