- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Lynch syndrome is an autosomal dominant hereditary cancer syndrome in which many cancers develop, the main one being colorectal cancer. Germline pathogenic variants in one of four
mismatch repair (MMR) genes are known to be causative of this disease. Accurate diagnosis using genetic testing can greatly benefit the health of those affected. Recently, owing to the
improvement of sequence techniques, complicated variants affecting the functions of MMR genes were discovered. In this study, we analyzed insertions of a retrotransposon-like sequence in
exon 5 of the _MSH6_ gene and exon 3 of the _MSH2_ gene found in Japanese families suspected of having Lynch syndrome. Both of these insertions induced aberrant splicing, and these variants
were successfully identified by mRNA sequencing or visual observation of mapping results, although a standard DNA-seq analysis pipeline failed to detect them. The insertion sequences were
~2.5 kbp in length and were found to have the structure of an SVA retrotransposon (SVA). One SVA sequence was not present in the hg19 or hg38 reference genome, but was in a Japanese-specific
reference sequence (JRGv2). Our study illustrates the difficulties of identifying SVA insertions in disease genes, and that the possibility of polymorphic insertions should be considered
when analyzing mobile elements. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS _TP53_ MINIGENE ANALYSIS OF 161 SEQUENCE
CHANGES PROVIDES EVIDENCE FOR ROLE OF SPATIAL CONSTRAINT AND REGULATORY ELEMENTS ON VARIANT-INDUCED SPLICING IMPACT Article Open access 08 May 2025 A 39 KB STRUCTURAL VARIANT CAUSING LYNCH
SYNDROME DETECTED BY OPTICAL GENOME MAPPING AND NANOPORE SEQUENCING Article Open access 29 November 2023 WIDESPREAD SOMATIC L1 RETROTRANSPOSITION IN NORMAL COLORECTAL EPITHELIUM Article Open
access 10 May 2023 INTRODUCTION Lynch syndrome is an autosomal dominant hereditary cancer syndrome in which colon cancer is particularly prominent. It is caused by a pathogenic variant in
one of the mismatch repair genes (MMR genes; _MLH1_, _MSH2_, _MSH6_, or _PMS2_) [1,2,3,4,5]. In recent years, owing to the advent of next-generation sequencing (NGS) and the progression of
analytical techniques, many variants affecting the functions of MMR genes have been revealed [6]. Lynch syndrome accounts for about 3% of all colon cancer cases [7, 8] and its prevalence in
the general population has been estimated at 1:440 [9]. Because various cancers besides colon cancer occur in those affected by this syndrome, determination of the responsible variant and
appropriate surveillance are important to the healthcare of patients [10]. Retrotransposons are “mobile genetic elements” that move in the genome. Transposon insertion causes a change in the
gene at or near the insertion point. Such a change is considered to provide room for the evolution of the genome [11]. Retrotransposons occupy ~40% of the human genome sequence and are
classified into two groups: long terminal repeat (LTR) and non-LTR. LTR retrotransposons do not have transposable element activity, while non-LTR retrotransposons, including LINE-1
(constitute 16.9% of the human genome), Alu (10.6%), and SVA (0.2%) elements, have such activity; this latter group covers approximately one-third of the human genome and causes various
hereditary diseases via its insertion [12]. SVA retrotransposons, hominid-specific retrotransposons, consist of SINE (short interspersed repetitive elements), VNTR (variable number of tandem
repeats), and Alu, and are ~3 kb in length. This type of retrotransposon is rare, with ~2700 copies of it in the human genome [13]. Regarding its relationship with disease, it was reported
that a cause of Fukuyama-type congenital muscular dystrophy was splice abnormality by the insertion of an SVA-type retrotransposon [14]. With regard to Lynch syndrome, an Alu insertion
variant in MSH2 was reported [15, 16], while a report of a single case of SVA insertion in _PMS2_ was also published [17]. In this study, insertion of an SVA-type retrotransposon was found
in exonic regions of the _MSH2_ or _MSH6_ gene. One of the inserted sequences aligned only with JRGv2 (Japanese Reference Genome V2) provided by the Tohoku Medical Megabank but not with
standard human reference genomes. RNA sequencing revealed an aberrant alternative splicing event associated with these variants. MATERIALS AND METHODS PATIENTS Patients enrolled in Study for
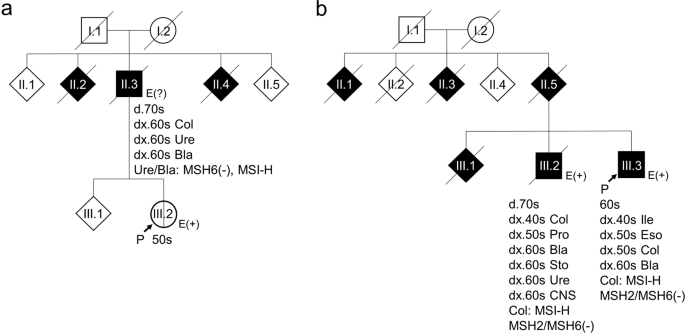
the Establishment of Effective Screening and Diagnosis of Lynch Syndrome (Dial study) were analyzed. Case 1 (Fig. 1a) had never been diagnosed with cancer. However, his father had developed
three metachronous Lynch syndrome-associated cancers (colorectal, ureteral, and bladder cancers). Microsatellite instability (MSI) and immunohistochemical testing revealed that the ureteral
and bladder cancers showed microsatellite instability-high (MSI-H) and loss of MSH6. Case 2 (Fig. 1b) was diagnosed with metachronous multiple cancers and his older brother showed a similar
phenotype. In both siblings, the colorectal cancer was MSI-H, and loss of MSH2/MSH6 was observed (Fig. 1b, III.2, 3). All procedures were performed in accordance with the ethical standards
of the responsible committee on human experimentation and with the 1964 Helsinki Declaration, as revised in 2013, as well as the Japanese ethical guidelines for human genome/gene analysis
research. This study was approved by the Institutional Review Boards of Saitama Cancer Center (no. 729). Written consent was obtained from the patient before inclusion in the study. DNA AND
RNA EXTRACTION FROM PERIPHERAL BLOOD MONONUCLEAR CELLS (PBMCS) Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood collected in heparinized vacutainer tubes using
Ficoll®-Paque Premium (GE Healthcare, Chicago, IL, USA). These cells were resuspended in KBM502 (KOHJIN BIO, Sakado, Japan) supplemented with 10% FBS (GE Healthcare) and
penicillin–streptomycin (FUJIFILM Wako Pure Chemical, Osaka, Japan), and plated and cultured in tissue culture tubes (TPP, Trasadingen, Switzerland) at 37 °C in a 5% CO2 humidified
atmosphere. After 1 week, cells were divided into culture tubes for DNA extraction and RNA extraction with or without puromycin (Thermo Fisher Scientific, Waltham, MA, USA) treatment, as
reported previously [18]. DNA and RNA (with/without puromycin treatment) were extracted using AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany), in accordance with the manufacturer’s
instructions. NEXT-GENERATION SEQUENCING ANALYSIS FOR GENETIC TESTING Genetic testing for LS was conducted with both DNA and RNA. DNA was sequenced using QIAseq Targeted DNA Custom Panel
(Qiagen) including MMR genes (_MLH1_, _MSH2_, _MSH6_, _PMS2_, and _EPCAM_), in accordance with the manufacturer’s instructions. Transcripts of MMR genes were amplified by PCR with cDNA
synthesized from RNA and sequenced using Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA, USA), in accordance with the manufacturer’s instructions with slight modifications. [The
library amplification was carried out using KAPAHiFi DNA Polymerase (Kapa Biosystems, Wilmington, DE, USA), not NPM.] Sequencing was performed on Miseq (Illumina). The sequence reads were
analyzed with CLC Genomics Workbench (Qiagen, RRID: SCR_011853) using hg19 as a reference. The Japanese reference genome was obtained from JMORP (https://jmorp.megabank.tohoku.ac.jp/, [19]).
The accession numbers for the _MSH2_ and _MSH6_ genes (transcripts) were NG_007110.2 (LRG218t1) and NG_007111.1 (LRG219t1), respectively. Exons are numbered according to the accession
number of LRG. To confirm the inserted sequences, amplified PCR products were sequenced using the same method as in the RNA sequencing. Because there was difficulty amplifying the insertion
sequence of Case 2 using standard PCR enzyme, we used PrimeSTAR GXL DNA Polymerase (Takara Bio, Kusatsu, Japan). The list of primers designed for the amplification of MMR transcripts and
confirmation of the inserted sequences is given in Table 1. Visualization of mapping results was performed using Integrative Genomics Viewer software
(http://software.broadinstitute.org/software/igv/, RRID: SCR_011793). Sequence data resulted from this study is already submitted to the DNA DataBank of Japan (DDBJ) repository, accession
DRA009831 and DRA009891. RESULTS FINDING OF INSERTION IN EXONIC REGIONS OF _MSH6_ Multigene panel testing on the DNA sample of Case 1 using our standard sequencing pipeline detected neither
a pathogenic single-nucleotide variant nor copy number variation. However, sequencing of the _MSH6_ transcript revealed a deletion of the 5′ region of exon 5 in 27% of the reads of
transcripts isolated from puromycin treated PBMCs (23% in untreated cells) (Fig. 2a), suggesting that aberrant splicing was induced by the use of a cryptic 3′ splice site. Via careful
evaluation of the mapping of the DNA sequence around this region, we found a soft-clipped sequence in the middle of exon 5 consisting of repetitive “GGGAGA” units and accounted for 304 out
of a total of 749 reads covering this site (Fig. S2a). These results suggested the presence of a larger inserted sequence with the sequence at the 3′ end although the 5′ end of the insertion
had not been detected. According to this assumption, we attempted to amplify the inserted DNA fragment using a forward primer (_MSH6_ F) located in exon 4 together with an
insertion-specific reversed primer (_MSH6_ R) (Table 1, Fig. 3a). Since the wild-type sequence between this primer pair is 2.6 kb, amplification of an ~5 kb PCR fragment revealed that the
inserted sequence is ~2.4 kb (Fig. 3c). INSERTION OF SVA-TYPE RETROTRANSPOSON CAUSES ABERRANT SPLICING To examine the inserted fragment, we sequenced the whole 5 kbp amplicon as described in
“Materials and methods” section. The results revealed the presence of an insertion sequence in exon 5 with a target site duplication starting at a poly-T tract (Fig. S1a). Together with the
“GGAGA” repeats at the 3′ end, this suggested that the insertion resulted from retrotransposition of an SVA element into exon 5 (Fig. S1b). The characteristics of this sequence are
reminiscent of an SVA-type retrotransposon (SVA). However, our standard mapping method failed to pinpoint the position in the reference genome, probably because of the repetitive sequence.
Using de novo assembly software, we obtained the whole sequence of the insert. By re-mapping on the reference genome, the sequence starting from poly-T turned out to be unique to chr12:
96233959–96236309 and this region was annotated as SVA E by Repeat Masker (http://www.repeatmasker.org/). This variant was considered to be represented as NC_000002.11:
g.48030698_48030699ins[SVA;48030684_48030698]. In addition, detailed RNA analysis revealed the deletion of the initial 174 bp sequence of exon 5, which caused an in-frame deletion of the
MSH6 protein. Taking these findings together, we concluded that the insertion of a ~2.4 kbp SVA E retrotransposon into exon 5 changes its splicing acceptor site (Fig. 4a, Table 2). SVA
INSERTION MAY BE A MORE FREQUENT CAUSE OF LYNCH SYNDROME THAN WE ASSUMED As described above, aberrant splicing induced by SVA insertion has been reported for _PMS2_. Case 1 in this study led
us to the assumption that SVA insertion is not a very rare causes of Lynch syndrome. Thus, we performed a thorough investigation of cases in which one of the Lynch syndrome genes was
apparently mutated, but the nature of the variant had not been determined. The RNA sequence of _MSH2_ from Case 2 showed an aberrant transcript lacking 88 bp in the middle of exon 3 (Fig.
2b). Carefully evaluating the read mapping exon 3 revealed soft-clipped sequence similar to Case 1 consisting of repetitive “GGGAGA” (234 out of a total of 406 reads) and poly-T tract (234
out of 394 reads) (Fig. S2b). We assumed that this insertion sequence was also an SVA retrotransposon, but this insertion was not amplified by our standard PCR reactions. Therefore, we
employed PrimeSTAR GXL DNA polymerase (Takara Bio) instead of our standard PCR polymerase EX Taq (Takara Bio) to achieve better extension of “difficult to replicate” regions such as AT- or
GC-rich regions and attempted both two-step and three-step PCR cycles with two kinds of primer pairs, _MSH2_ F1/R1 and _MSH2_ F2/R2 (Table 1, Fig. 3b). Then, the insert was successfully
amplified (Fig. 3d) and the product was subjected to NGS. Unfortunately, our de novo assembly program failed to construct a single sequence as in Case 1. Therefore, we manually created
contigs from adjacent reads and merged them into a single sequence. However, we could not map the merged sequence on either hg19 or hg38. Only a slightly similar sequence in chromosome 16 of
hg19 was present (Fig. S3a). We thus assumed that the sequence is specific to the Japanese. By searching GGGenome (https://gggenome.dbcls.jp/en/), the sequence was revealed to be unique to
chr6: 111170450–111172854 of JRGv2, a Japanese-specific reference sequence [19] (Fig. S3b). Coverage analysis using the JRGv2 decoy sequence also suggested that this insertion sequence may
be specific to the sequence (Fig. S4). Furthermore, this sequence was thought to be inserted into NC_000006.11:g.111278198_111278199 in hg19 (Fig. S5). This sequence was annotated as SVA F
by Repeat Masker. Thus, in Case 2, an SVA F retrotransposon was integrated into exon 3 of _MSH2_ and created an extra intron in the middle of this exon (Fig. 4b). This variant was considered
to be represented as NC_000002.11: g.47637427_47637428ins[SVA;47637413_47637427]. As a result, shorter mRNA causing a frameshift was formed (Table 2). DISCUSSION In the genetic testing of
patients suspected of having Lynch syndrome in a multi-facility collaborative investigation, we found two novel exonic insertions of SVA in _MSH2_ and _MSH6_, and showed the splice effect of
these variants. Insertion of SVA has been reported only in an intron of the PMS2 gene [17], so this study is the first to identify it in other MMR genes and in an exonic region of these
four genes. As to the final evaluation of the pathogenicity of the two variants against Lynch syndrome, that in Case 1 is a variant of uncertain significance (VUS; PM2: absent in population
data, PM4: protein-length-changing variant) and that in Case 2 is likely pathogenic (PVS1: predicted null variant, PM2: absent in population data), according to ACMG criteria [20]. Regarding
Case 1, we tried to determine that the variant was pathogenic or likely pathogenic because it is a long deletion variant of 58 amino acids, but the variants registered in InSiGHT
(http://insight-database.org/) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) did not have an in-frame deletion variant that was determined to be pathogenic or likely pathogenic within
these 58 amino acids. For the diagnosis of Lynch syndrome, molecular tests, such as MSI testing and immunohistochemistry (IHC), are widely used, but those results are not included in the
criteria. In recent years, evaluation methods that include the results of MMR gene variants of a tumor and the results of IHC have been advocated [21, 22]. Our results indicate the
difficulty in finding SVA insertions (and possibly any other insertions of DNA sequence larger than a few kilobases) by standard methods of genetic testing. We had problems detecting SVA
insertion in both Case 1 and Case 2. In Case 1, our standard DNA analysis failed to detect the abnormality. Only a special tool, such as Scramble (https://github.com/GeneDx/scramble), could
detect this change (Table S1). However, we were able to notice the abnormality by analyzing RNA, which prompted us to re-examine the result of DNA sequencing. The DNA change was barely
detectable by visual inspection of the mapping results. This supports the idea that RNA analysis is often useful to discover variants that are difficult to detect and evaluate using DNA [23,
24]. However, RNA analysis does not always work well. In Case 2, quite a few reads (6%) supportive of aberrant splicing were detected in the RNA derived from PBMCs with puromycin treatment
(no reads in untreated cells). The ratio of aberrant-splicing reads was also similar in a relative (Fig. 1b, III-2), but was not recognized as an aberrant change by our standard
computational analysis. Because the loss of MSH2 protein was observed in IHC, we examined the whole region of this gene by manually viewing the mapping and eventually discovered SVA
insertion. It is unclear why the ratio of variant transcripts was low, but here are a various possible causes, including technical problems [25, 26]. In view of the difficulty of detection,
it is possible that there are a significant number of hidden Lynch syndrome patients who harbor an insertion of SVA (or any fragment extending a few kilobases) in any of the genes causative
of this condition. In support of this idea, variants involving the insertion of a mobile element such as Alu and LINE-1 sequences have been reported in genes causative of the hereditary
cancer-predisposing syndrome including NF1 and BRCA [27, 28]. Another problem that we encountered is that the insertion sequence of Case 2 did not exist in hg19 or hg38, and existed only in
JRGv2, the Japanese reference genome. Active retrotransposons are known to generate polymorphic insertions by themselves [29, 30]. In addition, it has been reported that an Alu sequence
moves to another locus once per 20 births [31]. Our data and these studies suggested that the consideration of ethnic and individual differences is important for identifying the origin of
inserted mobile elements. In this study, we detected insertions of SVA in exons of the _MSH2_ and _MSH6_ genes. To date, we have identified 137 pathogenic or likely pathogenic variants and
65 variants of uncertain significance (VUS) from 580 probands suspected of Lynch syndrome based on family history and molecular testing. In light of the difficulty in detecting them, the
insertion of mobile elements including SVA may not be a rare cause of Lynch syndrome. In addition, our results indicate that RNA analysis helps to increase the possibility of detection,
although it is not sufficient to detect all kinds of structural variants. To achieve precise diagnoses of genetic disorders and provide appropriate surveillance/treatment to patients,
technological advances to detect currently undetectable (or hardly detectable) variants are awaited. REFERENCES * Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, et al.
Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–25. Article CAS Google Scholar * Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe
MK, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–61. Article CAS Google Scholar *
Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80. Article CAS
Google Scholar * Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–9. Article CAS
Google Scholar * Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat
Genet. 1997;17:271–2. Article CAS Google Scholar * Espenschied CR, LaDuca H, Li S, McFarland R, Gau CL, Hampel H, et al. Multigene panel testing provides a new perspective on Lynch
syndrome. J Clin Oncol. 2017;35:2568–75. Article CAS Google Scholar * Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet. 1999;36:801–18.
CAS PubMed PubMed Central Google Scholar * Cunningham JM, Kim CY, Christensen ER, Tester DJ, Parc Y, Burgart LJ, et al. The frequency of hereditary defective mismatch repair in a
prospective series of unselected colorectal carcinomas. Am J Hum Genet. 2001;69:780–90. Article CAS Google Scholar * Chen S, Wang W, Lee S, Nafa K, Lee J, Romans K, et al. Prediction of
germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296:1479–87. Article CAS Google Scholar * Dominguez-Valentin M, Sampson JR, Seppälä TT, Ten Broeke SW, Plazzer JP,
Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med.
2020;22:15–25. Article CAS Google Scholar * Kazazian HH Jr, Moran JV. Mobile DNA in Health and Disease. N. Engl J Med. 2017;377:361–70. Article CAS Google Scholar * Cordaux R, Batzer
MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. Article CAS Google Scholar * Hancks DC, Kazazian HH Jr. SVA retrotransposons: evolution and
genetic instability. Semin Cancer Biol. 2010;20:234–45. Article CAS Google Scholar * Taniguchi-Ikeda M, Kobayashi K, Kanagawa M, Yu CC, Mori K, Oda T, et al. Pathogenic exon-trapping by
SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature. 2011;478:127–31. Article CAS Google Scholar * Kloor M, Sutter C, Wentzensen N, Cremer FW, Buckowitz A, Keller M, et
al. A large MSH2 Alu insertion mutation causes HNPCC in a German kindred. Hum Genet. 2004;115:432–8. Article CAS Google Scholar * Baert-Desurmont S, Coutant S, Charbonnier F, Macquere P,
Lecoquierre F, Schwartz M, et al. Optimization of the diagnosis of inherited colorectal cancer using NGS and capture of exonic and intronic sequences of panel genes. Eur J Hum Genet.
2018;26:1597–602. Article CAS Google Scholar * van der Klift HM, Tops CM, Hes FJ, Devilee P, Wijnen JT. Insertion of an SVA element, a nonautonomous retrotransposon, in PMS2 intron 7 as a
novel cause of Lynch syndrome. Hum Mutat. 2012;33:1051–5. Article Google Scholar * Nomura S, Sugano K, Kashiwabara H, Taniguchi T, Fukayama N, Fujita S, et al. Enhanced detection of
deleterious and other germline mutations of hMSH2 and hMLH1 in Japanese hereditary nonpolyposis colorectal cancer kindreds. Biochem Biophys Res Commun. 2000;271:120–9. Article CAS Google
Scholar * Nagasaki M, Kuroki Y, Shibata TF, Katsuoka F, Mimori T, Kawai Y, et al. Construction of JRG (Japanese reference genome) with single-molecule real-time sequencing. Hum Genome Var.
2019;6:27. https://doi.org/10.1038/s41439-019-0057-7. Article PubMed PubMed Central Google Scholar * Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and
guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med. 2015;17:405–24. Article Google Scholar * Gray PN, Tsai P, Chen D, Wu S, Hoo J, Mu W, et al. TumorNext-Lynch-MMR: a comprehensive next generation sequencing assay for the
detection of germline and somatic mutations in genes associated with mismatch repair deficiency and Lynch syndrome. Oncotarget. 2018;9:20304–22. Article Google Scholar * Shirts BH, Konnick
EQ, Upham S, Walsh T, Ranola JMO, Jacobson AL, et al. Using somatic mutations from tumors to classify variants in mismatch repair genes. Am J Hum Genet. 2018;103:19–29. Article CAS Google
Scholar * Pagenstecher C, Wehner M, Friedl W, Rahner N, Aretz S, Friedrichs N, et al. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Genet. 2006;119:9–22.
Article CAS Google Scholar * Brandão RD, Mensaert K, López-Perolio I, Tserpelis D, Xenakis M, Lattimore V, et al. Targeted RNA-seq successfully identifies normal and pathogenic splicing
events in breast/ovarian cancer susceptibility and Lynch syndrome genes. Int J Cancer. 2019;145:401–14. Article Google Scholar * Thakur PK, Rawal HC, Obuca M, Kaushik S. Bioinformatics
Approaches for Studying Alternative Splicing. In: Ranganathan S, Gribskov M, Nakai K, Schönbach, editors. Encyclopedia of Bioinformatics and Computational Biology (volume 2). Amsterdam,
Netherland: Elsevier B.V.; 2019, p. 221–34. * Tajaddod M, Tanzer A, Licht K, Wolfinger MT, Badelt S, Huber F, et al. Transcriptome-wide effects of inverted SINEs on gene expression and their
impact on RNA polymerase II activity. Genome Biol. 2016;17:220. Article Google Scholar * Katharina W, Tom C, Annekatrin W, Ludwine M. The NF1 gene contains hotspots for L1
endonuclease-dependent de novo insertion. PLos Genet. 2011;7:e1002371. Article Google Scholar * Qian Y, Mancini-DiNardo D, Judkins T, Cox HC, Brown K, Elias M, et al. Identification of
pathogenic retrotransposon insertions in cancer predisposition genes. Cancer Genet. 2017;216–217:159–69. Article Google Scholar * Wang L, Norris ET, Jordan IK. Human retrotransposon
insertion polymorphisms are associated with health and disease via gene regulatory phenotypes. Front Microbiol. 2017;8:1418. https://doi.org/10.3389/fmicb.2017.01418. Article PubMed PubMed
Central Google Scholar * Puurand T, Kukuškina V, Pajuste FD, Remm M. AluMine: alignment-free method for the discovery of polymorphic Alu element insertions. Mob DNA. 2019;10:31. Article
Google Scholar * Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, et al. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009;19:1516–26.
Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Edanz (www.edanzediting.co.jp) for editing the English text of a draft of this manuscript. IM received generous
support from M. Kagawa, T. Ito, A. Yamamoto, and N. Kamae for providing clinical information. FUNDING This research was supported by Japan Agency for Medical Research and Development (AMED)
under grant JP 18kk0205004, JSPS KAKENHI Grant Number JP18K07339 and National Cancer Center Research and Development Found Grant Number 31-A-2. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS *
Department of Molecular Diagnosis and Cancer Prevention, Saitama Cancer Center, 780 Komuro, Ina-machi, Kitaadachi-gun, Saitama, 362-0806, Japan Gou Yamamoto, Izumi Miyabe, Keisuke Tanaka,
Miho Kakuta & Kiwamu Akagi * Department of Clinical Genetics, Juntendo University Graduate School of Medicine, Tokyo, Japan Motoko Watanabe * Department of Urology, Saitama Medical
Center, Saitama Medical University, Kawagoe, Saitama, Japan Satoru Kawakami * Department of Digestive Tract and General Surgery, Saitama Medical Center, Saitama Medical University, Kawagoe,
Saitama, Japan Hideyuki Ishida Authors * Gou Yamamoto View author publications You can also search for this author inPubMed Google Scholar * Izumi Miyabe View author publications You can
also search for this author inPubMed Google Scholar * Keisuke Tanaka View author publications You can also search for this author inPubMed Google Scholar * Miho Kakuta View author
publications You can also search for this author inPubMed Google Scholar * Motoko Watanabe View author publications You can also search for this author inPubMed Google Scholar * Satoru
Kawakami View author publications You can also search for this author inPubMed Google Scholar * Hideyuki Ishida View author publications You can also search for this author inPubMed Google
Scholar * Kiwamu Akagi View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Kiwamu Akagi. ETHICS DECLARATIONS CONFLICT
OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL FIGURE 1, 2, 3, 4, 5 RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Yamamoto, G., Miyabe, I., Tanaka, K. _et al._ SVA retrotransposon insertion in exon of MMR genes results in aberrant RNA splicing and causes Lynch syndrome. _Eur J Hum Genet_ 29,
680–686 (2021). https://doi.org/10.1038/s41431-020-00779-5 Download citation * Received: 06 April 2020 * Revised: 30 October 2020 * Accepted: 17 November 2020 * Published: 08 December 2020 *
Issue Date: April 2021 * DOI: https://doi.org/10.1038/s41431-020-00779-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative






