- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The infection and outbreak of _Salmonella_ typhimurium (_S_. typhimurium) highlight the need for developing a reliable on-site detection strategy fitting to various settings.
However, due to the requirement of specialized instruments and trained personnel, traditional detection methods have to be implemented in laboratories and are not ideal for on-site
applications. To achieve a sample-to-answer and field-deployable detection for _S_. typhimurium, we developed an integrated nucleic acid detection platform combining single-vial of
loop-mediated isothermal amplification (LAMP)-clustered regularly interspaced short palindromic repeat (CRISPR)/Cas12a system, portable device and smartphone app. This platform enables the
extraction, concentration, and purification of DNA, amplification of the target, and output of visual fluorescent signals within 1 h. With this detection platform, 102 CFU/mL of _S_.
typhimurium in food and environmental matrix was able to be accurately detected. This method demonstrated excellent selectivity. Also, an auxiliary smartphone application was developed to
achieve simplified result interpretation. Our method exhibited potential to better control and respond to outbreaks of foodborne diseases, especially in low-resource settings. SIMILAR
CONTENT BEING VIEWED BY OTHERS DNAZYME-BASED COLORIMETRIC BIOSENSOR FOR RAPID DETECTION OF _SHIGELLA FLEXNERI_ Article Open access 29 April 2025 A TAQMAN QPCR FOR PRECISE DETECTION AND
QUANTIFICATION OF DIARRHEAGENIC _ESCHERICHIA COLI_ Article Open access 14 May 2025 DEVELOPMENT OF HIGH-RESOLUTION MELTING (HRM) ASSAY TO DIFFERENTIATE THE SPECIES OF _SHIGELLA_ ISOLATES FROM
STOOL AND FOOD SAMPLES Article Open access 10 January 2022 INTRODUCTION _Salmonella_ typhimurium (_S_. typhimurium) is one of the leading foodborne pathogens responsible for human
gastroenteritis and diarrhea, posing a significant health hazard in many countries1,2. The infection by this pathogen can result in hospitalizations and deaths3. Human infections with _S_.
typhimurium are typically acquired through the consumption of contaminated poultry, vegetables, fruit and eggs4. The wide spread of _S_. typhimurium, its resistance to drugs, as well as the
similarity of salmonellosis symptoms with other diseases, makes difficulty for the effective management of disease outbreaks caused by this bacterium5. Thus, the effective implementation of
rapid detection for _S_. typhimurium plays a significant role in preventing and controlling outbreaks of foodborne illnesses. The currently available detection methods for _S_. typhimurium,
such as traditional bacterial culture, enzyme-linked immunosorbent assay (ELISA), and polymerase chain reaction (PCR) analyses, all have their own limitations6. Traditional bacterial culture
methods are time-consuming and labor-intensive, and require precise conditions7. ELISA stands as a labor-intensive procedure, requires skilled personnel, and suffers inefficient mass
transfer and high cost of reagents, along with requirement of spectrophotometers to quantify the results8. PCR necessitates advanced equipment, skilled personnel, and precise temperature
control during cycling9. Given the aforementioned limitations of traditional detection methods and the need for rapid on-site detection, it is imperative to establish an efficient, robust
and convenient detection method. In response to this demand, a promising detection strategy has been reported by combining loop-mediated isothermal amplification (LAMP) with clustered
regularly interspaced short palindromic repeat (CRISPR)/Cas12a10. This LAMP-CRISPR/Cas12a system has exhibited excellent performance in detecting SARS-CoV-211, _Mycobacterium tuberculosis_
complex12 and _Escherichia coli_ O157:H713. Compared to PCR, LAMP does not require sophisticated temperature control devices, simplifies the operation steps, and shortens the detection
time14. Due to its high sensitivity and isothermal amplification capabilities, LAMP is widely applied in the field of on-site detection of pathogenic microorganisms. However, non-specific
amplification due to improper operation or suboptimal conditions in the LAMP reaction may lead to false positive results. CRISPR/Cas system specifically and precisely recognizes the
amplicons by guide RNA (gRNA) and cleave single-stranded DNA reporter (labeled with 5′-Fluorophore/3′-Quencher) to produce fluorescent signal15. CRISPR/Cas12a-mediated detection enhances the
specificity of the LAMP assay, avoiding false positive results caused by LAMP reaction by recognizing the specific sequence of the amplicons. This provides a pathway for both
high-sensitivity and high-specificity detection. In previous works, Gong et al.16 reported a rapid, sensitive, and specific _S_. typhimurium detection method by integrating LAMP with
CRISPR/Cas12b system within a single tube. The platform’s limit of detection (LOD) is 12.5 copies per reaction. Zhang et al.17 developed a novel detection machinery based on DNA walker and
CRISPR-Cas12a technologies. _Nt. AlwI_ nicking endonuclease was triggered by the _S_. typhimurium specific sequence to produce particular signaling nucleotide, activating Cas12a for strong
fluorescence signal output. The LOD of DNA walker was successfully decreased by 2,000 folds to 5 CFU/mL. So-Young et al.18 developed a lateral flow biosensor (LFB) based on
LAMP/CRISPR-Cas12a. False positives were prevented by synthesizing CRISPR ribonucleic acid (crRNA) with a selected 21-base pair protospacer. The detection limit was 1.22×100 CFU/mL in pure
culture. However, the reported target DNA required for LAMP-CRISPR/Cas12a system is generally extracted by commercial kits, which requires laboratory instruments supporting19. To achieve
on-site detection of _S_. typhimurium throughout the analysis process, a portable and easy-to-operate nucleic acid extraction device is needed as an upstream procedure of the
LAMP-CRISPR/Cas12a assay. Fronczek et al.20 reported a rapid, paper microfluidic- and smartphone-based protocol developed for the extraction and direct fluorescent identification of the
nucleic acids of _S_. typhimurium. While offered the advantages of portability, efficiency, and cost-effectiveness, this paper-based microfluidic device faced challenges in precisely
controlling fluid flow and laborious operational procedures. Chen et al.21 developed a portable origami microfluidic device that enabled rapid detection of _S. enterica_. Compared to
paper-based device, this polyethersulfone-based device improved the efficiency of nucleic acid amplification. However, the origami structure may impede the sufficient interaction between
reagents and DNA templates. Additionally, the extraction process, being non-enclosed, was vulnerable to the risk of cross-contamination. Magnetic silica bead-based nucleic acid extraction is
characterized by simple operation, low risk of cross-contamination and high efficiency to extract target bacterial DNA from complex matrices22. Under certain concentrations of Polyethylene
Glycol (PEG) and with the presence of Sodium Chloride (NaCl), the conformation of DNA molecules undergoes a drastic change, leading to the exposure of numerous negatively charged phosphate
groups on the phosphate backbone, which can form ionic bridges with the functional groups on the surface of the magnetic beads (such as carboxyl groups or hydroxyl groups) through
dissociated cations (such as Na+), thereby achieving specific adsorption of DNA onto the magnetic beads23,24. This process of DNA adsorption onto the surface of the hydroxyl-functionalized
magnetic beads is reversible, and under appropriate conditions, the bound DNA molecules can be desorbed and recovered25. Therefore, in this study, we developed a magnetic silica bead-based
nucleic acid extraction device for conveniently and rapidly extracting nucleic acid. This device incorporated a syringe and a polytetrafluoroethylene (PTFE) tube. The syringe was filled with
a pre-loaded lysis solution, and the PTFE tube housed both the washing buffer and elution buffer, with an air barrier separating them. The entire extraction process occurred within the
closed tube, which minimized potential aerosol contamination. Furthermore, we have successfully developed a single-vial LAMP-CRISPR/Cas12a assay for the detection of _S_. typhimurium (Fig.
S1). We lyophilized LAMP and CRISPR/Cas12a reagents into a reaction vial. The DNA extracted from our device was amplified in the vial. With just a portable ultraviolet (UV) lamp, the
detection results can be read out by the naked eyes. We also developed a smartphone App for auxiliary discrimination of fluorescence results. Technology roadmap was provided in Supporting
Information (Fig. S2). From nucleic acid extraction to results readout, all the gears and reagents were integrated within a portable suitcase. The detection results can be observed within 1
h without needing any sophisticated instruments. Its operation is simple and suitable for untrained personnel. We anticipate that this proposed method could enhance on-site nucleic acid
detection capability and enable better control and response to outbreaks of foodborne diseases, especially in low-resource settings. RESULTS ESTABLISHMENT OF LAMP FOR _S_. TYPHIMURIUM Seven
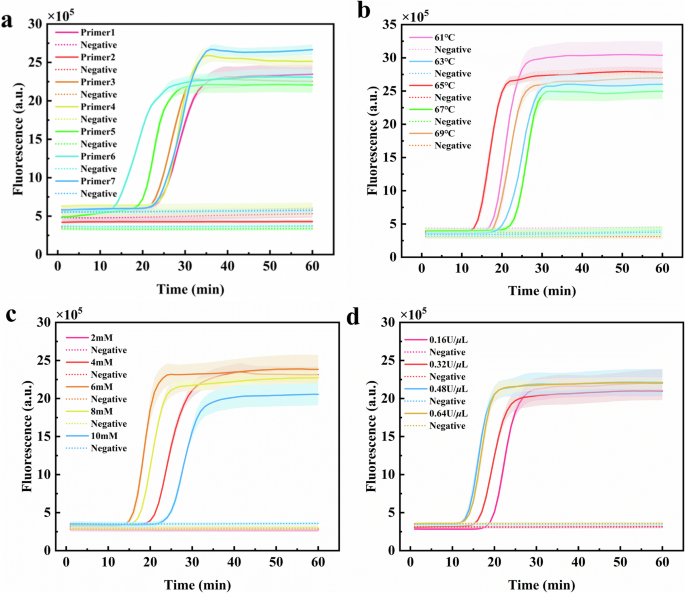
sets of candidate LAMP primers were self-designed and tested using genomic DNA of _S_. typhimurium as the target (Table S2). According to the position of the inflection point of the
amplification curve, primer set 6 showed better amplification efficiency (Fig. 1a). Then, we synthesized the target sequence corresponding to primer set 6 and optimized three key conditions
of the LAMP reaction. The optimal amplification temperature was 65 °C, the optimal Mg2+ concentration was 6 mM, and the optimal _Bst_ DNA polymerase concentration was 0.48 U/_μ_L (Fig.
1b–d). The gRNA sequence was designed according to the LAMP amplicon (Table S3). To facilitate the application of LAMP in on-site detection, we explored to freeze-dry a portion of LAMP
reagents at the bottom of the PCR vial after adding saturated D- (+) -trehalose dihydrate. As a comparison, 25 _μ_L of LAMP liquid mixture was used for normal LAMP reaction. The
amplification curves showed that despite slight differences in the plateau phase, the two methods yielded similar fluorescence curves, indicating similar amplification efficiency (Fig. S3).
This verified the feasibility of using lyophilized reagents for LAMP reactions. LYSIS SOLUTION OPTIMIZATION Based on previous work, we developed a lysis solution formulation for rapid
nucleic acid extraction26. Among that, PEG 8000 played a crucial role in separating the nucleic acid from other impurities and concentrating the nucleic acid of sample27. TrionX-100, a
nonionic surfactant, disrupted the cell membrane, facilitating bacteria lysis and releasing nucleic acids. It acted on the hydrophobic regions of the bacterial membrane, destabilizing the
lipid bilayer and allowing nucleic acids to dissolve in the extraction buffer28. Proteinase K efficiently digested various proteins in the sample, including nucleases, histones, and other
contaminants. Moreover, by degrading proteins, proteinase K promoted the release of nucleic acids bound with protein complexes29. Glycogen, as a carrier for nucleic acids and a
co-precipitant, enhanced the precipitation of nucleic acids, leading to better recovery and increased yield of nucleic acids30. These components possessed a significant impact on extraction
efficiency and quality of nucleic acids26. Thus, we systematically optimize their concentrations in the lysis solution. The developed LAMP reaction was applied to amplify and produce the
observable signal. The optimization results were as follows: (1) the optimal concentration of PEG 8000 was 18%; (2) the optimal concentration of TrionX-100 was 2.5%; (3) the optimal
concentration of Proteinase K was 150 ng/_μ_L; (4) the optimal concentration of Glycogen was 20 ng/_μ_L (Fig. 2). LAMP-CRISPR/CAS12A ASSAY DEVELOPMENT We combined the CRISPR/Cas12a system
with LAMP after rapid nucleic acid extraction to enhance the specificity and achieve visual on-site detection. In LAMP reaction, excessive or insufficient amplification temperature, Mg2+
concentration and primer specificity are among the factors that can lead to non-specific amplification. In the negative control group, we intentionally altered the amplification temperature
and Mg2+ concentration to induce instability in the entire amplification system, thereby simulating the non-specific amplification that might occur in actual processes. The LAMP reaction
under non-optimal reaction conditions was conducted. We compared real-time fluorescence detection using traditional Eva Green nucleic acid dye and CRISPR/Cas12a-mediated detection. In the
group using Eva Green, two curves from the negative controls produced fluorescence within 30 min due to non-specific amplification. However, in the group using CRISPR/Cas12a-mediated
detection to detect the same LAMP reaction products, the negative controls did not produce fluorescence. Due to the fact that the non-specific LAMP products typically lacked both the PAM
site and the specific sequence complementary to gRNA, CRISPR/Cas12a ribonucleoprotein (RNP) complex cannot recognize and bind to these products, thereby failing to activate the
trans-cleavage activity of Cas12a enzyme. As a result, the reporter was not non-specifically cleaved to excite fluorescence. This proved that CRISPR/Cas12a-mediated detection indeed enhanced
the specificity of the LAMP assay, which avoided false positive results caused by non-specific amplification in the LAMP reaction through recognizing the specific sequence of the amplicons
(Fig. 3a, b). Subsequently, we integrated LAMP and CRISPR/Cas12a reaction in a single vial by placing lyophilized LAMP reagent at the bottom and freeze-dried CRISPR/Cas12a reagent inside the
cap. Different concentrations of _S_. typhimurium, ranging from 0 to 3.7 × 107 CFU/mL, were tested using this vial, followed by our developed nucleic acid extraction method. The detection
results consistently showed positive outcomes when the sample contained 3.7 × 102 CFU/mL _S_. typhimurium or more (Fig. 3d). And this detection method exhibited excellent specificity, as
shown in Fig. 3f. We also tested the same batch of samples using traditional Eva Green detection with optimized reaction conditions (Fig. 3c, e). The results were coherent with those of
CRISPR/Cas12a-mediated detection. PORTABLE NUCLEIC ACID EXTRACTION DEVICE Our portable nucleic acid extraction device consisted of a long nozzle syringe made of PP, a PTFE tube, and a plug.
The syringe, PTFE tube, and plug were assembled together to provide a sealed environment for nucleic acid extraction (Fig. 4a). Both PP and PTFE materials possessed excellent hydrophobicity,
chemical inertness, and low price31,32. Their ability to load different chemical reagents made them attractive33,34. Given their low cost and easy accessibility, the syringes, PTFE tubes
and plugs were disposable to prevent cross-contamination of nucleic acids during the extraction process. The syringe was loaded with lysis solution (a mixture of lysis buffer and magnetic
bead buffer). The PTFE tube was loaded with washing buffer and elution buffer separated by air (Fig. 4b). The nucleic acids released from the lysed bacteria were captured by the magnetic
beads. By using an external magnet, the magnetic beads were moved, facilitating the washing and elution steps in the extraction process. Tween 20 was used to reduce friction between the
magnetic beads and the tube, allowing for smoother movement. We customized a small temperature controller and heating sleeve to enhance the efficiency of nucleic acid lysis. The heating
system, with its capability for precise temperature control from 20 °C to 100 °C, satisfied the lysis requirements within the range of 55 °C to 75 °C25. It was powered by a 24 V mobile power
supply. When fully charged, this power can support the portable device to work for three weeks, making the entire device utterly independent of the external environment and more suitable
for on-site detection. The overall dimension of this portable device was only palm-size (6 cm × 12 cm × 12 cm, Fig. 4c). We integrated this device, a 24 V mobile power supply, an UV lamp, a
test vial for LAMP-CRISPR, and a metal heater into a suitcase (Fig. 4d). This suitcase was compact and easy to transport, greatly facilitating the implementation and application of on-site
detection. COMPARISON OF THE DNA EXTRACTION PERFORMANCE OF DEVICE AND COMMERCIAL KITS We applied our nucleic acid extraction device and commercial TIANamp Bacteria DNA Kit (TIANGEN
Biochemical Technology Co., Ltd, Beijing, China) to extract DNA from the same _S_. typhimurium sample at a concentration of 3.1 × 108 CFU/mL. The concentration and purity were measured using
a microplate reader. The experiment was repeated three times to ensure accuracy and reliability. The results indicated that both extraction methods yielded high-concentration and
high-purity DNA (A260/A280 value > 1.8, Table S4 and S5). The device-based extraction method resulted in slightly lower DNA concentration than the kit-based extraction method (177.589
ng/_μ_L vs. 186.364 ng/_μ_L), but the difference was within an acceptable range. Furthermore, 200 _μ_L of _S_. typhimurium at different concentrations were extracted by using our nucleic
acid extraction device and commercial kit, followed by LAMP reactions. A low concentration of _S_. typhimurium (3.7 × 102 CFU/mL) was detected by both assays within 30 min. These
similarities regarding the detection limit and the time to reach the plateau phase suggested that both methods were comparable in terms of their extraction purity and concentration (Fig.
S4). The gel electrophoresis of the amplification products yielded similar bands in terms of the size and quantity of the DNA fragments, which also demonstrated the comparability of the two
methods. (Fig. S5). We further verified the on-site application potential of the nucleic acid extraction device. Two extraction methods (device- and commercial kit-based) were combined with
single-vial LAMP-CRISPR/Cas12a system. 200 _μ_L of _S_. typhimurium (3.7 × 107, 3.7 × 106, 3.7 × 105, 3.7 × 104, 3.7 × 103, 3.7 × 102, 3.7 × 101 and 3.7 × 100 CFU/mL) was tested as target.
200 _μ_L of nuclease-free water was tested as negative control. For both assays, consistent and accurate detection can be achieved when the target was present at a concentration of 3.7 × 102
CFU/mL, as shown in Fig. 5. We hereby reported a consistent 100% true positive rate with 3.7 × 102 CFU/mL of _S_. typhimurium. These results suggested that our method was effective for
extracting DNA and detecting _S_. typhimurium. Also, the ability to accurately detect 10² CFU/mL of _S_. typhimurium was enough for monitoring, as the infectious dose for _S_. typhimurium
was 1 × 103 CFU/mL35,36. Our nucleic acid extraction device offered the advantage of portability and convenience, making it well suited for on-site use. ANALYSIS OF FOOD AND ENVIRONMENTAL
SAMPLES Due to the wide existence of _S_. typhimurium, we simulated various application scenarios. Contaminated food samples (cabbage and chicken samples) and environmental samples (lake
water and soil samples) were tested. The gold standard method, bacterial plate counting, was simultaneously applied to verify the accuracy and practicality of our method. The detection
results have been listed in the Supporting Information (Tables S6–S9.). A total of 54 mock samples, including 42 contaminated samples and 12 negative controls, were tested. Analysis of all
samples accurately yielded the expected results. _S_. typhimurium was detected consistently at a concentration level of 102 CFU/mL or 102 CFU/g in different matrices. RESULT READOUT USING
SMARTPHONE APP We developed an auxiliary smartphone app to provide simplified result interpretation (positive or negative). The experimental results were assessed qualitatively by the
observed green fluorescence. We developed a Java-based algorithm that utilized the green channel (G value) of the RGB value as the criteria. First, we prepared standard bacteria samples with
known concentrations and measured the range of G values. The calibration results are listed in Table S10. The results were interpreted as negative when the G value ranged from 0 to 199, and
as positive when the G value ranged from 200 to 255. Then, we implemented the codes of the corresponding range of G values into the app as the standard for sample detection. Images were
collected using a smartphone camera, with vials positioned at the bottom of the suitcase to minimize the impact of ambient light. The app automatically generates a qualitative result of
“positive” or “negative” by matching the G value range in codes. _S_. typhimurium in the food matrix was successfully detected (Fig. 6). The detection results were in concordance with the
results derived from single vial detection using the LAMP-CRISPR/Cas12a assay. The app-based method was able to accurately detect _S_. typhimurium at a concentration of 102 CFU/mL.
DISCUSSION In this study, we have developed an on-site sample-to-answer detection platform, combining LAMP-CRISPR/Cas12a technology, a portable device, and a smartphone app. This platform
minimized the requirements for external equipment by using an all-in-one suitcase, making on-site detection straightforward and efficient. By using this platform, the whole process of DNA
extraction, amplification and output of visual fluorescent signals was able to be completed within 1 h, and 102 CFU/mL of _S_. typhimurium in food and environmental matrix was able to be
accurately detected, demonstrating its tremendous potential for on-site applications. Compared to commercial extraction kit, our device had similar DNA extraction concentrations and
purities, meeting the requirements for subsequent experiments. The tube system, including a syringe, a PTFE tube and a plug, was disposable and had a low price (0.28 $). The extraction
process was conducted within the closed tube, which minimized the potential aerosol contamination. We successfully established a single-vial lyophilized LAMP-CRISPR/Cas12a system.
CRISPR/Cas12a-mediated detection enhanced the specificity of the LAMP assay, avoiding false positive results caused by non-specific amplification. Furthermore, we developed a smartphone app
to provide auxiliary interpretation of detection results, minimizing the subjectivity and error by recognizing changes in color values. By designing specific LAMP primers and gRNA targeting
different bacteria, this proposed platform could be extended to the detection of other foodborne pathogens. While this study promoted the applications of on-site detection of _S_.
typhimurium, the current assay is limited in its capacity for accurate quantification and multiplex detection. In future further research, we will place a significant emphasis on the
development of assays tailored for quantitative detection and simultaneous detection of multiple targets. METHODS REAGENTS AND MATERIALS Polyethylene glycol (PEG) 8000, Tris-HCl, Ethylene
Diamine Tetraacetic Acid (EDTA), SanMag Si-OH magnetic beads and TrionX-100 were purchased from Sangon Biotech (Shanghai, China). 2-Mercaptoethanol (2-ME) was bought from BioShop Canada
(Burlington, Ontario, Canada). MgSO4, _Bst_ 2.0 WarmStart DNA polymerase, EnGen Lba Cas12a enzyme, 10× NE Buffer r2.1 and 10× Isothermal Amplification Buffer were obtained from New England
BioLabs (Ipswich, MA, USA). D- (+) -Trehalose dihydrate, Proteinase K, Tween-20, Sodium acetate anhydrate, Betaine and deoxynucleotide (dNTP) were bought from Sangon Biotech (Shanghai,
China). Eva Green nucleic acid dye (20× in water) was bought from YeSen (Shanghai, China). RLT lysis buffer was obtained from QIAgen (Germantown, MD, USA). RNA-grade Glycogen was purchased
from ThermoFisher Scientific (New York, USA). BACTERIAL CULTURE _S_. typhimurium (ATCC 13311), _Enterococcus faecalis_ (_E. faecalis_, ATCC 29212), _Escherichia coli_ O157:H7 (_E. coli_
O157:H7, ATCC 25922), _Vibrio parahaemolyticus_ (_V. parahaemolyticus_, ATCC 17802), _Listeria monocytogenes_ (_L. monocytogenes_, ATCC 17802) and _Staphylococcus aureus_ (_S. aureus_,
ATCC49775) were stored at −80 °C in School of Public Health, Jilin University. After being revived on Luria–Bertani (LB) agar plates, these strains were incubated in LB medium at 37 °C for
16 h and shaken at 180 rpm/min. Then, bacteria were harvested during the exponential growth phase. LOOP-MEDIATED ISOTHERMAL AMPLIFICATION Design protocol of the primer, sequence of the
amplification region of _S_. typhimurium and sequences of the LAMP primer sets are summarized in Supporting Information (Table S1). All primers were synthesized by Sangon Biotech (Shanghai,
China). Optimization protocol of the LAMP reaction is provided in Supporting Information. After optimization, 25 _μ_L of LAMP reaction system contained 1.6 mM dNTP, 1× isothermal
amplification buffer, 6 mM MgSO4, 0.4 M Betaine, 0.8× Eva Green nucleic acid dye, 0.2 _μ_M F3 primer, 0.2 _μ_M B3 primer, 1.6 _μ_M FIP primer, 1.6 _μ_M BIP primer, 0.8 _μ_M LF primer, 0.8
_μ_M LB primer, 0.48 U/_μ_L _Bst_ 2.0 WarmStart DNA polymerase and 2 _μ_L of sample (2 _μ_L of nuclease-free water as negative control). LAMP reactions were performed at 65 °C for 30 min.
CONSTRUCTION OF LABORATORY NUCLEIC ACID EXTRACTION METHOD 10 mL of the initial magnetic bead buffer was prepared containing 20 mM Tris-HCl, 2 M NaCl, 15% PEG 8000, 2 mM EDTA, 125 _μ_L of 50
mg/mL SanMag Si-OH. 10 mL of the initial lysis buffer was prepared by adding Glycogen (final concentration 17 ng/_μ_L), 2-ME (final concentration 1%), Proteinase K (final concentration 175
ng/_μ_L), TrionX-100 (final concentration 2%), 100 _μ_L of Tween-20 into the RLT lysis buffer. One portion of the lysis solution comprised 400 _μ_L magnetic beads buffer, 600 _μ_L lysis
buffer and 200 _μ_L anhydrous ethanol. The washing buffer was prepared by mixing 30 mL of 75% anhydrous ethanol, 10 mL of double distilled water, and 100 _μ_L of 0.1 M Sodium acetate. The
elution buffer was 50–100 _μ_L of nuclease-free water or Tris-EDTA buffer. A nucleic acid extraction method operated in laboratory was constructed. 200 _μ_L of samples was mixed with one
portion of the lysis solution in a 1.5 mL of Eppendorf (EP) vial. The mixture was heated at 65 °C for 10 min and shaken for 2 min. Thereafter, magnetic beads were captured by a magnetic rack
and washed twice with 600 _μ_L of washing buffer. After drying to remove the residual ethanol for 5 min, 50 _μ_L of elution buffer was added to liberate the DNA from the magnetic beads to
obtain a high-quality DNA extract. OPTIMIZATION OF NUCLEIC ACID EXTRACTION BUFFER The constructed laboratory nucleic acid extraction method was combined with the lyophilized LAMP system to
screen for the optimal concentrations of key components in the lysis solution based on the cycle threshold. Reaction conditions were as follows: There was no change in the amount of lysis
solution and washing buffer. 50 _μ_L of elution buffer included 0.4 M betaine, 0.8× Eva Green nucleic acid dye and the LAMP primers. 50 _μ_L of lyophilized LAMP reagents were added in the
bottom of the 200 _μ_L PCR vial, including 1.6 mM dNTP, 1× isothermal amplification buffer, 6 mM MgSO4, 0.48 U/_μ_L of _Bst_ 2.0 WarmStart DNA polymerase, and 10 _μ_L of saturated D- (+)
-Trehalose dihydrate. The vial was placed in a −80 °C refrigerator for 30 min, and then in a vacuum freeze-drying machine at −80 °C and 30 Pa for 1 h. After nucleic acid extraction and
elution, 50 _μ_L of the elution buffer was transferred to this vial containing the freeze-dried LAMP mixtures to hydrate the reagents. Amplification was conducted at 65 °C. Optimization
protocol of the lysis solution is provided in Supporting Information. After optimization, 10 mL of lysis buffer was prepared by adding Glycogen (final concentration 20 ng/_μ_L), 2-ME (final
concentration 1%), Proteinase K (final concentration 150 ng/_μ_L), TrionX-100 (final concentration 2.5%), 100 _μ_L of Tween-20 into the RLT lysis buffer. Subsequently, 10 mL of magnetic bead
buffer was prepared containing 20 mM Tris-HCl, 2 M NaCl, 18% PEG 8000, 2 mM EDTA, 125 _μ_L of 50 mg/mL SanMag Si-OH. LAMP-CRISPR/CAS12A DETECTION SYSTEM IN A SINGLE VIAL The sequence of the
gRNA specifically identifying the LAMP amplicon was listed in the Supporting Information (Table S3). The reporter was designed as a single strand DNA with a length of 8 nucleotides labeled
with 6-carboxyfluorescein (6-FAM) and BlackHoleQuencher1 (BHQ1, Table S3). 1 _μ_M EnGen®Lba Cas12a was preincubated with 3 _μ_M gRNA in 1× NE Buffer r2.1 for 30 min to form a
ribonucleoprotein (RNP) complex in a 1:1 ratio. A 20 _μ_L-mixture of 0.55 _μ_M RNP complex, 40 mM MgSO4, and 50 mM Tris-HCl with 10 _μ_L of saturated D- (+) -trehalose dihydrate was placed
on the cap of a 1.5 mL EP vial. 50 _μ_L of freeze-dried LAMP reagents were lyophilized in the bottom of the same 1.5 mL EP vial. During the lyophilization process, kept the vial cap open and
upward, and the reagents in the cap were adhered. After nucleic acid extraction and elution, 50 _μ_L of elution buffer consisting 0.4 M betaine, 5 _μ_M reporter probe, LAMP primers,
nuclease-free water was added to hydration. The bottom of the vial was heated at 65 °C for 30 min to perform the LAMP reaction. Subsequently, the vial was turned upside down so that the LAMP
amplicons contacted with the freeze-dried CRISPR/Cas12a reagents at the top. Then, after flicking, the vial was placed at room temperature for 10 min and the fluorescence results were
visualized under the excitation of a portable ultraviolet lamp. DEVICE FOR ON-SITE NUCLEIC ACID EXTRACTION To better apply nucleic acid extraction in on-site detection, we designed a device
and integrated our developed nucleic acid extraction method into the device. This device included a long nozzle syringe made of Polypropylene (Henghongqian E-commerce Co., Ltd, Suzhou,
China), a PTFE tube (Yuantong E-commerce Co., Ltd, Taizhou, China) with an inner diameter of 2 mm, a heating sleeve with a temperature controller (Zhenglong Electric Heating Technology Co.,
Ltd, Yancheng, China) and a plug. A 24 V mobile power bank provided the power. The frame of the device was printed with black high-toughness photosensitive resin (Yuewu Technology Co., Ltd,
Jiaxing, China) through stereo lithography appearance technology of 3D printing. One portion of the lysis solution was added to the syringe. 600 _μ_L of washing buffer and 50 _μ_L of elution
buffer (0.4 M betaine, 5 _μ_M reporter, the primers, and nuclease-free water) were added into the PTFE tube and separated by air. After the solution was loaded, a plug sealed the end of the
PTFE tube and the long nozzle of the syringe was connected to the beginning of the PTFE tube, ensuring air tightness. Under air pressure, the solution did not move, making the device easy
to transport. ON-DEVICE DETECTION OF _S_. TYPHIMURIUM On-device method for detecting _S_. typhimurium was as follows: (1) 200 _μ_L of sample was sucked by the syringe and mixed thoroughly
with the pre-stored lysis solution. Then the syringe was placed back into the heating sleeve, and the nozzle was connected to the PTFE tube. The syringe was heated by the heating sleeve at
65 °C to lysis the bacteria (10 min). (2) The magnetic beads were transferred from the syringe nozzle with a magnet to the PTFE tube for washing (2 min) and eluting (5 min). After elution,
the magnetic beads were moved back to the washing buffer. (3) The plug was bunged off. By pressing the syringe, the elution buffer flowed from the device to the tube with lyophilized
LAMP-CRISPR/Cas12a reagents. (4) The bottom of this tube was maintained at 65 °C for 30 min to conduct LAMP reaction. Subsequently, the tube was inverted, followed by incubating at room
temperature for 10 min to complete the CRISPR/Cas12a-mediated fluorescence detection. A UV flashlight was used to excite green fluorescence for visual detection. The whole experimental
process of the on-site detection of _S_. typhimurium could be completed within 1 h (Fig. 7). The operation example was provided in the Supporting Information (Fig. S6). PERFORMANCE OF THE
NUCLEIC ACID EXTRACTION DEVICE The device and commercial TIANamp Bacteria DNA Kit were utilized to extract DNA from 200 _μ_L of _S_. typhimurium with a concentration of 3.1 × 108 CFU/mL. The
usage protocol of the commercial DNA extraction kit was provided in Supporting Information. A microplate reader was utilized to quantify the concentration and assess the purity of DNA
samples. The amount of nucleic acid can be directly determined by the reader. The purity of DNA can be assessed by examining the A260/A280 ratio of nucleic acids. A ratio exceeding 1.9
suggested the presence of ribonucleic acid contamination, whereas a ratio below 1.6 indicated potential contamination with proteins, phenolic compounds, or other impurities. 200 _μ_L of _S_.
typhimurium with concentrations ranging from 3.7 × 107 CFU/mL to 3.7 × 100 CFU/mL were utilized to extract DNA by the device and commercial kit. The extracted DNA were then applied to the
LAMP freeze-dried method, with a reaction time of 60 min. The fluorescence curves were monitored using the QuantStudio™ 5 Real-Time PCR system, and the extraction efficiency of the nucleic
acid extraction device was evaluated based on the cycle threshold. Then, the amplification products were tested using gel electrophoresis. Furthermore, the same extraction products were
applied to the single-vial LAMP-CRISPR/Cas12a system. The extraction efficiency of the nucleic acid extraction device was evaluated by determining the minimum concentration of _S_.
typhimurium that could be detected through the CRISPR/Cas12a-mediated detection. DETECTION OF _S_. TYPHIMURIUM IN FOOD AND ENVIRONMENTAL SAMPLES The contaminated cabbage, chicken, lake
water, and soil samples were prepared following the previous literature37. The cabbage and chicken were purchased from the local supermarkets in Changchun, China, and the lake water and soil
were taken from the South Lake Park in Changchun, China. 1 g of cabbage was cut into small pieces and ground in 10 mL sterile double distilled water for 5 min. The mixture was filtered
through a 0.22 _μ_m filter to obtain the sterile cabbage filtrate. 5 g of chicken was thoroughly ground into minced meat. After spreading on a flat surface, it was exposed to ultraviolet
light for 5 h to remove the natural accumulation of pathogenic bacteria. 1 g of sterilized mince was thoroughly mixed with 10 mL of sterile double distilled water to form homogenate,
followed by filtering through a 0.22 _μ_m filter to obtain the sterile chicken filtrate. 50 g of soil was autoclaved and dissolved with sterile double distilled water. The mixture was
filtered with filter paper to obtain the soil filtrate. 1 Liter of lake water sample was filtered with a 0.22 _μ_m filter. 200 _μ_L of _S_. typhimurium in the mid-logarithmic growth phase
was mixed with the sample filtrate mentioned above, and cultured at 37 °C and 180 rpm shaking. Part of the culture liquid was plated and bacterial counted, and the liquid in the same culture
tube was detected by using our developed device and assay. SMARTPHONE APP FOR RESULT READING To minimize the subjectivity and error in the interpretation of results, an Android Studio-based
smartphone App was developed to provide auxiliary interpretation of detection results. The app was capable of image extraction, data collection, and result analysis using Java algorithms.
In practical application, the operator only needed to (1) take a picture of the test tube, (2) click the “Analyze” button, (3) select the image and (4) click the detection region. A brief
result, “negative” or “positive”, would display automatically. The smartphone used was a vivo X80 model, equipped with the Android 13 operating system. The core code has been attached to the
Part 8 of the Supporting Information. To enhance the precision of APP-based result interpretation, the method established in this study was used to detect 200 _μ_L of different
concentrations of _S_. typhimurium (102, 103, 104, 105, 106 CFU/mL), with 200 _μ_L of nuclease-free water serving as the negative control. Each experiment was repeated three times. The
experimental results were captured using a smartphone, and the RGB color values were read using an APP, which served as the basis for interpretation, implemented in Java code. To validate
the feasibility of the APP-based detection for _S_. typhimurium, chicken filtrate was spiked with a known concentration of _S_. typhimurium to prepare simulated samples of varying
concentrations (102 CFU/mL, 104 CFU/mL, and 106 CFU/mL). The detection method established in this study was applied to test 200 _μ_L of the aforementioned simulated samples at different
concentrations, with 200 _μ_L of nuclease-free water serving as the negative control. For each concentration, three replicates were set up, and the smartphone APP was utilized to assist in
interpreting and analyzing the results. DATA AVAILABILITY The data supporting the finding reported herein are available on reasonable request from the corresponding author. CODE AVAILABILITY
Code for data analysis is available in Supplementary Information. REFERENCES * Chatterjee, R. et al. _Salmonella_ Typhimurium PgtE is an essential arsenal to defend against the host
resident antimicrobial peptides. _Microbiol. Res._ 271, 127351 (2023). CAS PubMed Google Scholar * Majee, S. et al. Spatiotemporal evaporating droplet dynamics on fomites enhances long
term bacterial pathogenesis. _Commun. Biol._ 4, 1173 (2021). CAS PubMed PubMed Central Google Scholar * Srisa-Art, M., Boehle, K. E., Geiss, B. J. & Henry, C. S. Highly sensitive
detection of _Salmonella_ typhimurium using a colorimetric paper-based analytical device coupled with immunomagnetic separation. _Anal. Chem._ 90, 1035–1043 (2018). CAS PubMed Google
Scholar * Jia, S., McWhorter, A. R., Andrews, D. M., Underwood, G. J. & Chousalkar, K. K. Challenges in vaccinating layer hens against _Salmonella_ Typhimurium. _Vaccines (Basel)_ 8,
696 (2020). CAS PubMed Google Scholar * Angelopoulou, M. et al. Rapid detection of _Salmonella_ typhimurium in drinking water by a white light reflectance spectroscopy immunosensor.
_Sensors (Basel)_ 21, 2683 (2021). CAS PubMed Google Scholar * Wen, C. Y. et al. Colorimetric and photothermal dual-mode lateral flow immunoassay based on Au-Fe3O4 multifunctional
nanoparticles for detection of _Salmonella_ typhimurium. _Mikrochim. Acta_ 190, 57 (2023). CAS PubMed PubMed Central Google Scholar * Lu, Z. et al. _Salmonella_ typhimurium strip based
on the photothermal effect and catalytic color overlap of PB@Au nanocomposite. _Food Chem._ 385, 132649 (2022). CAS PubMed Google Scholar * Ahirwar, R., Bhattacharya, A. & Kumar, S.
Unveiling the underpinnings of various non-conventional ELISA variants: a review article. _Expert Rev. Mol. Diagn._ 22, 761–774 (2022). CAS PubMed Google Scholar * Feng, W. et al.
Molecular diagnosis of COVID-19: challenges and research needs. _Anal. Chem._ 92, 10196–10209 (2020). CAS PubMed Google Scholar * Pang, B. et al. Isothermal amplification and ambient
visualization in a single tube for the detection of _SARS-CoV-2_ using loop-mediated amplification and CRISPR technology. _Anal. Chem._ 92, 16204–16212 (2020). CAS PubMed Google Scholar *
Wang, R. et al. opvCRISPR: one-pot visual RT-LAMP-CRISPR platform for _SARS-cov-2_ detection. _Biosens. Bioelectron._ 172, 112766 (2021). CAS PubMed Google Scholar * Wang, Y. et al.
LAMP-CRISPR-Cas12-based diagnostic platform for detection of Mycobacterium tuberculosis complex using real-time fluorescence or lateral flow test. _Mikrochim. Acta_ 188, 347 (2021). CAS
PubMed Google Scholar * Lee, S. Y. & Oh, S. W. Filtration-based LAMP-CRISPR/Cas12a system for the rapid, sensitive and visualized detection of _Escherichia coli_ O157:H7. _Talanta_
241, 123186 (2022). CAS PubMed Google Scholar * Das, D., Lin, C. W. & Chuang, H. S. LAMP-based point-of-care biosensors for rapid pathogen detection. _Biosensors (Basel)_ 12, 1068
(2022). CAS PubMed Google Scholar * Paul, B. & Montoya, G. CRISPR-Cas12a: functional overview and applications. _Biomed. J._ 43, 8–17 (2020). PubMed PubMed Central Google Scholar *
Gong, J. et al. One-tube detection of _Salmonella_ Typhimurium using LAMP and CRISPR-Cas12b. _Microbiol. Spectr._ 12, e0127124 (2024). PubMed Google Scholar * Zhang, H. et al. A cascade
amplification strategy for ultrasensitive _Salmonella_ typhimurium detection based on DNA walker coupling with CRISPR-Cas12a. _J. Colloid Interface Sci._ 625, 257–263 (2022). CAS PubMed
Google Scholar * So-Young, L. & Se-Wook, O. Lateral flow biosensor based on LAMP-CRISPR/Cas12a for sensitive and visualized detection of _Salmonella_ spp. _Food Control_ 145, 109494
(2023). Google Scholar * Yu, Z. et al. A rapid, ultrasensitive, and highly specific method for detecting fowl adenovirus serotype 4 based on the LAMP-CRISPR/Cas12a system. _Poult. Sci._ 9,
104048 (2024). Google Scholar * Fronczek, F. C. et al. Paper microfluidic extraction and direct smartphone-based identification of pathogenic nucleic acids from field and clinical samples.
_RSC Adv._ 4, 11103 (2014). CAS Google Scholar * Chen, Y., Hu, Y. & Lu, X. Polyethersulfone-based microfluidic device integrated with DNA extraction on paper and recombinase polymerase
amplification for the detection of _Salmonella_ enterica. _ACS Sensors_ 8, 2331–2339 (2023). CAS PubMed Google Scholar * Klein, S. et al. SARS-CoV-2 RNA extraction using magnetic beads
for rapid large-scale testing by RT-qPCR and RT-LAMP. _Viruses_ 12, 863 (2020). CAS PubMed PubMed Central Google Scholar * Hawkins, T. L., O’Connor-Morin, T., Roy, A. & Santillan, C.
DNA purification and isolation using a solid-phase. _Nucleic Acids Res._ 22, 4543–4544 (1994). CAS PubMed PubMed Central Google Scholar * Ali, N., Rampazzo, R. C. P., Costa, A. D. T.
& Krieger, M. A. Current nucleic acid extraction methods and their implications to point-of-care diagnostics. _Biomed. Res. Int._ 2017, 9306564 (2017). PubMed PubMed Central Google
Scholar * Li, Y. et al. Research on a magnetic separation-based rapid nucleic acid extraction system and its detection applications. _Biosensors (Basel)_ 13, 903 (2023). CAS PubMed Google
Scholar * Liu, Y. et al. On-site viral inactivation and RNA preservation of gargle and saliva samples combined with direct analysis of SARS-CoV-2 RNA on magnetic beads. _ACS Meas. Sci. Au_
2, 224–232 (2022). CAS PubMed PubMed Central Google Scholar * Bar-Or, I. et al. Regressing SARS-CoV-2 sewage measurements onto COVID-19 burden in the population: a proof-of-concept for
quantitative environmental surveillance. _Front Public Health_ 9, 561710 (2022). PubMed PubMed Central Google Scholar * Paul, R., Ostermann, E. & Wei, Q. Advances in point-of-care
nucleic acid extraction technologies for rapid diagnosis of human and plant diseases. _Biosens. Bioelectron._ 169, 112592 (2020). CAS PubMed PubMed Central Google Scholar * Allweiss, L.
et al. Quantification of the hepatitis B virus cccDNA: evidence-based guidelines for monitoring the key obstacle of HBV cure. _Gut_ 72, 972–983 (2023). CAS PubMed Google Scholar *
Pecorini, S., Camurri, G., Torrini, L. & Ferraresi, R. Highly sensitive real-time PCR method to identify species origin in heparinoids. _Anal. Bioanal. Chem._ 412, 289–298 (2020). CAS
PubMed Google Scholar * Wu, H. et al. Carrying out pseudo dual nucleic acid detection from sample to visual result in a polypropylene bag with CRISPR/Cas12a. _Biosens. Bioelectron._ 178,
113001 (2021). CAS PubMed Google Scholar * Yin, J., Hu, J., Sun, J., Wang, B. & Mu, Y. A fast nucleic acid extraction system for point-of-care and integration of digital PCR.
_Analyst_ 144, 7032–7040 (2019). CAS PubMed Google Scholar * Dhanumalayan, E. & Joshi, M. G. Performance properties and applications of polytetrafluoroethylene (PTFE)—a review. _Adv.
Compos. Hybrid. Mater._ 2, 247–268 (2018). Google Scholar * Zhong, Z., Yao, X., Gao, X. & Jia, L. Polydopamine-immobilized polypropylene microfuge tube as a pH-responsive platform for
capture/release of DNA from foodborne pathogens. _Anal. Biochem._ 534, 14–18 (2017). CAS PubMed Google Scholar * Wei, S. et al. On-site colorimetric detection of _Salmonella_ typhimurium.
_NPJ Sci. Food_ 6, 48 (2022). PubMed PubMed Central Google Scholar * Fang, S. et al. Simultaneous and sensitive determination of _Escherichia coli_ O157:H7 and _Salmonella_ Typhimurium
using evanescent wave dual-color fluorescence aptasensor based on micro/nano size effect. _Biosens. Bioelectron._ 185, 113288 (2021). CAS PubMed Google Scholar * Pang, B. et al.
Colorimetric detection of Staphylococcus aureus using gold nanorods labeled with yolk immunoglobulin and urease, magnetic beads, and a phenolphthalein impregnated test paper. _Mikrochim.
Acta_ 186, 611 (2019). PubMed Google Scholar Download references ACKNOWLEDGEMENTS This study was funded by [the National Natural Science Foundation of China] [grant numbers 82373578,
82204101, 82103892] and [the Science and Technological Research Project of Education Department of Jilin Province][grant number JJKH20250210KJ] and [the Program of “Medicine+X”
Interdisciplinary Innovation Team of Bethune Medical Department of Jilin University] [grant number 2022JBGS09]. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * School of Public Health, Jilin
University, 130021, Changchun, P.R. China Liang Zhang, Mengfan Zhang, Caihong Yin, Nan Wang, Beibei Fan, Yanli Fu, Yanwen Liu, Liangyun Bu, Zhenyue Su, Bo Pang, Jinhua Li & Xiuling Song
* Disease Control and Prevention of Liaoning Province, 110000, Shenyang, P.R. China Liang Zhang * Health Monitoring and Inspection Center of Baishan City, 134300, Baishan, P.R. China Mengfan
Zhang * Department of Cadre Ward, The First Hospital of Jilin University, 130021, Changchun, P.R. China Xingxing Liu * Department of Thyroid Surgery, General Surgery Center, The First
Hospital of Jilin University, 130021, Changchun, P.R. China Jia Wei Authors * Liang Zhang View author publications You can also search for this author inPubMed Google Scholar * Mengfan Zhang
View author publications You can also search for this author inPubMed Google Scholar * Xingxing Liu View author publications You can also search for this author inPubMed Google Scholar *
Jia Wei View author publications You can also search for this author inPubMed Google Scholar * Caihong Yin View author publications You can also search for this author inPubMed Google
Scholar * Nan Wang View author publications You can also search for this author inPubMed Google Scholar * Beibei Fan View author publications You can also search for this author inPubMed
Google Scholar * Yanli Fu View author publications You can also search for this author inPubMed Google Scholar * Yanwen Liu View author publications You can also search for this author
inPubMed Google Scholar * Liangyun Bu View author publications You can also search for this author inPubMed Google Scholar * Zhenyue Su View author publications You can also search for this
author inPubMed Google Scholar * Bo Pang View author publications You can also search for this author inPubMed Google Scholar * Jinhua Li View author publications You can also search for
this author inPubMed Google Scholar * Xiuling Song View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS L.Z.: Methodology, Data curation, Formal
analysis, Software, Writing-original draft; M.Z.: Methodology, Formal analysis; X.L.: Methodology, Formal analysis; J.W.: Writing - original draft; C.Y.: Writing - original draft; N.W.:
Methodology; B.F.: Writing-original draft; Y.F.: Formal Analysis; Y.L.: Methodology; L.B.: Methodology; Z.S.: Software; B.P.: Conceptualization, Project administration, Supervision, Funding
acquisition, Writing-review & editing; J.L.: Supervision, Funding acquisition, Writing-review & editing; X.S.: Resources, Investigation, Funding acquisition, Validation, Supervision;
CORRESPONDING AUTHORS Correspondence to Bo Pang, Jinhua Li or Xiuling Song. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use,
sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or
other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Zhang, L., Zhang, M.,
Liu, X. _et al._ Portable DNA extraction integrated with LAMP-CRISPR/Cas12a technology for on-site detection of _Salmonella_ Typhimurium. _npj Sci Food_ 9, 39 (2025).
https://doi.org/10.1038/s41538-025-00401-2 Download citation * Received: 17 April 2024 * Accepted: 26 February 2025 * Published: 24 March 2025 * DOI:
https://doi.org/10.1038/s41538-025-00401-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative