- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Spirocyclic tetrahydronaphthyridines (THNs) are valuable scaffolds for drug discovery campaigns, but access to this 3D chemical space is hampered by a lack of modular and scalable
synthetic methods. We hereby report an automated, continuous flow synthesis of α-alkylated and spirocyclic 1,2,3,4-tetrahydro-1,8-naphthyridines (“1,8-THNs”), in addition to their
regioisomeric 1,6-THN analogues, from abundant primary amine feedstocks. An annulative disconnection approach based on photoredox-catalysed hydroaminoalkylation (HAA) of halogenated
vinylpyridines is sequenced in combination with intramolecular SNAr _N_-arylation. To access the remaining 1,7- and 1,5-THN isomers, a photoredox-catalysed HAA step is telescoped with a
palladium-catalysed C–N bond formation. Altogether, this provides a highly modular access to four isomeric THN cores from a common set of unprotected primary amine starting materials, using
the same bond disconnections. The simplifying power of the methodology is illustrated by a concise synthesis of the spirocyclic THN core of Pfizer’s MC4R antagonist PF-07258669. SIMILAR
CONTENT BEING VIEWED BY OTHERS DIRECT ACCESS TO SPIROCYCLES BY PD/WINGPHOS-CATALYZED ENANTIOSELECTIVE CYCLOADDITION OF 1,3-ENYNES Article Open access 27 September 2021 ACCESSING LADDER-SHAPE
AZETIDINE-FUSED INDOLINE PENTACYCLES THROUGH INTERMOLECULAR REGIODIVERGENT AZA-PATERNÒ–BÜCHI REACTIONS Article Open access 16 February 2024 MODULAR ACCESS TO CHIRAL BRIDGED
PIPERIDINE-Γ-BUTYROLACTONES VIA CATALYTIC ASYMMETRIC ALLYLATION/_AZA_-PRINS CYCLIZATION/LACTONIZATION SEQUENCES Article Open access 02 January 2024 INTRODUCTION Bicyclic compounds featuring
saturated _N_-heterocycles fused to (hetero)aromatic units are highly prized in medicinal chemistry1,2,3,4, offering a combination of polar functionality, high Fsp3-content, and rigidly
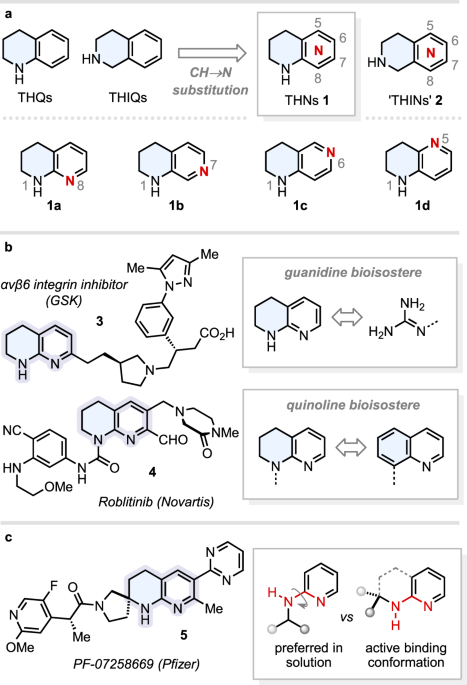
disposed groups on the aromatic core that can engage in key interactions with a protein target (e.g., H-bonds)5,6. Tetrahydronaphthyridines (THNs) are semi-saturated bicycles that ring-fuse
a piperidine with a pyridine—these in turn being the two most popular _N_-heterocycles deployed in small-molecule pharmaceuticals7,8. Positioning of the two THN nitrogen atoms generates
eight different structural isomers: four of which (1A–D) can be considered as CH → N bioisosteres of tetrahydroquinolines (THQs), and the remaining four (structures 2) as CH → N bioisosteres
of tetrahydroisoquinolines (THIQs) (Fig. 1a). The substitution of CH units for N atoms in (hetero)aromatic systems can impart orders of magnitude improvements in key physicochemical (e.g.,
solubility) and pharmacological parameters9, and synthetic strategies that could provide facile access to any THN isomer (e.g., 1A–D) would be highly enabling. Without a trivial naming
convention for THNs, we shall hereafter refer to structures 1 as “THNs” and their isomeric counterparts 2 as “THINs”10,11,12,13,14,15,16, by analogy to THQs and THIQs. Amongst other
applications17,18, THNs have found use as guanidine mimetics of the arginine binding motif in RGD-binding integrin inhibitors (e.g., 3)19,20. Scaffold morphing of quinolines to THNs can also
be an effective tactic to improve aqueous solubility, as exemplified during the development of the FGFR4 selective inhibitor Roblitinib (FGF401) 4 (Fig. 1b)21. Spirocyclisation of fused,
semi-saturated _N_-heterocycles is also emerging as a powerful design strategy for medicinal chemistry. When compared to their flat, all-aromatic counterparts, partial saturation and
installation of a spirocycle simultaneously increases Fsp3, reduces structural flexibility, and introduces alternative exit vectors for access to novel 3D chemical space22. In favourable
cases, this can lead to greatly enhanced potency, selectivity, solubility, and metabolic stability23,24. For instance, Pfizer have exploited a spirocyclic THN as the core of their MC4R
antagonist PF-07258669 5, which is currently in phase I clinical trials for the treatment of appetite loss (Fig. 1c)25. The spirocycle in 5 was rationally designed to enforce a
_cis_-relationship between the N–H bond and the adjacent N(sp2) lone pair, which is the optimal geometry for target binding but opposite to the (_trans_) conformer favoured in solution for
the non-constrained analogue. Despite the many opportunities for structural and physicochemical tuning that THNs can offer, their widespread adoption in early-stage drug discovery has likely
been hampered by their poor commercial availability, and the scarcity of THN synthesis approaches that are readily amenable to library generation. Other than the semi-hydrogenation of
naphthyridines26,27,28, which can present regio- and chemoselectivity challenges, several routes to THNs have been devised based on the annulation of 2-aminopyridines. These processes tend
to be relatively labour-intensive29,30, and whilst catalytic annulations do exist31,32, their functional group tolerance is low. Inverse electron demand, intramolecular, hetero-Diels-Alder
reactions of tethered imidazolyl33 or alkynyl34,35,36 triazines sequenced with N2 extrusion are also on record for THN synthesis, but the substrate syntheses require multiple steps.
Moreover, none of the above approaches are amenable to _spirocyclic_ THN synthesis. Another distinct strategy is to form THNs via the _N_-arylative cyclisation of γ-pyridyl amines, either by
intramolecular Pd-catalysed C–N coupling25,37,38, SNAr reactions37,38,39, or Chichibabin reactions40,41. γ-Pyridyl amines 6 can themselves be constructed via Sonogashira-hydrogenation
sequences25,41, _B_-alkyl Suzuki-Miyaura coupling40, or the SN2 ring-opening of cyclic sulfamidates 9 with _ortho_-lithiated halopyridines39 (Fig. 2a). Whilst these approaches can enable
access to spirocyclic THNs, the chemistry is not well suited to library synthesis, given the meagre commercial availability of α-(di)substituted propargylic amines (7), allylic amines (8),
or γ-hydroxy amines as starting materials. Yu and co-workers have developed a Pd-catalysed γ-C(sp3)–H arylation of primary alkylamines that can access γ-pyridyl amines, and applied this to a
single example of THN synthesis, but the amines amenable to this procedure are largely unfunctionalized and have limited commercial availability42. Recently, visible-light
photoredox-catalysed approaches have been reported by ourselves and Gaunt et al., respectively, for the modular synthesis of γ-aryl primary amines by the C–C bond-forming coupling of readily
available primary alkylamines 1037 or ketone-derived imines 1138 with styrenes. Between these two disclosures, four examples of spirocyclic 1,2,3,4-tetrahydro-1,8-naphthyridine synthesis
were showcased, proceeding via the SNAr cyclisation of (isolated) γ-pyridyl amines from the photoredox step. In this work, we show that photoredox-catalysed hydroaminoalkylation43,44,45
(HAA) of halogenated vinyl pyridines, followed by intramolecular _N_-arylation via SNAr, can be sequenced in continuous flow46,47,48,49,50,51,52,53,54,55,56 to enable an automated synthesis
of α-alkylated and spirocyclic 1,2,3,4-tetrahydro-1,8-naphthyridines (“1,8-THNs”) 13, in addition to their regioisomeric 1,6-THN analogues 14 (Fig. 2b). To access the corresponding 1,7- and
1,5-THN isomers—15 and 16, respectively—a photoredox-catalysed HAA step can be telescoped with a palladium-catalysed C–N bond formation. Altogether, this provides a highly modular approach
to four THN isomers 13–16 from a common set of unprotected primary amine starting materials 10, using the same bond disconnections. RESULTS AND DISCUSSION REACTION OPTIMISATION
Photoredox-catalysed hydroaminoalkylation (HAA) of 2-fluoro-3-vinylpyridine 17 with cyclohexylamine 10A gives γ-pyridyl amine 18A in 97% yield, and subjection of this purified material to
DIPEA (1.5 equiv) in DMF at 120 °C for 20 h delivers the corresponding THN 13A in 92% yield via intramolecular SNAr cyclisation37. In order to sequence these reactions together in continuous
flow, we transferred the chemistry to a Vapourtec R-series flow system equipped with a Uniqsis PhotoSyn LED photoreactor (420 nm LEDs, ~260 W radiant output power, 5-ml reactor coil) and a
high-temperature tube reactor (up to 250 °C). As per the batch procedure, the photoredox-catalysed HAA was initially carried out with 2,4,6-tris(diphenylamino)-3,5-difluorobenzonitrile
(3DPA2FBN) as the photocatalyst, and tetrabutylammonium azide (Bu4N+N3–) as the hydrogen atom transfer (HAT) catalyst57,58. However, we found that Bu4N+N3– could be replaced with cheaper and
far less hygroscopic sodium azide (NaN3), which is soluble in DMF at 0.06 M. For the SNAr step, a temperature of 180 °C with _t_R = 20 min proved sufficient for complete conversion (see
Supplementary Table 1). By running both steps in sequence in continuous flow, an overall yield of 98% of spirocyclic THN 13A could be obtained from 2-fluoro-3-vinylpyridine 17 and
cyclohexylamine 10A as feedstocks (in a 1:1 ratio). This corresponds to a productivity of 2.20 mmol h–1 (445 mg h–1). AUTOMATED CONTINUOUS FLOW SYNTHESIS OF THNS FROM PRIMARY ALKYLAMINES
With an optimised continuous flow protocol in hand, we next sought to execute an automated library synthesis of ‘lead-like’5,59 THN products 13, using an autosampler to sequentially load
different amine substrates into the Vapourtec flow system. The same autosampler also serves as a fraction collector, into which the steady-state solutions of each product 13 are dispensed
(Fig. 3a and see Supplementary Data 1 for NMR spectra of all compounds). Each run using 1.50 mmol of the vinylpyridine substrate takes ~90 min, which corresponds to 16 compounds in a 24 h
period, or 40 compounds total over 60 h if all rack positions are utilised. Cyclic primary amines 10A–D of varying ring sizes were well tolerated, and amine 10E bearing benzylic C–H bonds
also participated smoothly. Various functionalities including free hydroxyl groups (13F, O), ethers (13G, K), thioethers (13H), carbamates (13I, J, P), and imidazoles (13Q) proved compatible
with the process. For amines bearing electronegative atoms attached to the β- or γ-carbon (10G–K, O, P), a slightly elevated temperature of 200 °C proved necessary in most cases to drive
the SNAr step to completion within the 20 min residence time. Strained four-membered ring substrates 3-amino-_N_-Boc-azetidine 10J and 3-aminooxetane 10K proved especially challenging for
the photoredox step, on account of their α-C–H bonds being strengthened by ring strain and inductive effects57; amine 10J for example gave only 49% yield of 13J, along with 46% of unreacted
10J. By increasing the stoichiometry of amines 10J and 10K to 3 equivalents, however, the valuable spirocyclic THNs 13J and 13K could be obtained in 80% and 61% yields, respectively.
Non-spirocyclic THNs are also readily accessible via this methodology; isopropylamine 10L was used, for example, to generate α,α-dimethyl-substituted THN 13L in 75% yield. For
α-monoalkylated amines (10M–Q), it proved necessary to use 3.0 equivalents of the amine substrate, to mitigate against the formation of undesired dialkylated products during the photoredox
α-C–H alkylation step37. As ethylamine 10M is a gas at ambient pressure, it was dispensed as a 2.0 M solution in THF, affording the simple α-methylated THN 13M in 36% yield. Ethanolamine 10O
and _N_-Boc ethylenediamine 10P also proved to be effective substrates, generating α-hydroxymethyl- and α-aminomethyl-substituted THNs 13O and 13P, respectively. We next sought to extend
our automated synthesis protocol to the formation of isomeric 1,2,3,4-tetrahydro-1,6-naphthyridines (“1,6-THNs”) 14, using 4-chloro-3-vinylpyridine 18 as a radical acceptor (Fig. 3b). Whilst
the chlorinated compound 18 is far easier to access than its 4-fluoropyridine-derived60 counterpart, the decreased SNAr reactivity of the C–Cl bond necessitated that the temperature of the
flow SNAr step be raised still further to 220 °C. Under these conditions, a small library of spirocyclic 1,6-THNs 14A–E could be prepared in 46–64% yield. GRAM-SCALE REACTION AND RESOLUTION
OF THN ENANTIOMERS To demonstrate the scalability of our THN synthesis in flow, we executed the reaction of 4-aminopiperidine substrate 10I on gram scale on a 5-ml reactor coil, delivering
1.85 g of spirocyclic THN 13I in 87% yield (equating to a productivity of 600 mg h–1) (Fig. 4a). Whilst these reactions inevitably produce racemic materials, resolution of the THNs via
chiral preparative HPLC provides convenient access to both enantiomers, as exemplified for THN 13N on a 520 mg scale (Fig. 4b and see Supplementary Figs. 4–8 for HLPC traces). ACCESS TO THN
DERIVATIVES WITH FUNCTIONAL HANDLES ON THE PYRIDINE RING Another important objective was to demonstrate further elaboration of the THN products on the pyridine ring. One strategy, which is
especially useful for C(6) functionalisation, is to carry out electrophilic halogenation or catalytic C–H borylation61 reactions (i.e., 19–21) (Fig. 5a). In order to access THNs 23 and 25
halogenated _ortho_ or _para_ to the pyridine nitrogen, we utilised vinyl pyridines 22 and 24, respectively, with the necessary chloro handles preinstalled. Using this strategy, the
C(7)-chloro THN 23 was isolated in 68% yield, and the C(5)-chloro THN 25 in 31% yield (Fig. 5b). The latter isomer was anticipated to be the most challenging, requiring the amine nucleophile
to distinguish between a _para_-chloro and an _ortho_-fluoro site of attack during the SNAr step39. Taken together, these strategies enable vector growth from any ring position on the fused
pyridine moiety of 1,8-THNs, which is likely to be of significant value for fragment-based drug discovery1,2,3,4. STEPWISE SYNTHESIS OF OTHER THN ISOMERS Varying the position of the
pyridine nitrogen atom in these spirocyclic THN scaffolds is another highly desirable objective from a medicinal chemistry standpoint9. Having already demonstrated an automated flow
synthesis of 1,8- and 1,6-THNs 13 and 14 from primary alkylamine feedstocks, we were motivated to develop a practical catalytic solution to access 1,7- and 1,5-THN isomers, based on the same
photoredox-catalysed HAA disconnection approach. With intramolecular _N_-arylation via SNAr no longer being feasible, we instead opted to carry out this key step using palladium catalysis.
Following a flow photoredox HAA of amine 10I with 3-chloro-4-vinylpyridine 26, γ-pyridyl amine 27 was isolated in 25% yield. The low yield in this case was traced to extensive polymerisation
side reactions, for which vinylpyridine 26 seems to be particularly prone. Subsequent cyclisation via a Buchwald-Hartwig C–N coupling then gave 1,7-THN 28 in 79% yield (Fig. 6a). An
analogous sequence using 3-chloro-2-vinylpyridine 29 gave 1,5-THN in an overall 47% yield over the two steps (Fig. 6b). APPLICATION TO THE SYNTHESIS OF PFIZER’S MC4R ANTAGONIST PF-07258669 5
Finally, we sought to apply our methodology to a concise synthesis of the spirocyclic THN core (35) of Pfizer’s MC4R antagonist PF-07258669 5, which was previously synthesised in 15 total
steps (11 steps LLS) (Fig. 7a)25. In our case, starting from commercially available 3-amino _N_-Boc pyrrolidine 32, a photocatalytic HAA reaction with vinylpyridine 33 in continuous flow
gave γ-pyridyl amine 34 (427 mg) in 79% yield. Attempted thermal SNAr cyclisation of 34 at 220 °C in a high-temperature tubular reactor (_t_R = 20 min) gave only 22% yield of THN 35,
indicating that the methyl substituent α- to the pyridine nitrogen deactivates this pathway. Fortunately, an intramolecular, palladium-catalysed Buchwald-Hartwig _N_-arylation process (as
used in the Pfizer route) proved more efficacious, delivering the spirocyclic THN core 35 in 84% yield (Fig. 7b). Taking into account a 3-step synthesis of vinyl pyridine 33, the longest
linear sequence is five steps. The industrial route, whilst 11 steps in the longest linear sequence, is enantioselective, compared to a racemic synthesis in our case. Nevertheless, this
illustrates how dramatically the synthesis of complex spirocyclic amines can be streamlined when using a photoredox annulation strategy from unprotected amines57. CONCLUSION In summary, we
have developed an automated, continuous flow synthesis of α-alkylated and spirocyclic 1,2,3,4-tetrahydro-1,8-naphthyridines (“1,8-THNs”), in addition to their regioisomeric 1,6-THN
analogues, from abundant primary amine feedstocks. An annulative disconnection approach based on photoredox-catalysed hydroaminoalkylation (HAA) of halogenated vinylpyridines is sequenced in
combination with intramolecular SNAr _N_-arylation. To access the remaining 1,7- and 1,5-THN isomers, a photoredox-catalysed HAA step is telescoped with a palladium-catalysed C–N bond
formation. Altogether, this provides a highly modular access to four isomeric THN cores from a common set of unprotected primary amine starting materials, using the same bond disconnections.
The simplifying power of the methodology is illustrated by a concise synthesis of the spirocyclic THN core (35) of Pfizer’s MC4R antagonist PF-07258669 (5). METHODS A general procedure for
the flow chemistry protocol described in Fig. 3 can be found in Supplementary Methods (pages S4–5), plus photographs and schematics of the setup in Supplementary Figs. 1–3. Representative
procedure for the automated continuous flow synthesis of 1,2,3,4-tetrahydro-1,8-naphthyridine (13a): following the General Procedure (pages S4–5), 5 ml of reagent feed A
[2-fluoro-3-vinylpyridine 17 (185 mg, 1.50 mmol, 1.0 equiv) and 3DPA2FBN (9.6 mg, 15.0 μmol, 1 mol%) in anhydrous DMF], 5 ml of reagent feed B [cyclohexylamine 10A (149 mg, 1.50 mmol, 1.0
equiv) and NaN3 (19.5 mg, 300 μmol, 20 mol%) in anhydrous DMF], and 10 ml of reagent feed C [DIPEA (291 mg, 2.25 mmol, 1.5 equiv) in anhydrous DMF] were reacted in flow, setting the
high-temperature tube reactor to 180 °C. The steady-state mixture (10 ml) was collected and concentrated in vacuo on an Asynt spiral evaporator. Purification via automated flash column
chromatography on SiO2 gel (12 g) in 40–60 °C petroleum ether (5 CV) then 100:0 → 0:100 40–60 °C petroleum ether–EtOAc (over 20 CV) then EtOAc (5 CV) gave 13A as a colourless, crystalline
solid (149 mg, 98%, productivity = 2.20 mmol h–1). DATA AVAILABILITY Detailed experimental procedures and characterisation of compounds can be found in Supplementary Methods in the
Supplementary Information. NMR spectra are available as a separate Supplementary Data 1. All original data are available from the authors upon request. REFERENCES * Luise, N., Wyatt, E. W.,
Tarver, G. J. & Wyatt, P. G. A continuous flow strategy for the facile synthesis and elaboration of semi-saturated heterobicyclic fragments. _Eur. J. Org. Chem_. 2019, 1341–1349 (2019).
* Luise, N. & Wyatt, P. G. Generation of polar semi‐saturated bicyclic pyrazoles for fragment‐based drug‐discovery campaigns. _Chem. Eur. J._ 24, 10443–10451 (2018). Article CAS PubMed
Google Scholar * Palmer, N., Peakman, T. M., Norton, D. & Rees, D. C. Design and synthesis of dihydroisoquinolones for fragment-based drug discovery (FBDD). _Org. Biomol. Chem._ 14,
1599–1610 (2016). Article CAS PubMed Google Scholar * Twigg, D. G. et al. Partially saturated bicyclic heteroaromatics as an sp3‐enriched fragment collection. _Angew. Chem. Int. Ed._ 55,
12479–12483 (2016). Article CAS Google Scholar * Nadin, A., Hattotuwagama, C. & Churcher, I. Lead-oriented synthesis: a new opportunity for synthetic chemistry. _Angew. Chem. Int.
Ed._ 51, 1114–1122 (2012). Article CAS Google Scholar * Roughley, S. D. & Jordan, A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug
candidates. _J. Med. Chem._ 54, 3451–3479 (2011). Article CAS PubMed Google Scholar * Shearer, J., Castro, J. L., Lawson, A. D. G., MacCoss, M. & Taylor, R. D. Rings in clinical
trials and drugs: present and future. _J. Med. Chem._ 65, 8699–8712 (2022). Article CAS PubMed PubMed Central Google Scholar * Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis
of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. _J. Med. Chem._ 57, 10257–10274 (2014). Article CAS
PubMed Google Scholar * Pennington, L. D. & Moustakas, D. T. The necessary nitrogen atom: a versatile high-impact design element for multiparameter optimization. _J. Med. Chem._ 60,
3552–3579 (2017). Article CAS PubMed Google Scholar * Sierov, D., Nazarenko, K., Shvydenko, K., Shvydenko, T. & Kostyuk, A. Synthetic approaches to tetrahydro-2,7- and
-1,6-naphthyridines. _Tetrahedron Lett._ 61, 152194 (2020). Article CAS Google Scholar * Jouha, J. et al. Domino aza-Michael-_ih_-Diels−Alder reaction to various 3‑vinyl-1,2,4-triazines:
access to polysubstituted tetrahydro-1,6-naphthyridines. _Org. Lett._ 19, 4770–4773 (2017). Article CAS PubMed Google Scholar * Jackl, M. K., Kreituss, I. & Bode, J. W. Synthesis of
tetrahydronaphthyridines from aldehydes and HARP reagents via radical Pictet−Spengler reactions. _Org. Lett._ 18, 1713–1715 (2016). Article CAS PubMed Google Scholar * Sirakanyan, S. N.,
Spinelli, D., Geronikaki, A., Hovakimyan, A. A. & Noravyan, A. S. New heterocyclic systems derived from pyridine: new substrates for the investigation of the azide/tetrazole
equilibrium. _Tetrahedron_ 70, 8648–8656 (2014). Article CAS Google Scholar * Johnson, R. J., O’Mahony, D. J. R., Edwards, W. T. & Duncton, M. A. J. A concise one-pot synthesis of
trifluoromethyl-containing 2,6-disubstituted 5,6,7,8-tetrahydroquinolines and 5,6,7,8-tetrahydronaphthyridines. _Org. Biomol. Chem._ 11, 1358–1366 (2013). Article CAS PubMed Google
Scholar * Mailyan, A. K., Peregudov, A. S., Dixneuf, P. H., Bruneau, C. & Osipov, S. N. Cyclobutene ring-opening of bicyclo[4.2.0]octa-1,6-dienes: access to CF3-substituted
5,6,7,8-tetrahydro-1,7-naphthyridines. _J. Org. Chem._ 77, 8518–8526 (2012). Article CAS PubMed Google Scholar * Zhou, Y., Porco, J. A. Jr & Snyder, J. K. Synthesis of
5,6,7,8-tetrahydro-1,6-naphthyridines and related heterocycles by cobalt-catalyzed [2 + 2 + 2] cyclizations. _Org. Lett._ 9, 393–396 (2007). Article CAS PubMed Google Scholar *
Milkiewicz, K. L. et al. Synthesis and structure–activity relationships of 1,2,3,4-tetrahydropyrido[2,3-_b_]pyrazines as potent and selective inhibitors of the anaplastic lymphoma kinase.
_Bioorg. Med. Chem._ 18, 4351–4362 (2010). Article CAS PubMed Google Scholar * Nam, T.-g et al. Tetrahydro-1,8-naphthyridinol analogues of α-tocopherol as antioxidants in lipid membranes
and low-density lipoproteins. _J. Am. Chem. Soc._ 129, 10211–10219 (2007). Article CAS PubMed Google Scholar * Slack, R. J., Macdonald, S. J. F., Roper, J. A., Jenkins, R. G. &
Hatley, R. J. D. Emerging therapeutic opportunities for integrin inhibitors. _Nat. Rev. Drug Discov._ 21, 60–78 (2022). Article CAS PubMed Google Scholar * Procopiou, P. A. et al.
Discovery of (_S_)-3-(3-(3,5-dimethyl-1_H_-pyrazol-1-yl)phenyl)-4-((_R_)-3-(2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-l)ethyl)pyrrolidin-1-yl)butanoic Acid, a nonpeptidic αvβ6 integrin
inhibitor for the inhaled treatment of idiopathic pulmonary fibrosis. _J. Med. Chem._ 61, 8417–8443 (2018). Article CAS PubMed Google Scholar * Fairhurst, R. A. et al. Discovery of
roblitinib (FGF401) as a reversible-covalent inhibitor of the kinase activity of fibroblast growth factor receptor 4. _J. Med. Chem._ 63, 12542–12573 (2020). Article CAS PubMed Google
Scholar * Hiesinger, K., Dar’in, D., Proschak, E. & Krasavin, M. Spirocyclic scaffolds in medicinal chemistry. _J. Med. Chem._ 64, 150–183 (2021). Article CAS PubMed Google Scholar
* Chan, B. K. et al. Discovery of spiro-azaindoline inhibitors of hematopoietic progenitor kinase 1 (HPK1). _ACS Med. Chem. Lett._ 13, 84–91 (2022). Article CAS PubMed Google Scholar *
Velázquez, F. et al. Design and synthesis of P2−P4 macrocycles containing a unique spirocyclic proline: a new class of HCV NS3/4A inhibitors. _ACS Med. Chem. Lett._ 7, 1173–1178 (2016).
Article PubMed PubMed Central Google Scholar * Garnsey, M. R. et al. Discovery of the potent and selective MC4R antagonist PF-07258669 for the potential treatment of appetite loss. _J.
Med. Chem._ 66, 3195–3211 (2023). Article CAS PubMed Google Scholar * Wang, W., Feng, X. & Du, H. Borane-catalyzed metal-free hydrogenation of 2,7-disubstituted 1,8-naphthyridines.
_Org. Biomol. Chem._ 14, 6683–6686 (2016). Article CAS PubMed Google Scholar * Ma, W., Chen, F., Liu, Y., He, Y.-M. & Fan, Q.-H. Ruthenium-catalyzed enantioselective hydrogenation of
1,8-naphthyridine derivatives. _Org. Lett._ 18, 2730–2733 (2016). Article CAS PubMed Google Scholar * Yasuda, N. et al. An efficient synthesis of an αVβ3 antagonist. _J. Org. Chem._ 69,
1959–1966 (2004). Article CAS PubMed Google Scholar * Wijtmans, M. et al. 6-Amino-3-pyridinols: towards diffusion-controlled chain-breaking antioxidants. _Angew. Chem. Int. Ed._ 42,
4370–4373 (2003). Article CAS Google Scholar * Wang, X. et al. Rorγ inhibitor having sulfonyl structure. CN Patent WO 2020108538 (2020). * Xiong, B. et al. Ruthenium-catalyzed
straightforward synthesis of 1,2,3,4-tetrahydronaphthyridines via selective transfer hydrogenation of pyridyl ring with alcohols. _Org. Lett._ 17, 4054–4057 (2015). Article CAS PubMed
Google Scholar * Hofmann, N., Homberg, L. & Hultzsch, K. C. Synthesis of tetrahydroquinolines via borrowing hydrogen methodology using a manganese PN3 pincer catalyst. _Org. Lett._ 22,
7964–7970 (2020). Article CAS PubMed PubMed Central Google Scholar * Lahue, B. R., Lo, S.-M., Wan, Z.-K., Woo, G. H. C. & Snyder, J. K. Intramolecular inverse-electron-demand
Diels−Alder reactions of imidazoles with 1,2,4-triazines: a new route to 1,2,3,4-tetrahydro-1,5-naphthyridines and related heterocycles. _J. Org. Chem._ 69, 7171–7182 (2004). Article CAS
PubMed Google Scholar * Haenel, F., John, R. & Seitz, G. Trifluormethyl-substituierte, heterocyclisch anellierte pyridine durch intramolekulare Diels-Alder-cycloaddition mit inversem
elektronenbedarf. _Arch. Pharm._ 325, 349–352 (1992). Article CAS Google Scholar * Seitz, G. & Richter, J. Inverse intramolecular [4+2]-cycloaddition with activated
3-alkynylamino-1,2,4-triazines. _Chem. Ztg._ 113, 252–254 (1989). CAS Google Scholar * John, R. & Seitz, G. 3-Methylthio-5-trifluormethyl-1,2,4-triazin als edukt zur synthese
heteroanellierter pyridine durch intramolekulare Diels-Alder-cycloaddition mit inversem elektronenbedarf. _Arch. Pharm._ 322, 561–564 (1989). Article CAS Google Scholar * Askey, H. E. et
al. Photocatalytic hydroaminoalkylation of styrenes with unprotected primary alkylamines. _J. Am. Chem. Soc._ 143, 15936–15945 (2021). Article CAS PubMed PubMed Central Google Scholar *
Blackwell, J. H., Harris, G. R., Smith, M. A. & Gaunt, M. J. Modular photocatalytic synthesis of α-trialkyl-α-tertiary amines. _J. Am. Chem. Soc._ 143, 15946–15959 (2021). Article
Google Scholar * Schrader, T. O. et al. Asymmetric syntheses of (_R_)-4-halo-6,6_a_,7,8,9,10-hexahydro-5_H-_pyrazino[1,2-_a_][1,_n_]naphthyridines, important 5-HT2C agonist precursors.
_Tetrahedron Lett._ 59, 2030–2033 (2018). Article CAS Google Scholar * Breslin, M. J. et al. Nonpeptide αVβ3 antagonists. Part 10: in vitro and in vivo evaluation of a potent 7-methyl
substituted tetrahydro-[1,8]naphthyridine derivative. _Bioorg. Med. Chem. Lett._ 14, 4515–4518 (2004). Article CAS PubMed Google Scholar * Hartner, F. W. et al. Methods for the synthesis
of 5,6,7,8-tetrahydro-1,8-naphthyridine fragments for αVβ3 integrin antagonists. _J. Org. Chem._ 69, 8723–8730 (2004). Article CAS PubMed Google Scholar * Wu, Y., Chen, Y.-Q., Liu, T.,
Eastgate, M. D. & Yu, J.-Q. Pd-Catalyzed γ-C(sp3)–H arylation of free amines using a transient directing group. _J. Am. Chem. Soc._ 138, 14554–14557 (2016). Article CAS PubMed PubMed
Central Google Scholar * DiPucchio, R. C., Rosca, S.-C. & Schafer, L. L. Hydroaminoalkylation for the catalytic addition of amines to alkenes or alkynes: diverse mechanisms enable
diverse substrate scope. _J. Am. Chem. Soc._ 144, 11459–11481 (2022). Article CAS PubMed Google Scholar * Manßen, M. & Schafer, L. L. Early transition metal-catalyzed
hydroaminoalkylation. _Trends Chem._ 3, 428–429 (2021). Article Google Scholar * Trowbridge, A., Walton, S. M. & Gaunt, M. J. New strategies for the transition-metal catalyzed
synthesis of aliphatic amines. _Chem. Rev._ 120, 2613–2692 (2020). Article CAS PubMed Google Scholar * Wang, G., Ang, H. T., Dubbaka, S. R., O’Neill, P. & Wu, J. Multistep automated
synthesis of pharmaceuticals. _Trends Chem._ 5, 432–445 (2023). Article CAS Google Scholar * Buglioni, L., Raymenants, F., Slattery, A., Zondag, S. D. A. & Noël, T. Technological
innovations in photochemistry for organic synthesis: flow chemistry, high-throughput experimentation, scale-up, and photoelectrochemistry. _Chem. Rev._ 122, 2752–2906 (2022). Article CAS
PubMed Google Scholar * Brandão, P., Pineiro, M. & Pinho e Melo, T. M. V. D. Flow chemistry: sequential flow processes for the synthesis of heterocycles. In _Heterocycles: Synthesis,
Catalysis, Sustainability, and Characterization_ (eds Brandão, P., Pineiro, M. & Pinho e Melo, T. M. V. D.) (Wiley‐VCH, 2022). * Rehm, T. H. Photochemistry in flow for drug discovery. In
_Flow Chemistry in Drug Discovery_ (eds Alcazar, J., de la Hoz, A. & Díaz-Ortiz, A.) (Springer, 2021). * Sambiagio, C. & Noël, T. Flow photochemistry: shine some light on those
tubes! _Trends Chem._ 2, 92–106 (2020). Article CAS Google Scholar * Baumann, M., Moody, T. S., Smyth, M. & Wharry, S. A perspective on continuous flow chemistry in the pharmaceutical
industry. _Org. Process Res. Dev._ 24, 1802–1813 (2020). Article CAS Google Scholar * Rehm, T. H. Flow photochemistry as a tool in organic synthesis. _Chem. Eur. J._ 26, 16952–16974
(2020). Article CAS PubMed Google Scholar * Gioiello, A., Piccinno, A., Lozza, A. M. & Cerra, B. The medicinal chemistry in the era of machines and automation: recent advances in
continuous flow technology. _J. Med. Chem._ 63, 6624–6647 (2020). Article CAS PubMed PubMed Central Google Scholar * Brandão, P., Pineiro, M. & Pinho e Melo, T. M. V. D. Flow
chemistry: towards a more sustainable heterocyclic synthesis. _Eur. J. Org. Chem_. 2019, 7188–7217 (2019). * Bogdan, A. R. & Dombrowski, A. W. Emerging trends in flow chemistry and
applications to the pharmaceutical industry. _J. Med. Chem._ 62, 6422–6468 (2019). Article CAS PubMed Google Scholar * Sharma, U. K. & Van der Eycken, E. V. (eds) _Flow Chemistry for
the Synthesis of Heterocycles. Topics in Heterocyclic Chemistry_ (Springer, 2018). * Ryder, A. S. H. et al. Photocatalytic α-tertiary amine synthesis via C−H alkylation of unmasked primary
amines. _Angew. Chem. Int. Ed._ 59, 14986–14991 (2020). Article CAS Google Scholar * Grayson, J. D. & Cresswell, A. J. γ-Amino phosphonates via the photocatalytic α-C–H alkylation of
primary amines. _Tetrahedron_ 81, 131896 (2021). Article CAS Google Scholar * Foley, D. J., Nelson, A. & Marsden, S. P. Evaluating new chemistry to drive molecular discovery: fit for
purpose? _Angew. Chem. Int. Ed._ 55, 13650–13657 (2016). Article CAS Google Scholar * Desai, P. B. Preparation and stability of 4-fluoropyridine. _J. Chem. Soc. Perkin Trans._ 1,
1865–1866 (1973). Article Google Scholar * Preshlock, S. M. et al. A traceless directing group for C–H borylation. _Angew. Chem. Int. Ed._ 52, 12915–12919 (2013). Article CAS Google
Scholar Download references ACKNOWLEDGEMENTS This work was supported by the Engineering and Physical Sciences Research Council (EP/X026566/1). A.J.C. thanks the Royal Society for a
University Research Fellowship (UF150533), the University of Bath for a Global Doctoral Scholarship (Q.C.) and both AstraZeneca and UCB for generous financial support. The authors gratefully
acknowledge the technical staff within Chemistry at the University of Bath for technical support and assistance in this work, including the Material and Chemical Characterisation Facility
(MC²) (https://doi.org/10.15125/mx6j-3r54). We also acknowledge Dr Michelle Garnsey at Pfizer for helpful discussions. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Chemistry,
University of Bath, Claverton Down, Bath, BA2 7AY, UK Qiao Cao, Joshua D. Tibbetts & Alexander J. Cresswell * Medicinal Chemistry, Oncology R&D, AstraZeneca, Cambridge, CB4 0WG, UK
Gail L. Wrigley * UCB, 216 Bath Road, Slough, SL1 3WE, UK Adam P. Smalley Authors * Qiao Cao View author publications You can also search for this author inPubMed Google Scholar * Joshua D.
Tibbetts View author publications You can also search for this author inPubMed Google Scholar * Gail L. Wrigley View author publications You can also search for this author inPubMed Google
Scholar * Adam P. Smalley View author publications You can also search for this author inPubMed Google Scholar * Alexander J. Cresswell View author publications You can also search for this
author inPubMed Google Scholar CONTRIBUTIONS A.J.C. designed and supervised the project, with additional input and supervisory support provided by G.L.W. and A.P.S. Q.C. planned and carried
out all of the experimental work, with the exception of the syntheses of compounds 29 and 30, which were prepared by J.D.T. A.J.C. wrote the manuscript with assistance from the other
co-authors. CORRESPONDING AUTHOR Correspondence to Alexander J. Cresswell. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW
INFORMATION _Communications Chemistry_ thanks the anonymous reviewers for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION DESCRIPTION OF ADDITIONAL SUPPLEMENTARY
FILES SUPPLEMENTARY DATA 1 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Cao, Q., Tibbetts, J.D., Wrigley, G.L. _et al._ Modular, automated synthesis of spirocyclic tetrahydronaphthyridines from primary alkylamines. _Commun Chem_ 6, 215 (2023).
https://doi.org/10.1038/s42004-023-01012-2 Download citation * Received: 13 July 2023 * Accepted: 22 September 2023 * Published: 04 October 2023 * DOI:
https://doi.org/10.1038/s42004-023-01012-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative