- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
The embellishing of the macrocycle core with sulfur substituents of varied sterical requirements changes the structural dynamics of chiral, triangular polyimines. Despite their formal high
symmetry, these compounds adopt diverse conformations, in which the macrocycle core represents a non-changeable unit. DFT calculations reveal that the mutual arrangement of sulfur-containing
substituents is controlled mainly by sterical interactions. The presence of sulfur atoms affects the chiroptical properties of these compounds and causes a red shift of respective
absorption bands compared to the basic trianglimine. Unexpectedly, the aromatic fragments attached to the sulfur atom have less impact on ECD spectra, visible only in particular spectral
regions. Such a possibility to adapt various conformations is also seen in the crystalline phase; however, a stiff basic unit – the triangular macrocycle core – caused macrocycles’
self-assembly into columnar-like aggregates. In the crystal lattice, around the macrocycle having bulky SCPh3 groups, a space filled with solvent is formed; however, the macrocycle’s
internal cavity is closed and unavailable for guest molecules. Titration of the solutions of basic SBn-substituted imine and amine macrocycles by AgOTf results in significant changes in the
ECD spectra, confirming possible binding interactions between macrocycle and metal cations.
Due to their wide-ranging applications, macrocyclic compounds are highly sought-after synthetic targets1,2. Unfortunately, the challenge of obtaining cyclic macromolecules with defined
molecular weights and shapes persists. On the other hand, the molecule’s shape persistency and the use of reactions based on dynamical bond formation led to obtaining compounds with regular
and symmetrical structures resembling geometrical figures3,4,5.
The synthesis of triangular chiral hexaimines (trianglimines) involves the direct formation of macrocycles from vicinal diamine (usually optically pure trans-1,2-diaminocyclohexane, DACH, 1)
and aromatic 1,4-dialdehyde6. In the simplest case, terephthalaldehyde (2) serves this function, and the products’ high, usually quantitative yields are an added advantage7. Further
functionalization of the macrocycle is achieved by reducing C = N imine bonds and subsequently modifying nitrogen atoms. These modifications lead to the formation of more chemically
resistant materials, and the proper N-functionalization allows for control over molecular dynamics8.
The nature of the π-electronic system and functional groups in the aromatic linkers and control of the conformational liability of a molecule is crucial for applications of the macrocycles.
Recently, it has been shown that simple trianglimines and trianglamines enable efficient separation of linear over branched hydrocarbons, constitutional and geometrical isomers, and
enantiomers. Trianglamine-containing membranes have turned out to be effective nanofiltration agents that allow for the purification of organic
solvents9,10,11,12,13,14,15,16,17,18,19,20,21,22.
The particular case of the molecule of properties differs in the solid state and the solution, representing trianglsalen containing two hydroxyl groups in the para position of each aromatic
linker. In one of the polymorphs, the trianglsalen molecules form a tubular architecture composed of supramolecular segments, each consisting of six macrocycles. These nanoporous,
chromogenic crystals can release water even at − 70 °C, which is associated with the crystal’s color change, allowing for the quantification of water uptake23.
It has been known that polyaza-macrocycles characterized by different structural properties, containing or not heteroatoms attached to the molecule core, may form metal complexes and
metallogels. Zinc(II)- or copper(II)-trianglamine complexes used in asymmetric catalytic reactions and N,N’-thiourea-derived trianglamine supergelators, forming chiral metallogels with
Ag(I), Cu(I), and Cu(II) salts are two distinct examples24,25,26,27,28,29.
Feeling that the chemistry of chiral and shape-persistent macrocyclic polyimines containing heteroatoms attached to the skeleton is still at an early stage of research, we have decided to
expand our interest on trianglimine derivatives embellished with sulfur derivatives in each aromatic linker.
In principle, the incorporation of thioether moieties into the trianglimine skeleton would affect the structural preferences of the mother compound. The trianglimine and its reduced congener
represent extreme cases of compounds with contrasting structural dynamics. Therefore, incorporating flexible “arms” into the skeleton will represent a missing link connecting rigid
trianglimine macrocycle with highly flexible trianglamine. As the macrocycles planned to be synthesized are chiral, the impact of sulfur atoms on the chiroptical properties, namely
electronic circular dichroism (ECD), of these compounds is worth interest. ECD spectroscopy is a convenient method that provides structural information regarding the structure of chiral
macrocyclic species and their supramolecular assemblies30,31. Additionally, ECD spectroscopy may be used to directly observe macrocyclic receptors’ interactions with metal cations.
In most cases known so far, the heteroatoms (oxygen-containing groups) are already present in the aromatic unit, and subsequent modifications rely on introducing formyl groups to the
molecule skeleton32,33,34. The bromine is an exception, which may be conveniently introduced to the terephthalaldehyde through direct bromination, using N-bromosuccinoimide (NBS) in
concentrated sulfuric acid. The product of the reaction, namely 2,5-dibromoterephthalaldehyde (3, see Scheme 1), is obtained with a high yield. Further transformations are based on
well-established palladium-catalyzed reactions that form aryl- or alkynyl-substituted terephthalaldehydes35.
The general synthetic route provides chiral thio-modified macrocycles.
Having established a convenient procedure for introducing bromine atoms into the terephthaldehyde skeleton, we used bromination product 3 as the starting point in the synthetic path to
obtaining macrocycle compounds (Scheme 1). Subsequent substitution of the bromine atom(s) by thiol will lead to the formation of respective thioethers 536. Using thiols of the higher
molecular mass as nucleophiles yielded respective thioethers, even if the substituent attached to the sulfur atom was as bulky as the trityl group. Some monosubstituted thioether 5 g was
isolated alongside the double-substituted product in the selected case. The thioether 5 h was synthesized intentionally for comparison purposes, starting from mono-brominated
terephthalaldehyde 4. The latter is either isolated in small amounts as by-products of terephthalaldehyde dibromination or might be synthesized when only half of NBS has been used. Although
the thioethers thus obtained are stable, there is one exception – thioether 5f, which changed the color from yellow to deep red upon standing. It is worth noting that during the synthesis of
5f, some trityl peroxide was formed, which is barely separable from the aldehyde by crystallization.
With the semi-products in hands, the macrocycles 6 were synthesized at the next stage through cyclocondensation reactions between equimolar amounts of dialdehyde and (R,R)-17. The cyclic
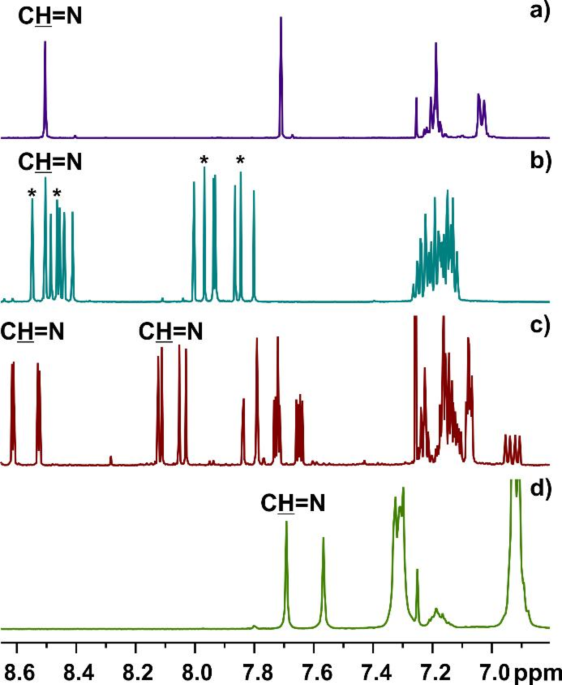
structure of a given product was confirmed by NMR spectroscopy and mass spectra. The number of peaks on the NMR spectra directly reflects the symmetry of the given compound (see Fig. 1a-c).
For macrocycles obtained from symmetrical, double-substituted aldehydes, the highest available symmetry is D3. On the other hand, when non-symmetrical aldehydes 5 g and 5 h were used for
reactions, a mixture of C3-symmetrical and non-symmetrical (trivial, C1-symmetrical) products 6 g and 6 h, respectively, was obtained. The proportion of the products that differ in symmetry
has not been altered upon the change of reaction conditions. Additionally, the 1H NMR spectra measured for 6 g during two weeks and in the time intervals have shown no changes in the
composition of the mixture. In both cases, the non-symmetrical product amount was three times as big as the symmetrical one, as indicated by the integration of diagnostic signals originating
from imine CH = N protons.
Diagnostic part of 1H NMR spectra (CDCl3) of (a) symmetrical macrocycle 6a, a mixture of symmetrical and non-symmetrical macrocycles: (b) 6 g and (c) 6 h, and (d) sterically crowded
macrocycle 6f. Asterisks indicate signals of protons originating from the C3-symmetrical macrocycle 6 g.
While for most macrocyclic compounds discussed here, the diagnostic protons appeared at around 8.5 ppm, and there is one exception. For the particular case of 6 h, the imine CH = N protons
appeared in a wide range of chemical shifts. The deshielded signals are observed between 8.7 and 8.5 ppm and originated from CH = N imine protons near the heteroatoms. In contrast, more
shielded signals (observed between 8.15 and 8.05 ppm) correspond to the resonances of the remaining azomethine protons.
Having neglected the symmetry, the reactions mostly provided chemically homogeneous products. Some additional, very small peaks on 1H NMR spectra were visible for macrocycle 6a. Although the
macrocyclic product exhibited trimeric and triangular structure, resulting from [3 + 3] cyclocondensation reaction, additional peaks originating from expanded [4 + 4] congener were also
observed on the mass spectra measured for crude reaction mixture. These peaks have vanished after purification. It should be noted that the temperature-dependent 1H NMR measurements did not
exhibit any spectacular changes. Apart from minimal shifts in the downfield region, the respective pics remain sharp.
The particular case represents a macrocycle 6f embellished with six S-trityl groups. The presence of bulky substituents in the aldehyde skeleton may cause some problems with the
macrocyclization reaction. However, the reaction proceeded smoothly, providing a triangular macrocycle with an almost quantitative yield. Significant crowding within the macrocycle ring has
not been revealed in the shape of the 1H NMR spectrum. The broadening of the signals is visible mainly for aliphatic protons. Unexpectedly, the presence of trityl groups caused a shielding
effect for azomethine protons, which appeared at 7.69 ppm (see Fig. 1d). This is apparently due to the possible perpendicular (or close to it) orientation of one of the phenyl rings from the
neighboring trityl group to the azomethine proton. The same trend is observed in the 1H NMR spectrum of parent dialdehyde 5f, where the CHO proton appeared at 9.5 ppm.
As mentioned earlier, the presence of STr moieties has caused problems with the stability of the compounds, which is revealed by naked-eye visible changes in the color of the samples, both
aldehyde and macrocycle. Protecting the product against moisture and direct light slows the decomposition process but does not eliminate it.
The post-synthetic modification of the model macrocycle 6a, which involved reducing C = N imine bonds, proceeded without complications and led to the quantitative formation of trianglamine
78.
Due to their poor shape complementarity, we were fortunate to obtain crystals suitable for X-ray study only for few examples. These included symmetrical macrocycles 6b and 6f and the
macrocycle 6 g (Fig. 2).
Top and side views of the molecular structure of macrocycles: (a) 6b, (b) 6f, and (c) 6 g. Hydrogen atoms were omitted for clarity. The proper distances are shown as dashed lines and are
given in Å.
Generally, the triangular backbone of macrocycles 6b, 6f, and 6 g display similar conformations in the crystal structure. The C*-N = C-Cipso torsion angles have similar values that
concentrate around 180°. Furthermore, in all cases, the molecule assumes a bowl-like configuration.
Compound 6b crystallizes in monoclinic space group P21 with one macrocycle molecule in an asymmetric unit. The molecule has the D3 symmetry, but due to the conformational lability of the
substituents, it assumes the C1 symmetry in the crystal lattice. The molecule takes a bowl-like shape with an upper rim diameter of approximately 8.15 Å and a lower rim of 4.80 Å. The
position of S atoms of substituents determines both rims. The size of the internal gap is determined by the distances between the centroids of the phenyl rings in the macrocycle skeleton,
and they range from 6.06 to 6.49 Å. However, it should be emphasized that the macrocycle molecules fill the space, and no gaps are available for the solvent molecules in the crystal.
As it has been emphasized earlier, while the triangular skeleton of the molecule is relatively rigid, the carbon chains of the substituents are flexible. This means that their conformation
can change and adapt to the available space in the crystal lattice. As a result, the substituents on the triangular core of the molecule are disordered. Notably, despite the disorder that
results from the flexibility of the aliphatic linker in the substituent, the bowl in the upper part remains open. A phenyl ring in the neighboring molecule’s lower part is inserted into the
macrocycle cavity in the crystal structure. The phenyl ring of the substituent is arranged almost parallel to the aromatic system of the macrocycle skeleton (the angle between the rings is
about 5.3°), and a system is stabilized by the π∙∙∙π interaction (the distance between the planes of the aromatic rings is about 3.5 Å) (Figure S129 in SI). In the crystal structure,
supramolecular columns are formed, which are additionally stabilized by C-H∙∙∙π and C-H∙∙∙S interactions.
The three-dimensional structure is stabilized by weak interactions involving aromatic systems (C-H∙∙∙π interactions) and van der Waals interactions.
Compound 6f crystallizes in the monoclinic system in the P21 space group, comprising two macrocyclic molecules and solvents: hexane, dichloromethane, and water. The host-guests ratio in the
crystal structure is 1:1:1:3. The guest molecules are situated within the extrinsic space – in a void with an elongated shape created by the macrocyclic host molecules. Therefore, the
disorder in the solvent-accessible space was quantified using a solvent mask instruction, as implemented in Olex2 software37, resulting in the identification of 256 electrons within a volume
of 1930 ų in a void per unit cell. This finding is consistent with one hexane, one dichloromethane, and three water molecules per asymmetric unit, accounting for 244 electrons per unit
cell.
In the molecular structure of 6f, the C*-N = C-Cipso torsion angles exhibit similar values, approximately 180°. The triangular skeleton assumes a bowl-like shape. The inner cavity size of
the molecular bowl is determined by the rigid building blocks that comprise the macrocyclic skeleton. The distances between the three centroids of the phenyl rings that define the cavity
size range from 6 to 6.5 Å. Macrocycle 6f assumes a rigid pillararene-like conformation with three -STr substitutes at aromatic linkers on both sides of the skeleton. The presence of bulky
-STr groups precludes the rotation of the aromatic linkers, compelling the macrocycle to assume a conformation that optimizes the size of the cavity. Consequently, in 6f, the mean cavity
diameter is 6.5 Å, with a depth of 18 Å. Notably, guest molecules do not penetrate into the macrocycle cavity despite the bulky and partially open structure.
Although macrocycle 6f formally exhibits D3 symmetry, it remains in C1 symmetry in the solid state. It should be noted that not all of the -STr substituents present in the crystalline
structure exhibit the same arrangement. Four groups are oriented towards the center of the cavity, one group is oriented towards the exterior, and another is partially twisted, resulting in
a partially open macrocycle structure with three distinct arrangements of aromatic building blocks. The configuration of the aromatic linkers within the macrocycle skeleton can be described
by pseudotorsion angles, specifically C(Tr)-S-(Ar)-S-C(Tr). The indicated angles have values of 163°, 16°, and 91°, respectively. Additionally, each trityl group attached to the linker
exhibits a distinct helicity (Figure S130 and S131 in SI). This observed ‘asymmetry’ may be attributed to the limited potential for intermolecular interactions (the ‘asymmetry’ of the
molecule can be reinforced by creating supramolecular interactions).
Imine macrocycles in the 6f crystalline form establish a supramolecular network through the utilization of weak C-H∙∙∙π and C-H∙∙∙S intermolecular interactions facilitated by the -STr groups
of neighboring molecules. In the 6f crystal structure, the macrocycles self-assemble into columnar-like aggregates, and the angle of inclination of the stack axis to the plane of the
macrocycle ring is approximately 45°. The distinguished columnar motif is also observed in simple trianglimines and trianglsalenes crystal structures (See Fig. 3)34,38.
Supramolecular columns in crystals of compound (top and side view) (a) 6b, (b) 6f and (c) 6 g.
Compound 6 g crystallizes in triclinic space group P1 with one macrocycle molecule. The crystal structure contains some disordered solvent molecules caged in intermolecular holes. A solvent
mask was calculated, and 46 electrons were found in a volume of 203 Å3 in 1 void per unit cell. This is consistent with diethyl ether (C4H10O) per asymmetric unit, which accounts for 42
electrons per unit cell. Therefore, the estimated host-guest ratio in the crystal structure of compound 6 g is 1:1.
The macrocycle molecule shows C1 symmetry, with one of the three bromine atoms on the opposite side of the ring plane. However, as shown in the structural analysis, the molecule is
disordered and, alternatively, takes a conformation where all three Br atoms are situated on the same side of the molecule (the refined occupancy factor is 0.19). As in macrocycles 6b and 6f
crystals, the molecule takes a bowl-like shape. The rims are determined by the position of S and Br atoms of substituent, and their lengths vary depending on the arrangement of the
substituents on the triangular backbone. For a molecule with two bromine atoms at the bottom of the bowl, the upper rim diameter is approximately 7.68 Å, and the lower rim is 5.02 Å. For the
molecule where all three bromine atoms are located on the lower part of the bowl, the upper rim diameter is approximately 8.18 Å, and the lower rim is 4.59 Å. The size of the internal gap
is determined by the distances between the centroids of the phenyl rings in the macrocycle skeleton, and they range from 6.04 to 6.59 Å.
As it has been mentioned, the crystal contains some solvent but is located in the spaces between the molecules. The top of the molecular bowl is covered by a benzyl substituent, which makes
its interior inaccessible to guest molecules. In addition, the aromatic ring is slightly inserted into the cavity of the macrocycle, and C-H∙∙∙π interactions stabilize its position (Figure
S132 in SI).
In the crystal structure, the molecules are arranged in columns, and the triangular skeleton of the molecule is significantly inclined to the column axis (the nitrogen atoms of
diaminocyclohexane determined the plane, and the angle between the column axis and the plane normal is about 48°). The structure of the column is stabilized by interactions of the bromine
atom with the imine N-atom (Br∙∙∙N 3.3 Å) as well as C-H∙∙∙Br interactions (distances H∙∙∙Br are 2.94 Å and 2.99 Å). The molecules are stacked so that the cyclohexane ring of the next
macrocycle covers the lower part of the bowl of one of them. The three-dimensional structure of the crystal is stabilized by C-H∙∙∙N interaction as well as C-H∙∙∙π interactions between
adjacent stacks of molecules.
As the compounds in hands are chiral and optically active, we have taken advantage of determining their chiroptical properties, emphasizing ECD as the method of choice39. The ECD
spectroscopy, supported by theoretical calculations, would shed light on the structure in the solution of these compounds and, therefore, may be considered complementary to X-ray
crystallography40,41. A feature that significantly facilitates the experimental and theoretical analyses is the solubility of these compounds in non-polar solvents, such as cyclohexane.
Thus, routine calculations performed for isolated molecules in the gas phase will reflect experimental conditions.
An analysis of experimental ECD data led to the formulation of some general conclusions. Regardless of the substitution patterns, the measured CD spectra are similar (see Fig. 4 for
representative examples and SI for the remaining data).
Examples of ECD spectra of macrocycles 6a, 6c, 6f, and 6 h: experimental, measured in cyclohexane (black lines) and calculated at the TD-DFT level (red dashed lines). The calculated ECD
spectra have been Boltzmann averaged. Wavelengths have been corrected to match the experimental UV maxima (Δε values are given in mol−1 cm−1 dm3 units).
The lowest-energy positive signed Cotton effects (C.es) in ECD spectra are visible at around 370 nm. This band is associated with a low-intense UV band, which appears in the 380 − 360 nm
energy range. For most of the examined macrocyclic imines, the magnitude of the lowest-energy C.e. oscillates around Δε = 10 mol− 1 cm− 1 dm3. The exception is 6 h, for which this particular
band is barely visible.
The following ECD bands form a negative exciton-like couplet. The first negative C.e. is visible at around 280 nm, whereas the second, the positive one, appears at around 260 nm. The exciton
effect is related to the UV band of middle intensity, which is seen in the UV spectrum of a given compound at around 275 nm. The amplitude (A) of these exciton couplets varied, depending on
the type of substituent attached to the sulfur atom. The lowest amplitude was found for the most crowded macrocycle 6f (A = -70), whereas macrocycle 6d of A = -400 is placed on the opposite
pole.
Interestingly, the highest intensity UV band, which appears at the higher-energy region of the spectrum (usually at around 190 nm), does not reflect in high magnitude C.es in the ECD
spectrum of a given compound, as one can expect. Usually, the negative/positive C.es sequence is repeated in the higher-energy region of the spectrum. The magnitude of the positive C.e.,
which appears in this spectral region, is up to Δε = 100 mol− 1 cm− 1 dm3, while a negative one rarely exceeds Δε = -25 mol− 1 cm− 1 dm3.
One feature of these compounds is their indifference to solvent polarity. For example, the ECD spectra measured for model compound 6a have shown no significant alternations when the solvent
polarity was changed from non-polar cyclohexane through dichloromethane to highly polar acetonitrile. Even using methanol as a co-solvent did not alter the spectra’ shape or the magnitudes
of respective bands. An exception is STr-substituted macrocycle 6f, where the magnitude of respective Cotton effects increases with solvent polarity.
In contrast to the chiroptical properties of parent imine 6a, the ECD spectrum of its reduced congener 7 is characterized by a shallow magnitude of C.es. The first low-energy ECD band of the
negative sign appeared at 275 nm and is associated with a middle-intense UV band visible in the same spectral region. The following C.es form sequence +/-/+. The increase of the solvent
polarity is reflected in the rise in the magnitude of respective C.es, and the highest values are found in the ECD spectrum measured in dichloromethane containing 20% methanol. This suggests
that the hydrogen bonds may significantly stabilize the structure, and these non-covalent interactions may be broken or weakened in a more polar solvent.
It is worth noting that the ECD spectra of the imines 6a-6 h are entirely different in shape from those of their oxygen-containing congeners42.
To determine the structural dynamics of the macrocyclic compounds and the chiroptical properties of the compounds under study, we have performed extensive calculations at the DFT level of
their structure and UV/ECD spectra. Before these, we chose representative examples for these calculations. This set includes symmetrical and non-symmetrical macrocycles 6a, 6c, 6 h, 6 g, and
the most challenging example, 6f. Details regarding the calculation scheme and the method used are provided in Experimental Section and the SI.
Without going into the structure details, one significant trend is visible when one analyzes the results of calculations – the number of thermally available structures (i.e., these having
relative energies within the range 0 ÷ 2 kcal mol− 1) varied depending on the functional used. The highest number of stable conformers was found for structures calculated using the B3LYP
functional43,44. However, considering empirical dispersion correction led to calculating only a few stable structures45. The results obtained by the pure M06L functional were placed
between46, albeit closer to, B3LYP-GD3BJ, at least in terms of the number of stable structures.
The “nature” of a method is reflected in the structure of the stable conformers, i.e., the more non-covalent interactions are considered, the more “compact” the structure becomes. The latter
may be understood as the tendency to maximize the interactions and, in this way, the mutual approach of these structurally labile aromatic fragments attached directly or through a carbon
atom to a sulfur atom. Conversely, in the case of a functional such as B3LYP, the structure of the given conformer is mainly controlled by sterical interactions. Figure 5 compares structures
of the lowest energy conformers of macrocycles 6a, 6c, 6 g, and 6 h, calculated using different methods.
Top and side view on overlays of the structures of the lowest energy conformers of macrocycles (a) 6a; (b) 6c; (c) 6 g, and (d) 6 h. Green-colored structures were optimized using the B3LYP
functional, orange-colored structures were optimized using the B3LYP-GD3BJ method, and blue-colored structures were optimized using the M06L functional. Hydrogen atoms were omitted for
clarity.
It is worth adding that the calculated structures only partially reflect the structures determined experimentally. While the macrocycle core remains (almost) unchanged, the differences
between experimental and calculated structures rely on the conformation of backbones, as one may expect.
Since there is no direct possibility to asses which method provides the correct results, we have done it by the comparison of experimental ECD spectra with those calculated for low-energy
conformers and Boltzmann averaged (see Fig. 4 and SI).
Unexpectedly, in most cases, the best agreement between experimental and theoretical results was seen when the ECD spectra were calculated for geometries previously optimized using B3LYP
hybrid functional. On the opposite pole are the results obtained for geometries optimized when the B3LYP-GD3BJ functional was used. The pure M06L functional shows its advantage over others
for two extremal cases, the simplest mono-substituted 6 h macrocycle and the most complex molecular system 6f. All the methods gave similar results among the functionals used for excited
state calculations. Therefore, we limited the discussion to the ECD spectra calculated using CAM-B3LYP functional47.
The direct inspection of the obtained data led to the conclusion that the conformations of the backbones have only a limited impact on the low-energy region of the ECD spectra. The
comparison of the ECD data obtained after Boltzmann averaging of spectra calculated for individual conformers with those calculated for the lowest energy structures show their almost perfect
overlay (see SI). Therefore, we have limited the further structural discussion to the lowest energy conformers of the given compounds.
A set of 5 torsion angles can conveniently describe each optimized conformer. The first three, α = N-C*-C*-N, β = H-C*-N = C, and γ = N = C-C-C(X), are related to the conformation of the
macrocycle core. In contrast, the remaining two, φ = (H)C-C-X-C and χ = C-X-C-C describe the conformation of the substituent concerning the macrocycle core. In the case of 6f, the angles ω =
S-C-Cipso-Cortho determine conformation, therefore P (90°>ω > 0°) or M (-90°