- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Pancreatic ductal adenocarcinoma (PDAC) remains a highly fatal malignancy partially due to the acquired alterations related to aberrant protein glycosylation that pathologically
remodel molecular biological processes and protect PDAC cells from death. Ferroptosis driven by lethal lipid peroxidation provides a targetable vulnerability for PDAC. However, the crosstalk
between glycosylation and ferroptosis remains unclear. Here, we identified 4F2hc, a subunit of the glutamate-cystine antiporter system Xc–, and its asparagine (_N_)-glycosylation is
involved in PDAC ferroptosis by _N_- and _O_-linked glycoproteomics. Knockdown of _SLC3A2_ (gene name of 4F2hc) or blocking the _N_-glycosylation of 4F2hc potentiates ferroptosis
sensitization of PDAC cells by impairing the activity of system Xc– manifested by a marked decrease in intracellular glutathione. Mechanistically, we found that the glycosyltransferase
B3GNT3 catalyzes the glycosylation of 4F2hc, stabilizes the 4F2hc protein, and enhances the interaction between 4F2hc and xCT. Knockout of _B3GNT3_ or deletion of enzymatically active B3GNT3
sensitizes PDAC cells to ferroptosis. Reconstitution of 4F2hc-deficient cells with wildtype 4F2hc restores ferroptosis resistance while glycosylation-mutated 4F2hc does not. Additionally,
upon combination with a ferroptosis inducer, treatment with the classical _N_-glycosylation inhibitor tunicamycin (TM) markedly triggers the overactivation of lipid peroxidation and enhances
the sensitivity of PDAC cells to ferroptosis. Notably, we confirmed that genetic perturbation of _SLC3A2_ or combination treatment with TM significantly augments ferroptosis-induced
inhibition of orthotopic PDAC. Clinically, high expression of 4F2hc and B3GNT3 contributes to the progression and poor survival of PDAC patients. Collectively, our findings reveal a
previously unappreciated function of _N_-glycosylation of 4F2hc in ferroptosis and suggest that dual targeting the vulnerabilities of _N_-glycosylation and ferroptosis may be an innovative
therapeutic strategy for PDAC. SIMILAR CONTENT BEING VIEWED BY OTHERS _O_-GLCNACYLATION PROMOTES PANCREATIC TUMOR GROWTH BY REGULATING MALATE DEHYDROGENASE 1 Article 25 July 2022
STT3-MEDIATED ABERRANT N-GLYCOSYLATION OF CD24 INHIBITS PACLITAXEL SENSITIVITY IN TRIPLE-NEGATIVE BREAST CANCER Article 12 December 2024 O-GLCNACYLATION OF FBP1 PROMOTES PANCREATIC CANCER
PROGRESSION BY FACILITATING ITS LYS48-LINKED POLYUBIQUITINATION IN HYPOXIC ENVIRONMENTS Article Open access 22 April 2025 INTRODUCTION Ferroptosis is a novel form of regulated cell death
characterized by the iron-dependent unrestricted toxic accumulation of lipid peroxidation products and plasma membrane rupture and was termed by Dixon et al. in 2012 [1]. Research in this
field has revealed various targetable vulnerabilities in cell metabolism, redox homeostasis, and iron handling, which may provide potential intervention targets for anticancer therapy [2,
3]. Among the numerous regulatory pathways involved in ferroptosis defense, the glutamate-cystine antiporter system Xc– is widely known and consists of two subunits, namely, the heavy chain
subunit 4F2hc (also known as CD98hc, encoded by the SLC3A2 gene) and the light chain subunit xCT (encoded by the SLC7A11 gene), which mediate the synthesis of cysteine-derived antioxidants
[4]. _SLC7A11_ has been identified as the target gene of nuclear factor erythroid 2-related factor 2 (NRF2), which is responsible for maintaining redox homeostasis during oxidative stress
[5, 6]. xCT predominantly functions in the intracellular transfer of extracellular cystine, and xCT seems to be more critical than 4F2hc in ferroptosis regulation. However, we noticed that
4F2hc, a glycoprotein, is extremely important for maintaining xCT protein stability and appropriate membrane localization. In terms of structural biology, 4F2hc controls the intracellular
trafficking and membrane topology of its heterodimerization partner [7] through polar and hydrophobic interactions. For example, residues in 4F2hc form a short helix to fix xCT on the
intracellular side [8]. Thus, 4F2hc is required for the recruitment of xCT to the plasma membrane, its degradation ultimately results in xCT functional inactivation [9, 10]. In addition,
recent studies have shown that deletion of 4F2hc causes the decompensation of system Xc–, indicating that 4F2hc plays an equally important role in inhibiting ferroptosis [11,12,13]. However,
as a membrane glycoprotein, 4F2hc has not been extensively studied in the context of ferroptosis. Disease-associated stress may pathologically remodel the proteome or cause protein
connectivity dysfunction, especially molecular chaperones [14]. Glycosylation, a widespread posttranslational modification (PTM) [15], is a finely tuned enzymatic reaction process that
occurs mainly in the endoplasmic reticulum (ER) and Golgi apparatus, where glycosyltransferases and glycosidases add glycans to proteins and lipids. There are two types of glycosylation:
_N_-glycosylation and _O_-glycosylation [16]. Glycosylation of proteins stabilizes proteins, helps proteins move to the right location, and guides molecular chaperones to fold properly.
Additionally, emerging evidence indicates that aberrant protein glycosylation plays a critical role in cell death evasion, sustained proliferative signaling, and chemoresistance in various
malignancies, including pancreatic ductal adenocarcinoma (PDAC) [17, 18]. Both glycosylation and ferroptosis are physiological metabolic processes that may provide new therapeutic
opportunities for cancer treatment. However, hitherto the crosstalk between glycosylation and ferroptosis has not yet been largely revealed. PDAC accounts for more than 90% of pancreatic
cancer cases and is often life-threatening, with a 5-year relative survival rate of approximately 11%. According to its higher incidence and mortality rates, PDAC is projected to rank as the
second leading cause of cancer-related death by 2040 [19, 20]. Accumulating evidence indicates that targeting ferroptosis could exploit a vulnerability in cancer, especially PDAC, which is
closely intertwined with _KRAS_ mutant-driven activation of the antioxidant system and is rich in iron [21]. Previous preclinical studies have indicated that suppressing system Xc– in the
extrinsic pathway (such as system Xc– inhibition and cysteine depletion [22]) or directly reducing the activity or expression of the core antioxidant molecule in the intrinsic pathway (such
as inhibitors targeted for GPX4 [23], FSP1 [24] and DHODH [25]) might effectively render PDAC cells susceptible to ferroptosis. But the success of such therapeutic strategies remains limited
because of ferroptosis resistance and some other unknown underlying mechanisms [26]. Therefore, a deeper understanding of ferroptosis resistance may reveal new ferroptosis-related
mechanisms and provide more optimized treatment options for PDAC. In this study, we performed integrative _N_- and _O_-linked glycoproteomics and functional analyses to reveal a previously
unrecognized coupling between PDAC ferroptosis and _N_-glycosylation of 4F2hc, and demonstrate that disturbs the process of 4F2hc glycosylation can induce susceptibility of PDAC to
ferroptosis. MATERIALS AND METHODS PATIENT SPECIMENS AND TISSUE MICROARRAY (TMA) CONSTRUCTION A total of 291 patients diagnosed with primary PDAC between January 2015 and July 2019 were
consecutively recruited from the pathology archives of the Peking Union Medical College Hospital (PUMCH) (Beijing, China). Patients who had received neoadjuvant therapy and who died owing to
postoperative complications or lacked follow-up information were excluded from this retrospective study. Hematoxylin and eosin (HE)-stained slides from all patients were retrieved and
reviewed by two pathologists (ZL and SY) who were blinded to the patient’s clinical outcomes. In cases of disagreement, a third pathologist (JC) confirmed the histological diagnosis.
Detailed clinicopathological data and prognostic information of all patients were collected from the medical records and telephone interviews. The TMA with a single 2-mm core per case was
constructed using a Manual Tissue Microarrayer (MiniCore, Mitogen, Hertford, UK). Briefly, representative tumor areas were marked on a HE stained slide and then punched from each donor
formalin-fixed paraffin-embedded (FFPE) block for the recipient TMA blocks. CELL LINES AND REAGENTS Human cell lines HPNE, PANC-1, MIA PaCa-2, BxPC-3, and AsPC-1 were obtained from the
National Biomedical Cell Resource Center (Beijing, China) and identified by STR Profiling (D2081-2084) as well as tested negative for contamination. Cells were maintained at 37 °C in 5% CO2
incubator. HPNE were grown in 75% Dulbecco’s modified Eagle’s medium (DMEM) without glucose (Sigma, D5030-10 × 1 L; St. Louis, MO, USA) and 25% Medium M3 Base (Incell corp, M300F-500; Texas,
USA), supplemented with 1% penicillin and streptomycin (Gibco, 1514022; Massachusetts, USA), 10% fetal bovine serum (FBS) (Corning, 35-010-CV; New York, USA), 5.5 mM D-glucose
(Sigma-Aldrich, G7021-100G), 10 ng/ml human recombinant EGF (PeProtech, AF-100-15; New Jersey, USA), 1 × GlutaMaxTM Supplement (Gibco, 35050061), and 750 ng/ml puromycin(Gibco, A1113803).
The remaining cancer cells were maintained in DMEM (Corning, 10-013-CV) or RPMI 1640 (Corning, 10-040-CV) supplemented with 10% FBS. All cell lines were cultured in 6–10 cm dishes or plated
in 6–24 well plates for the indicated experiment. All chemical compounds used in cell culture experiments included RSL3 (Selleck, S8155; Houston, USA), erastin (Selleck, S7242), imidazole
ketone erastin (Selleck, S8877), Liproxstatin-1 (Selleck, S7699), ±-α-tocopherol (Selleck, S6104), UAMC-3203 (Selleck, S8792), Z-VAD-FMK (Selleck, S7023), Necrostatin-1 (Selleck, S8037),
cycloheximide (Selleck, S7418), tunicamycin, and protein glycosylation inhibitor (Abcam, ab120296). LC-MS/MS FOR GLYCOPROTEOMICS ANALYSIS 1 × 106 PANC-1 cells were plated in 75 cm2 cell
culture flasks (Corning, 430640, New York, USA) until they reached 90% cell confluence and then incubated with the indicated concentration of RSL3 or not for 12 h (_n_ = 3 per group).
Following treatment, cells were lysed using 300 μL 8 M urea with 1% protease inhibitor and centrifuged at 14,000 × g for 20 min at 4 °C, and the supernatant was collected. The protein
concentrations of the lysates were determined using the Bradford methods (Thermo Scientific, PierceTM BCA protein assay kit, 23225, Massachusetts, USA). The details are provided in the
“Supplementary Materials and Methods”. CYCLOHEXIMIDE CHASE ANALYSIS To evaluate the stability of the 4F2hc protein (including but not limited to), cycloheximide (CHX) (20 μM) was added to
indicate cells that had been treated with tunicamycin or transfected with sg_B3GNT3_ for 0, 2, 4, and 8 h. Then collection each sample was collected and subjected to western blotting, and
the relative intensities of 4F2hc protein were quantified using Image J software. ENZYMATIC DEGLYCOSYLATION ANALYSIS OF INTERESTED GLYCOPROTEIN To validate the glycosylation of 4F2hc
proteins, PNGase F (New England BioLabs, P0704S, USA) and _O_-glycosidase (New England BioLabs, P0733, USA) were used according to the manufacturer’s protocol. Briefly, 20 μg protein from
the whole-cell lysates was added to a 10 μL total reaction volume comprising 1 μL of 10 × glycoprotein denaturing buffer and 9 μL water and then subjected to denaturation by heating at 100
°C for 10 min. After cooling to room temperature, 2 μL of NP-40, 2 μL of 10 × GlycoBuffer 2, and water were combined to make up a 20 μL reaction volume. Following incubation with or without
2 μL PNGase F at 37 °C overnight, the mixture was subjected to immunoblotting analysis with the 4F2hc antibody. RNA SEQUENCING Total RNA was extracted from PANC-1 cells treated with or
without RSL3 (n = 3 per group) using the TRIzol reagent (Invitrogen, 15596018, Carlsbad, CA, USA). The purity and concentration of RNA were determined using a NanoDrop spectrophotometer
(Thermo Scientific, Massachusetts, USA), and integrity was determined using an Agilent 2100 bioanalyzer (RNA 6000 Nano kit 5067-1511). The details are provided in the “Supplementary
Materials and Methods”. FERROPTOSIS-RELATED GENES (FRGS) AND GLYCOSYLTRANSFERASE GENES FRGs were retrieved from the FerrDb database (http://www.zhounan.org/ferrdb) [27], which included 255
drivers, 208 suppressors, and 125 markers (Fig. S1A and Table S1). After removing repetitive genes, 259 FRGs were eventually obtained and annotated into corresponding protein names for
subsequent intersection with differential glycoprotein. A total of 207 known glycosyltransferase genes were collected from the RNA-Seq data for TCGA pancreatic cancer subjects [28], which
were used to identify hub gene sets by intersecting with differentially expressed genes screened out from RNA sequencing for further analysis. MULTIPLEX
IMMUNOHISTOCHEMISTRY/IMMUNOFLUORESCENCE (MIHC/IF) mIHC/IF staining was performed on TMA slides using an Opal Multiplex IHC Assay Kit (Akoya Biosciences, MA, USA), according to the
manufacturer’s protocol. The details are provided in the “Supplementary Materials and Methods”. FLOW CYTOMETRIC DETECTION OF CELL DEATH AND LIPID ROS 1 × 105 cells were plated in 6-well
plates and incubated overnight, then treated with various compounds for 12 h. After treatment, for cell death analysis, viable cells and floating cells were collected and stained with 5 μL
FITC Annexin V and 5 μL propidium iodide (BD Bioscience, Apoptosis Detection Kit I, 556547) 15 min. For lipid ROS detection, 5 μM of C11-BODIPY 581/591 (Invitrogen, D3861) was added and
incubated with cells for 30 min at 37 °C, 5% CO2 in an incubator. Cells for both cell death analysis and lipid ROS detection were harvested and resuspended in 200 μL PBS, and then strained
through a 40 μm cell strainer (Corning Falcon, 352350) for flow cytometric analysis (BD LSRFortessa). A minimum of 10,000 cells per well were analyzed in the FITC channel or in combination
with the PI channel. The FlowJo v10 software was used to analyze cell death and lipid peroxidation. ANIMAL EXPERIMENTS Female and male BALB/c nude mice (4 weeks old) were purchased from the
VitalRiver Laboratory Animal Technology Co., Ltd. (Beijing, China), and housed in the Center for Experimental Animals Research at the Institute of Basic Medical Science, Chinese Academy of
Medical Science (CAMS). For the subcutaneous transplant tumor models, 2 × 106 of _Cas9_Control and _B3GNT3_KO PANC-1 cells were suspended in 100 μL PBS and injected subcutaneously into the
right side of immuno-deficient nude mice. Tumor growth was monitored using a Vernier caliper taken once per week for 28 days. For the orthotopic transplantation tumor models, 2 ×106 of
_Cas9_Control and _B3GNT3_KO PANC-1 cells, and 2 ×106 of shControl and sh_SLC3A2_ PANC-1 cells which stably expressed firefly luciferase were resuspended in PBS separately and were carefully
injected into the surgically exposed pancreatic tail on day 7, and the pancreatic peritoneal cavity abdominal wall and skin were stitched. Tumor progression (shControl and sh_SLC3A2_
groups) was monitored by bioluminescent imaging (IVIS Lumina III). To investigate the therapeutic effect of IKE (40 mg/kg) and its combination with tunicamycin (1 mg/kg) in nude mice bearing
sh_SLC3A2_ PANC-1 cells xenografts, the mice were treated with the above drugs every other day. The mice’s survival and tumor growth were also recorded. All tumors were surgically removed
and processed for IHC on day 28. The tumor volume was calculated using the following formula: tumor volume = 1/2 × tumor length × tumor width2. STATISTICS AND REPRODUCIBILITY All statistical
analyses were performed using the SPSS version. 22 or GraphPad Prism version 9. To compare two experimental conditions, the Mann-Whitney test or Student’s t-test was performed for unpaired
samples, and the Wilcoxon rank-sum test was applied for all paired t-test. A two-way analysis of variance (ANOVA) was performed to compare the multiple experimental groups. Two-sided
Pearson’s test or Spearman rank correlation test was used to identify correlations among the variables. The log-rank test and univariate and multivariate Cox regression analysis were
performed to estimate the survival and determine the independent prognostic factors, respectively. No statistical methods were used to predetermine the sample size. Each experiment was
repeated at least three times unless otherwise indicated in the figure legend. All data are presented as the mean ± SD unless otherwise stated. Statistical significance was set at a
threshold of _P_ value less than 0.05. RESULTS _N_-/_O_-GLYCOPROTEOMICS AND RNA-SEQ LINK _N_-GLYCOSYLATED 4F2HC AND GLYCOSYLTRANSFERASE B3GNT3 TO PDAC FERROPTOSIS Aberrant glycosylation of
the proteome protects pancreatic ductal adenocarcinoma (PDAC) from cell death induction [29]. However, its role in PDAC ferroptosis has not been well studied. To this end,
_N_-/_O_-glycoproteomics was performed to reveal the glycoproteomic characteristic of PDAC cells undergoing RSL3-induced ferroptosis (Fig. 1A). We first confirmed that most of the _N_- and
_O_-linked differentially expressed glycopeptides (DGPTs), including 530 (86.74%) _N_-DGPTs and 37 (6.06%) _O_-DGPTs, were upregulated in PANC-1 cells treated with RSL3 (Fig. 1B, S1B).
Accordantly, 191 (77.64%) _N_-linked differentially expressed glycoproteins (_N_-DGPs) and 21 (8.54%) _O_-linked DGPs were upregulated (Fig. S1C). Enrichment analyses of GeneOntology (GO)
terms indicated that those upregulated DGPs were enriched in transport, signal transduction, and cell death (Fig. S1D, F). Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway analysis of the upregulated _N_-DGPs highlighted the pathway in cancer (Fig. S1E, G). Finally, seven candidate DGPs were identified including 4F2hc, GDF15, ANO6, TFR1, LAMP2, EGFR,
and CD44 after the intersection of 211 _N_ + _O_ DGPs and 387 ferroptosis-related proteins (FRPs) (Fig. 1C, D). Remarkably, 4F2hc has drawn our attention, as it is widely reported to be
critical for ferroptosis defense but the role of 4F2hc glycosylation in ferroptosis is poorly understood. To decipher the specific differential modification sites of 4F2hc affected by
RSL3-induced ferroptosis, the tryptic glycopeptides of 4F2hc were analyzed by nanoscale LC–MS/MS. The results showed that Asn365 was identified as a unique differential upregulated
_N_-glycosylation site of 4F2hc (Fold change = 6.09; _P_-value = 0.02), and the intensity of Asn365-specific glycoforms increased dynamically after ferroptosis activation (Fig. 1E). 4F2hc
have four _N_-glycosylation sites include Asn365 predicted by NetNGlyc-1.0 (Fig. S1H). We confirmed that the _N_-glycosylation of endogenous 4F2hc was completely inhibited and the molecular
weight of 4F2hc was reduced from ~100 to ~70 kDa when PANC-1 cells lysates were treated with peptide-_N_-glycosidase F (PNGase F), which is a recombinant glycosidase was used to remove
asparagine-linked oligosaccharides from polypeptides, but not with recombinant _O_-glycosidase (Fig. 1F). Similar results were further confirmed in PANC-1, AsPC-1, and MIA PaCa-2 cells and
six human PDAC tissues when treated with PNGase F (Fig. 1G, H). Together, these results indicated that 4F2hc is a highly _N_-glycosylated protein in PDAC and its _N_-glycosylation might play
a potential role in ferroptosis execution. To identify which glycosyltransferases are critical for glycosylation initiation during ferroptosis execution, 207 glycosyltransferase genes
(GTGs) were collected from the reported literature. Parallel RNA sequencing (RNA-seq) was performed in PANC-1 cells treated with RSL3 or not. We identified approximately 5571 differentially
expressed genes (DEGs) (Fig. 1I). Then, 39 hub genes were identified after the intersection of the DEGs and GTGs, including 8 upregulated genes and 31 downregulated genes (Fig. 1I, J). KEGG
enrichment analysis demonstrated that ferroptosis was ranked among the top 10 pathways based on 2603 upregulated DEGs (Fig. S1I). Further analysis of 8 upregulated genes according to their
mRNA expression from the TCGA datasets revealed that just B3GNT3, B3GNT5, and ASGR1 were highly expressed in PDAC tissues compared to normal samples except for GCNT4 (Fig. S2A–D). High gene
expression levels of B3GNT3 and B3GNT5 were associated with a poor prognosis for patients with PDAC, but ASGR1 and GCNT4 showed the opposite pattern (Fig. S2E–H). The qRT-PCR validation
experiment showed that only B3GNT3 was elevated considerably in indicated cells treated with RSL3 (Fig. 1K). Importantly, we identified B3GNT3, but not other glucosyltransferases, as a bona
fide binding partner of 4F2hc protein in _SLC3A2_OE PANC-1 cells analyzed by immunoprecipitation coupled with liquid chromatography-mass spectrometry/mass spectrometry (IP–LC–MS/MS) (Fig.
1L, Fig. S1J). Accordantly, 4F2hc was also identified in _B3GNT3_OE PANC-1 cells (Fig. S1K). Co-immunoprecipitation results demonstrated that 4F2hc interacted with B3GNT3 in PANC-1 cells
treated with or without RSL3 (Fig. S1L). We further confirmed that the protein expression of 4F2hc and B3GNT3 gradually increased in PANC-1 cells treated with RSL3 in a dose-dependent
escalation manner (Fig. 1M). In contrast, the protein expression levels of xCT, DHODH, FSP1, and GPX4 showed the opposite trend (Fig. 1M). Finally, we confirmed that the protein levels of
4F2hc, B3GNT3, xCT, and GPX4 were highly expressed in PDAC cells compared to normal human pancreatic epithelial nestin-expressing (HPNE) cells (Fig. 1N). These results imply that the
glycosyltransferase B3GNT3 could be a potential modulator in the response to ferroptosis, which might involve _N_-glycosylation of 4F2hc. CLINICAL SIGNIFICANCE OF 4F2HC AND B3GNT3 IN PDAC
PATIENTS Inspired by these results, we sought to assess the clinical significance of 4F2hc and B3GNT3 in PDAC. We first confirmed that the protein levels of 4F2hc and B3GNT3 were highly
expressed in six paired PDAC tissues compared to adjacent normal tissues (Fig. 2A, B), which is consistent with the gene expression analysis from TCGA (Fig. S2I, J). We further confirmed
that 4F2hc and B3GNT3 were highly expressed (score of 2-3) in 59.92% (145/242) and 79.30% (203/256) of the 291 PDAC patient specimens, respectively (Fig. S2K, L). High expression of B3GNT3
positively correlated with 4F2hc in patients with PDAC (Fig. 2C). 4F2hc exhibited a membranous staining pattern, whereas B3GNT3 staining was visualized as tan granules scattered around the
cytoplasm (Fig. 2D). In addition, the correlation analysis showed that high expression of 4F2hc was more frequent in PDAC with poor differentiation, and positive protein expression of B3GNT3
was more significantly associated with American Joint Committee on Cancer (AJCC) III-IV stage and tumor stage in T1-2 (Fig. 2E, F) (Table S3). Notably, high protein expression of 4F2hc and
B3GNT3 was significantly associated with poor progression-free survival (PFS) and disease-specific survival (DSS) (Fig. 2G–J), which is consistent with the univariate analysis (Table S4).
Multivariate analyses indicated that the high protein expression of 4F2hc and B3GNT3 was an independent predictor of poor PFS and DSS (Table S5). Surprisingly, the prognostic value of 4F2hc
and B3GNT3 could be markedly modified by a combination of their different levels of expression (Fig. 2K, L). Finally, multiplex immunohistochemistry characterized 4F2hc and B3GNT3 were
detected in cytokeratin (CK)-positive epithelial cells, and both interacted spatially and were extensively mapped in similar areas. Remarkably, we observed a gradual increase in the
expression of B3GNT3 with the progression of glandular intraepithelial neoplasia but slightly declined in PDAC (Fig. 2M, Fig. S2M, N). Taken together, these results imply that both 4F2hc and
B3GNT3 contribute to PDAC development and prognosis. ABROGATION OF _N_-GLYCOSYLATION OF 4F2HC SENSITIZES PDAC CELLS TO FERROPTOSIS INSULT The interaction between 4F2hc and xCT hinders
ferroptosis by increasing intracellular cystine influx and the subsequent build-up of the glutathione (GSH)/GPX4 system (Fig. 3A). By analyzing the Cancer Therapeutics Response Portal (CTRP)
database, we found that the elevated gene expression of _SLC3A2_ was positively correlated with resistance to GPX4 inhibitors such as RSL3, ML162, and ML210, especially in RLS3-treated
pancreatic cancer cell lines (Fig. 3B, Fig. S3A, B). Notably, the expression of the high-glycosylation form of 4F2hc decreased gradually in a dose- and time-dependent manner of TM in PANC-1
cells and BxPC-3 cells, and the expression of GPX4 showed a similar decreasing trend (Fig. 3C, Fig. S3C, D). Furthermore, _N_-glycosylated 4F2hc disappeared from the cell membrane surface
after treatment with PNGase F, and the expression of xCT was moderately reduced (Fig. 3D), suggesting that inhibition of _N_-glycosylation of 4F2hc could accelerate its glycoprotein
degradation. Knockdown of _SLC3A2_ (sh_SLC3A2_) activated the antioxidant system by upregulating the gene and protein expression of _NFE2L2_, _SLC7A11_, and _DHODH_ but not GPX4 (Fig. 3E, F,
Fig. S3E, F). sh_SLC3A2_ markedly sensitized PANC-1 and MIA PaCa-2 cells to RSL3-induced ferroptosis (Fig. 3G), largely induced lipid peroxidation and decreased the amount of extracellular
glutamate and intracellular GSH in RSL3-treated PANC-1 cells (Fig. 3H–J). Notably, TM treatment dramatically enhanced the sensitivity of PANC-1 and MIA PaCa-2 cells (regardless of _SLC3A2_
knockdown or not) to RSL3-induced ferroptosis (Fig. 3K, L). There are four _N_-glycosylation sites in human 4F2hc protein, namely Asn365, Asn381, Asn424, and Ans506, which are located in the
extracellular domain of 4F2hc (Fig. S3G). To investigate the role of _N_-glycosylation of 4F2hc on ferroptosis resistance, a single Asn365 site mutant (asparagine (N) residue at 365 changed
to glutamine (Q), N365Q) and four glycosylation sites mutants (4NQ) were constructed. Results confirmed that reconstitution of wildtype (WT) 4F2hc exhibited a much more pronounced
protective effect against RSL3-induced ferroptosis in sh_SLC3A2_ PANC-1 and MIA PaCa-2 cells. While N365Q and 4NQ mutants failed to restore the cell viability and showed greater sensitivity
to RSL3-induced ferroptosis (Fig. 3M–O). Together, these results suggest that blocking the _N_-glycosylation of 4F2hc could sensitize PDAC cells to ferroptosis. IMPAIRED B3GNT3 SENSITIZES
PDAC CELLS TO FERROPTOSIS BY REGULATING THE STABILITY OF _N_-GLYCOSYLATED 4F2HC Glycotransferase B3GNT3 was initially identified as an enzyme that catalyzes the glycosylation of proteins.
However, its potential biological role in ferroptosis has not been reported. We first confirmed that high expression of _B3GNT3_ was positively correlated with resistance to ferroptosis
inducers including RSL3, ML162, and ML210, particularly in pancreatic cancer cell lines treated with RSL3 (Fig. 4A, Fig. S4A). Knockdown of _B3GNT3_ markedly sensitized PANC-1 and MIA PaCa-2
cells to RSL3-induced ferroptosis (Fig. 4B, C). CRISPR-_Cas_9-mediated _B3GNT3_ knockout (_B3GNT3_KO) markedly sensitized PANC-1 and MIA PaCa-2 cells to RSL3-induced ferroptosis (Fig. 4D,
E), which could be largely rescued by ferroptosis inhibitors (such as Liproxstatin-1, UAMC, and vitamin E), but could not be reversed by apoptosis inhibitor (Z-VAD-FMK) and necroptosis
inhibitor (Necrostatin-1) (Fig. S4B). _B3GNT3_KO potently decreased the levels of extracellular glutamate and intracellular GSH (Fig. 4F, G) and triggered the accumulation of lipid
peroxidation products in RSL3-challenged PANC-1 cells (Fig. 4H). Notably, we found that overexpression of _SLC3A2_, but not _B3GNT3_, largely rescued RSL3-induced ferroptosis in sh_SLC3A2_
PDAC cells (Fig. S4C, D). We further confirmed that _B3GNT3_KO substantially decreased the expression of xCT and GPX4, but compensatorily increased the expression of 4F2hc (Fig. 4I). To
elucidate the underlying mechanism, we further investigated the effect of B3GNT3 on the protein stability of 4F2hc. The results showed that _B3GNT3_KO accelerated the protein degradation of
4F2hc albeit with compensatory upregulation in PANC-1 and MIA PaCa-2 cells (Fig. 4J). Remarkably, the expression of GPX4 exhibited a faster turnover in _B3GNT3_KO cells than in _Cas9_Control
cells (Fig. 4J), implying that there is an underlying mechanism for the quality control of GPX4 regulated by B3GNT3-mediated glycosylation. Considering that _N_-glycosylation of 4F2hc
catalyzed by B3GNT3 depends on its enzymatic activity, we constructed two plasmids for overexpression and deletion of the enzyme activity of B3GNT3, denoted as 122-311OE and 122-311del,
respectively (Fig. S4E). Results showed that 122–311OE significantly delayed the protein attenuation of 4F2hc and GPX4 compared with the 122–311del in _B3GNT3_KO PANC-1 and MIA PaCa-2 cells
(Fig. S4F, G), suggesting overexpression of enzymatically active B3GNT3 had a moderate protective effect on the protein expression of 4F2hc and GPX4 in PDAC cells. We further confirmed that
reconstitution of enzymatically active 122-311OE partially restored the cell viability in _B3GNT3_KO PANC-1 and MIA PaCa-2 cells treated with RSL3, while reconstitution with a
glycosyltransferase-null 122–311del constructs could not (Fig. 4K–M). Together, our data suggest that B3GNT3 promotes the formation of non-glycosylated (NG) 4F2hc into glycosylated (G) 4F2hc
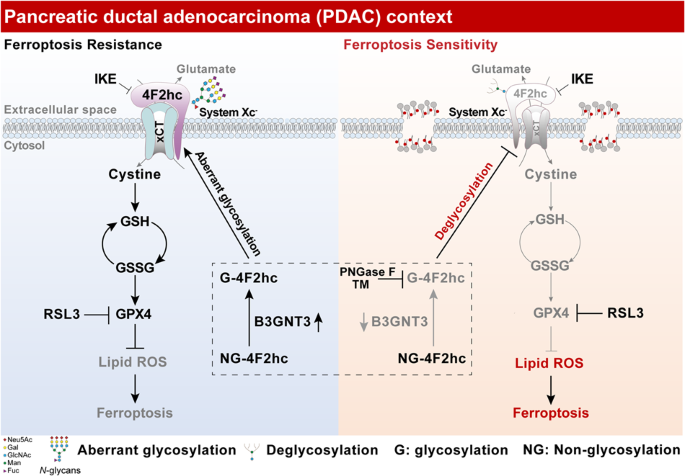
through _N_-glycosylation modification which stabilizes _N_-glycosylated 4F2hc, thereby enhancing 4F2hc-mediated ferroptosis resistance (Fig. 4N). _N_-GLYCOSYLATION STABILIZES 4F2HC IN PDAC
CELLS The crystal structure of the Erastin-bound 4F2hc-xCT complex (PDB: 7EPZ) was characterized very recently (Fig. 5A). Previous studies have shown that removing _N_-glycan from a
glycoprotein can lead to structural instability. However, whether _N_-glycosylation affects the stability of 4F2hc has not been further inspected. To do this, the _N_-glycosylation sites of
4F2hc were mutated and their changes in protein stability and flexibility were predicted by DynaMut and PredyFlexy based on MD simulations. Results showed that mutations of Asn365 and Asn424
led to instability of 4F2hc protein, manifested by reduction of the Gibbs free energy (ΔΔG) (Fig. 5B). The flexibilities (root-mean-square fluctuations (RMSFs)) were moderately elevated
when glycosylation sites N365, N381, and N424 were mutated, indicating that the mutation of _N_-glycosylation sites of 4F2hc increased its structural fluctuation which may lead to
instability of 4F2hc protein (Fig. 5C). Differential _N_-glycosylation site Asn365 in 4F2hc was conserved across multiple species (Fig. S5A). Since the _N_-glycosylation of 4F2hc mediated by
B3GNT3 is essential for its protein stability and the establishment of the xCT-GPX4-involved antioxidant system, we confirmed that re-expression of _N_-glycosylation mutant form N365Q and
4NQ significantly decreased the protein expression of xCT, GPX4, and B3GNT3 in sh_SLC3A2_ PANC-1 cells (Fig. S5B). This is probably due to the deglycosylation of 4F2hc accelerated its
protein instability and attenuation, reduced its demand for glycosyltransferase B3GNT3, downregulated the protein expression of its chaperone xCT, and ultimately decreased the expression of
downstream GPX4. We further confirmed that reconstitution of N365Q and 4NQ mutants accelerated the protein degradation rate of 4F2hc and GPX4 in sh_SLC3A2_ PANC-1 and MIA PaCa-2 cells
treated with protein synthesis inhibitor cycloheximide (CHX) for different times (Fig. 5D, E), suggesting that deglycosylated 4F2hc proteins are unstable and presumably more susceptible to
degradation. Moreover, CHX-chase assay revealed that the expression of less-glycosylated 4F2hc was faster decreased than high-glycosylated 4F2hc in PANC-1 and MIA PaCa-2 cells upon
_N_-linked glycosylation inhibitor tunicamycin (TM) treatment (Fig. 5F, G). Similarly, GPX4 exhibited a faster degradation rate in TM-treated PANC-1 and MIA PaCa-2 cells. In conclusion,
those data suggest that _N_-glycosylation of 4F2hc is responsible for 4F2hc protein stability. _N-_GLYCOSYLATION OF 4F2HC IS REQUIRED FOR ITS MEMBRANE LOCALIZATION AND THE INTERACTION WITH
XCT Aberrant _N_-glycosylation of a protein alters the interaction with its partner, leading to inappropriate membrane localization of its chaperone [14]. Given that xCT fixed at the cell
surface depends on a short helix (H1′) formed by the residues in 4F2hc [8], we wondered whether de-glycosylation of 4F2hc affects its cell membrane location and the interaction with xCT.
First, the co-expression and membrane colocalization of 4F2hc and xCT were observed in wild-type parental PANC-1 and MIA PaCa-2 cells but noticeably decreased with prolonged CHX treatment
time in TM-challenged cells (Fig. 6A). In addition, the fluorescence intensity of 4F2hc was enhanced in RSL3-challenged PANC-1 cells but its expression and membrane colocalization with xCT
were largely quenched in TM-treated PANC-1 cells with or without RSL3 co-treatment (Fig. S5C). Moreover, the upregulation of 4F2hc induced by RSL3 was mitigated by the treatment of TM in
PANC-1 cells, and the expression of NRF2, xCT, and B3GNT3 was decreased as well (Fig. S5D). The Co-IP results demonstrated that the knockdown of _SLC3A2_ indeed blocked the interaction
between 4F2hc and xCT, this physiological interaction was also disrupted in the TM-treated PANC-1 cells (Fig. S5E, F). We further confirmed that de-glycosylation of 4F2hc by re-expressing of
N365Q or 4NQ or treating with TM obviously decreased the interaction between 4F2hc and xCT in sh_SLC3A2_ PANC-1 cells (Fig. 6B, C). In addition, we confirmed that reconstitution with
enzymatically active 122-311OE plasmids retained the interaction between 4F2hc and xCT, while reconstitution with a glycosyltransferase-dead 122-311del mutant decreased this interplay in
_B3GNT3_KO PANC-1 cells (Fig. 6D, E). Notably, we found that N365Q or 4NQ or TM treatment obviously alleviated the colocalization of 4F2hc and xCT in sh_SLC3A2_ MIA PaCa-2 cells, and the
membrane protein expression level of xCT decreased significantly (Fig. 6F, G). We further confirmed that reconstitution of glycosylation-resistant N365Q or 4NQ mutant did not rescue the
membrane expression of 4F2hc and the colocalization with xCT in 4F2hc-deficient PANC-1 cells (Fig. S5G, H). In summary, blocking the _N_-glycosylation of 4F2hc attenuates its membrane
expression and the interaction with xCT, thereby conferring sensitivity to ferroptosis in PDAC cells. TM EXPOSURE INCREASED THE SENSITIVITY OF PDAC CELLS TO FERROPTOSIS PARTIALLY BY
SUPPRESSING _N_-GLYCOSYLATION Given the above findings, we aim to investigate the potential therapeutic values of TM in PDAC cells and evaluate whether the sensitivity of PDAC cells to
ferroptosis inducer could be largely enhanced by TM. Results showed that four human PDAC cell lines treated with TM were more sensitive to either RSL3 or imidazole ketone erastin
(IKE)-induced ferroptosis than those without TM treatment (Fig. 7A, B). Similarly, the cell viability of PANC-1 and MIA PaCa-2 cells was largely decreased under the combination treatment
with TM plus RSL3 or IKE (Fig. 7C, E). In addition, we observed relatively fewer annexin V/PI double-negative cells in indicated cells challenged with TM plus RSL3 or IKE than in RSL3- or
IKE-treated cells individually (Fig. 7D, F, G). Furthermore, the levels of lipid peroxidation products were remarkably increased in PANC-1 and MIA PaCa-2 cells treated with TM plus RSL3 or
IKE compared to those in RSL3- or IKE-treated counterparts (Fig. 7H, I). Gemcitabine (GEM) is a first-line chemotherapy regimen for pancreatic cancer but its clinical effect is limited owing
to chemoresistance [30, 31]. To investigate whether the sensitivity of PDAC to GEM can be largely improved by combining with ferroptosis inducer RSL3 or glycosylation inhibitor TM, we
confirmed that different concentrations of GEM plus RSL3 or TM significantly reduced the viability of PANC-1 cells compared to that in the monotherapy group, and GEM plus RSL3 plus TM was
lethal to PANC-1 cells at a lower concentration than either drug alone (Fig. S6A, B). Additionally, sh_SLC3A2_ or treated with TM greatly reduced the cell viability of GEM-challenged PANC-1
cells (Fig. S6C, D). Together, our results suggest that targeting the _N_-glycosylation pathway holds great potential for the combination therapy of PDAC. KNOCKOUT OF _B3GNT3_ LIMITED THE
PROLIFERATIVE CAPACITY OF PDAC CELLS IN VITRO AND IN VIVO To investigate whether _B3GNT3_ affects the cell biological behavior of PDAC cells, scratch assays were performed and results showed
that _B3GNT3_KO markedly attenuated the wound closure in PANC-1 and MIA PaCa-2 cells compared to that in wild-type cells (Fig. S7A). Transwell assay showed a noticeable reduction in the
number of migration and invasion of _B3GNT3_KO PANC-1 and MIA PaCa-2 cells (Fig. S7B). Similarly, _B3GNT3_KO suppressed the colony-forming abilities of PANC-1 and MIA PaCa-2 cells (Fig.
S7C). To further investigate whether B3GNT3 promotes the progression of PDAC depending on its glycosyltransferase activity, _B3GNT3_ knockout cells reconstituted with a
glycosyltransferase-dead 122-311del mutant were used in this concern. Results showed that there has no significant difference in the clone formation, wound closure, migration, and invasion
in _B3GNT3_KO PANC-1 and MIA PaCa-2 cells reconstituted with or without glycosyltransferase-null 122-311del mutant, suggesting that glycosyltransferase activity of B3GNT3 plays an important
role for PDAC cells proliferation (Fig. S8A–C). We next investigated the potential anticancer activity of _B3GNT3_KO in subcutaneous transplant tumors (STTs) and orthotopic transplantation
tumors (OTTs) implanted with PANC-1 cells (Fig. 8A). The results showed that _B3GNT3_KO markedly slowed the growth of STTs and OTTs compared to normal counterpart as indicated by the tumor
volume and tumor growth curves (Fig. 8B–F). These results suggested that _B3GNT3_ may be a crucial gene mainly involved in PDAC progression. _SLC3A2_ KNOCKDOWN RETARDS PDAC CELL GROWTH AND
POTENTIATES IKE OR TM-MEDIATED PDAC SUPPRESSION 4F2hc, as an important chaperone in amino acid transporter, few studies have reported its role in PDAC. We first confirmed that _SLC3A2_
knockdown significantly suppressed wound closure in PANC-1 and MIA PaCa-2 cells (Fig. S7D). Likewise, the transwell assay showed that the migration and invasion of sh_SLC3A2_ PANC-1 and MIA
PaCa-2 cells were remarkably inhibited (Fig. S7E). Relative colony formation ability was hampered in sh_SLC3A2_ PANC-1 and MIA PaCa-2 cells (Fig. S7F). Additionally, the OTTs model confirmed
that knockdown of _SLC3A2_ slowed the tumor growth (Fig. 8G, H), reduced the tumor weight and tumor volume (Fig. 8I, J), and had no effect on mouse weight (Fig. S8D). This is also supported
by the bioluminescent signals, HE staining (Fig. 8K), and the percentage of Ki-67 positive cells (Fig. 8L–M). Notably, this tumor suppression effect could be further dramatically
potentiated after treatment with IKE or TM (Fig. 8N–R). The combination of IKE and TM largely inhibited the tumor growth in sh_SLC3A2_ OTTs but caused undesirable toxicity manifested as
significant mice weight loss (Fig. 8O–R, S8E). Additionally, Ki-67 staining showed the same trend as that mentioned above (Fig. 8S, T). Taken together, these results indicated that knockdown
of _SLC3A2_ contributes to PDAC tumor suppression and this antitumor activity can be largely enhanced by combination with IKE or TM. DISCUSSION Aberrant glycosylation involves multifaceted
pathological and pathophysiological changes in PDAC, including but not limited to conferring tumor cells the ability to resist cell death. Ferroptosis and glycosylation are both
physiological metabolic processes and provide potential opportunities to target such vulnerabilities for PDAC treatment [32]. However, their crosstalk remains unclear. In this study, we
provided functional experimental and clinical evidence supporting an interesting and previously unexplored mechanism of _N_-glycosylation of 4F2hc in PDAC ferroptosis. In the context of
PDAC, (i) aberrant _N_-glycosylation of 4F2hc is catalyzed by glycosyltransferases B3GNT3, which stabilizes the _N_-glycosylated 4F2hc localization on the cell surface and recruits xCT
trafficking to the plasma membrane. Then, 4F2hc combined with xCT to support the proper assembly of system Xc–, which promotes the uptake of cystine and subsequent activation of the GSH-GPX4
axis, eventually enhancing PDAC to ferroptosis resistance. (ii) In contrast, deglycosylation of 4F2hc through PNGase F, protein _N_-glycosylation inhibitor TM, or _B3GNT3_KO accelerates
4F2hc degradation and impairs its membrane localization, limiting the membrane trafficking of xCT and potentially disrupts system Xc– assembly, ultimately promoting PDAC sensitivity to
ferroptosis (Fig. S9). Emerging evidence has suggested that metabolic reprogramming can confer ferroptosis resistance in malignancies [26, 33, 34]. The GPX4-GSH system functions primarily in
antioxidant defense through system Xc– mediated uptake of cystine. However, most studies have only focused on xCT as a key transporter for cysteine, and have ignored the important
prerequisite that the localization of xCT on the cell membrane requires the recruitment of 4F2hc [4, 35]. A recent study pointed out that 4F2hc plays an equally essential role as same as xCT
for preventing YTHDC2-induced ferroptosis [36]. Notably, high expression of 4F2hc was positively correlated with ferroptosis resistance in the CTRP analysis. 4F2hc has four glycosylation
sites [37, 38], we identified Asn365 as the most significant differential _N_-glycosylation site in RSL3-treated PANC-1 cells. This finding suggests that the ferroptosis stress-induced
upregulation of the Asn365 glycosylation site of 4F2hc might be responsible for 4F2hc stabilization. However, few studies have reported the role of _N_-glycosylation of 4F2hc in PDAC
ferroptosis. Thus, 4F2hc was the focus of our study. Protein glycosylation may alter the protein-protein interactions or affect the correct assembly of protein complexes [39]. Highlighted by
the migration of molecular weight and the attenuation of protein stability of 4F2hc in PDAC cell lines treated with PNGase F and TM, we found the abrogation of _N_-glycosylation of 4F2hc or
knockdown of _SLC3A2_ inhibited the activity of system Xc– and largely sensitized PDAC cells to ferroptosis insults. Notably, we observed that the membrane colocalization of 4F2hc and xCT
disappeared in TM-treated PANC-1 cells. Treatment with TM or knockdown of _SLC3A2_ both significantly inhibited the interaction between 4F2hc and xCT. This may symbolize the dysfunction of
system Xc– and is consistent with the notion that 4F2hc acts as a chaperone protein to support the function of xCT [7, 36]. Thus, we propose that _N_-glycosylated 4F2hc is critical for the
full functionality of system Xc– and that targeting this pathway may increase the susceptibility of PDAC to ferroptosis. _N_-glycosylation contributes to tumor heterogeneity and allows tumor
cells to resist cell death induced by chemotherapy [40]. In our study, we found that 77.64% of _N_-linked glycoproteins were upregulated as compared to their _O_-linked counterparts in
RSL3-induced PANC-1 cells. This indicates that _N_-glycosylation modifications are functionally significant for ferroptosis and require further in-depth investigation. Interestingly, GPX4, a
vital intracellular detoxifying enzyme, its expression was significantly decreased in the TM-mediated CHX-chase assay, suggesting that the membrane glycoprotein GPX4 undergoes
glycosylation, which is consistent with a previous study [41]. Further investigation of the glycosylation role of GPX4 in ferroptosis may be meaningful and warranted. The modification,
processing, and biosynthesis of glycoproteins depends on the participation of glycosyltransferases [42]. B3GNT3 was first identified as a type II transmembrane protein involved in the
glycosylation process [43]. High expression of B3GNT3 facilitates tumorigenesis and progression and is commonly associated with unfavorable survival in PDAC [44] and lung adenocarcinoma
[45], but not in neuroblastoma [46]. Similarly, our study revealed that _B3GNT3_KO suppressed PDAC cell proliferation, migration, invasion, and tumor suppression. Notably, we are the first
to show that B3GNT3 plays a role in ferroptosis resistance partially by glycosylating 4F2hc. TM was initially identified as a canonical inhibitor of _N_-linked glycosylation that disrupts
UDP-_N_-acetylglucosamine (GlcNAc) transfer to dolichol phosphate in the ER thus affecting the modification, location, and function of glycoproteins and triggering ER stress [47, 48]. We
found that TM treatment markedly increased the sensitivity of PDAC cells to RSL3 and IKE in vitro and in vivo. Although the role of ER stress induced by TM cannot be neglected, it is worth
noting that most of the roles played by TM are initially dependent on the targeting of GlcNAc which catalyzes the committed step of _N_-glycosylation in the ER [48]. In other words, TM
sensitized PDAC to ferroptosis at least in part by blocking _N_-glycosylation. Importantly, our findings established a potential link between ferroptosis and glycosylation by canonical
_N_-glycosylation inhibitor TM. Furthermore, as a promising anticancer compound, TM could markedly enhance the sensitivity of cancer cells to radiation therapy and chemotherapy [49,50,51].
In our study, we found that either IKE or TM alone, or a combination of both, could significantly inhibit tumor growth in sh_SLC3A2_ OTTs albeit with cytotoxicity caused by off-target
inhibition of the _N_-glycosylation process mediated by TM [52]. Accordingly, further exploration of more specific _N_-glycosylation inhibitors or structural modifications to reduce the
potential toxicity of TM may have extensive implications to attenuate ferroptosis resistance and increase the effectiveness of anti-PDAC therapeutics. In summary, the data from our study
demonstrate that the modulation of _N_-glycosylation of 4F2hc through the use of the _N_-glycosylation inhibitor TM or knockout of glycosyltransferase B3GNT3 can effectively increase the
sensitivity of PDAC cells to ferroptosis. This finding suggests that targeting the B3GNT3-4F2hc-_N_-glycosylation pathway, specifically in PDAC, holds promise as a potential strategy to
enhance the susceptibility of PDAC cells to ferroptosis. DATA AVAILABILITY The RNA sequencing raw data have been deposited in the Gene Expression Omnibus (GEO) database with the accession
number: GSE207741. Uncropped original western blots and relevant data are provided in the Supplementary files. The data generated in this study are available upon reasonable request from the
corresponding author. REFERENCES * Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell.
2012;149:1060–72. Article CAS PubMed PubMed Central Google Scholar * Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol.
2021;22:266–82. Article PubMed PubMed Central Google Scholar * Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381–96. Article CAS
PubMed PubMed Central Google Scholar * Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell.
2021;12:599–620. Article CAS PubMed Google Scholar * Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96.
Article CAS PubMed Google Scholar * Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107.
Article CAS PubMed PubMed Central Google Scholar * Nakamura E, Sato M, Yang H, Miyagawa F, Harasaki M, Tomita K, et al. 4F2 (CD98) heavy chain is associated covalently with an amino
acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J Biol Chem. 1999;274:3009–16. Article CAS PubMed Google Scholar * Yan R, Xie E, Li Y,
Li J, Zhang Y, Chi X, et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. 2022;32:687–90. Article CAS
PubMed PubMed Central Google Scholar * Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch.
2004;447:532–42. Article CAS PubMed Google Scholar * Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and
nutrient dependency of cancer. Cancer Commun (Lond). 2018;38:12. Article PubMed Google Scholar * Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate
tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4. Article CAS PubMed PubMed Central Google Scholar * Bi J, Yang S, Li L, Dai Q, Borcherding N, Wagner BA, et al.
Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis. 2019;10:682. Article PubMed PubMed Central Google Scholar * de la Ballina LR, Cano-Crespo S,
Gonzalez-Munoz E, Bial S, Estrach S, Cailleteau L, et al. Amino Acid Transport Associated to Cluster of Differentiation 98 Heavy Chain (CD98hc) Is at the Cross-road of Oxidative Stress and
Amino Acid Availability. J Biol Chem. 2016;291:9700–11. Article PubMed PubMed Central Google Scholar * Yan P, Patel HJ, Sharma S, Corben A, Wang T, Panchal P, et al. Molecular stressors
engender protein connectivity dysfunction through aberrant N-glycosylation of a chaperone. Cell Rep. 2020;31:107840. Article CAS PubMed PubMed Central Google Scholar * Very N, Lefebvre
T, El Yazidi-Belkoura I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget. 2018;9:1380–402. Article PubMed Google Scholar * Thomas D, Rathinavel AK,
Radhakrishnan P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim Biophys Acta Rev Cancer. 2021;1875:188464. Article CAS PubMed Google Scholar
* Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–55. Article CAS PubMed Google Scholar * Xu Y, Wang Y, Hoti N, Clark DJ,
Chen SY, and Zhang H. The next "sweet" spot for pancreatic ductal adenocarcinoma: Glycoprotein for early detection. Mass Spectrom Rev. 2021:e21748. * DeSantis CE, Ma J, Gaudet MM,
Newman LA, Miller KD, Goding Sauer A, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69:438–51. Article PubMed Google Scholar * Park W, Chawla A, O’Reilly EM. Pancreatic
cancer: A review. JAMA. 2021;326:851–62. Article CAS PubMed PubMed Central Google Scholar * Chen X, Zeh HJ, Kang R, Kroemer G, Tang D. Cell death in pancreatic cancer: from pathogenesis
to therapy. Nat Rev Gastroenterol Hepatol. 2021;18:804–23. Article PubMed Google Scholar * Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion
induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–9. Article CAS PubMed PubMed Central Google Scholar * Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R,
Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31. Article CAS PubMed PubMed Central Google Scholar * Doll S, Freitas FP, Shah R,
Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8. Article CAS PubMed Google Scholar * Mao C, Liu X, Zhang Y,
Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90. Article CAS PubMed PubMed Central Google Scholar * Wang
X, Chen Y, Wang X, Tian H, Wang Y, Jin J, et al. Stem cell factor SOX2 confers ferroptosis resistance in lung cancer via upregulation of SLC7A11. Cancer Res. 2021;81:5217–29. Article CAS
PubMed PubMed Central Google Scholar * Zhou N, Bao J. FerrDb: a manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database
(Oxford). 2020;2020. * Gupta R, Leon F, Thompson CM, Nimmakayala R, Karmakar S, Nallasamy P, et al. Global analysis of human glycosyltransferases reveals novel targets for pancreatic cancer
pathogenesis. Br J Cancer. 2020;122:1661–72. Article CAS PubMed PubMed Central Google Scholar * Lumibao JC, Tremblay JR, Hsu J, Engle DD. Altered glycosylation in pancreatic cancer and
beyond. J Exp Med. 2022;219. * Liang C, Shi S, Meng Q, Liang D, Ji S, Zhang B, et al. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: where we
are and where we are going. Exp Mol Med. 2017;49:e406. Article PubMed PubMed Central Google Scholar * Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX
or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med. 2018;379:2395–406. Article CAS PubMed Google Scholar * Wolpaw AJ, Dang CV. Exploiting metabolic vulnerabilities of
cancer with precision and accuracy. Trends Cell Biol. 2018;28:201–12. Article CAS PubMed Google Scholar * Brown CW, Chhoy P, Mukhopadhyay D, Karner ER, Mercurio AM. Targeting prominin2
transcription to overcome ferroptosis resistance in cancer. EMBO Mol Med. 2021;13:e13792. Article CAS PubMed PubMed Central Google Scholar * Liu W, Chakraborty B, Safi R, Kazmin D,
Chang CY, McDonnell DP. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat Commun. 2021;12:5103. Article CAS
PubMed PubMed Central Google Scholar * Fotiadis D, Kanai Y, Palacin M. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med. 2013;34:139–58. Article CAS PubMed Google
Scholar * Ma L, Zhang X, Yu K, Xu X, Chen T, Shi Y, et al. Targeting SLC3A2 subunit of system XC(-) is essential for m(6)A reader YTHDC2 to be an endogenous ferroptosis inducer in lung
adenocarcinoma. Free Radic Biol Med. 2021;168:25–43. Article CAS PubMed Google Scholar * Yan R, Zhao X, Lei J, Zhou Q. Structure of the human LAT1-4F2hc heteromeric amino acid
transporter complex. Nature. 2019;568:127–30. Article CAS PubMed Google Scholar * Lee Y, Wiriyasermkul P, Jin C, Quan L, Ohgaki R, Okuda S, et al. Cryo-EM structure of the human L-type
amino acid transporter 1 in complex with glycoprotein CD98hc. Nat Struct Mol Biol. 2019;26:510–7. Article CAS PubMed Google Scholar * Eichler J. Protein glycosylation. Curr Biol.
2019;29:R229–R231. Article CAS PubMed Google Scholar * Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15:346–66. Article PubMed
PubMed Central Google Scholar * Wang G, Wu Y, Zhou T, Guo Y, Zheng B, Wang J, et al. Mapping of the N-linked glycoproteome of human spermatozoa. J Proteome Res. 2013;12:5750–9. Article
CAS PubMed Google Scholar * Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. 2015;10:473–510. Article CAS PubMed PubMed Central Google Scholar *
Hennet T, Dinter A, Kuhnert P, Mattu TS, Rudd PM, Berger EG. Genomic cloning and expression of three murine UDP-galactose: beta-N-acetylglucosamine beta1,3-galactosyltransferase genes. J
Biol Chem. 1998;273:58–65. Article CAS PubMed Google Scholar * Zhuang H, Zhou Z, Zhang Z, Chen X, Ma Z, Huang S, et al. B3GNT3 overexpression promotes tumor progression and inhibits
infiltration of CD8(+) T cells in pancreatic cancer. Aging (Albany NY). 2020;13:2310–29. Article PubMed Google Scholar * Leng X, Wei S, Mei J, Deng S, Yang Z, Liu Z, et al. Identifying
the prognostic significance of B3GNT3 with PD-L1 expression in lung adenocarcinoma. Transl Lung Cancer Res. 2021;10:965–80. Article CAS PubMed PubMed Central Google Scholar * Ho WL, Che
MI, Chou CH, Chang HH, Jeng YM, Hsu WM, et al. B3GNT3 expression suppresses cell migration and invasion and predicts favorable outcomes in neuroblastoma. Cancer Sci. 2013;104:1600–8.
Article CAS PubMed PubMed Central Google Scholar * Surani MA. Glycoprotein synthesis and inhibition of glycosylation by tunicamycin in preimplantation mouse embryos: compaction and
trophoblast adhesion. Cell. 1979;18:217–27. Article CAS PubMed Google Scholar * Yoo J, Mashalidis EH, Kuk ACY, Yamamoto K, Kaeser B, Ichikawa S, et al. GlcNAc-1-P-transferase-tunicamycin
complex structure reveals basis for inhibition of N-glycosylation. Nat Struct Mol Biol. 2018;25:217–24. Article CAS PubMed PubMed Central Google Scholar * Hou H, Sun H, Lu P, Ge C,
Zhang L, Li H, et al. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse Xenograft models of human hepatocellular carcinoma. Mol Cancer Ther.
2013;12:2874–84. Article CAS PubMed Google Scholar * Wojtowicz K, Januchowski R, Nowicki M, Zabel M. Inhibition of protein glycosylation reverses the MDR phenotype of cancer cell lines.
Biomed Pharmacother. 2015;74:49–56. Article CAS PubMed Google Scholar * Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts
receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008;68:3803–9. Article CAS PubMed PubMed Central Google Scholar * Duksin D, Mahoney WC. Relationship of the structure and
biological activity of the natural homologues of tunicamycin. J Biol Chem. 1982;257:3105–9. Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We gratefully
appreciated Professor Wei-Min Tong (Chinese Academy of Medical Sciences and Peking Union Medical College, China) for his support of the animal experiments and expert advice on this study. We
thank Beijing Qinglian Biotech Co., Ltd, for their technical support in the _N_- and _O_-glycoproteomics and RNA-seq analysis. We thank Mrs. Qing Zhong (Department of Medical Science
Research Center, Translational Medicine Center, Peking Union Medical College Hospital) for her assistance with flow cytometry analysis. FUNDING This work was supported by the National
Natural Science Foundation of China (Grant Nos. 81472326 and 81672648), the Chinese Academy of Medical Sciences Initiative Medicine (CAMS-2016-I2M-1-001), the CAMS Innovation Fund for
Medical Sciences (CIFMS) (2021-I2M-C&T-B-023), the National High Level Hospital Clinical Research Funding (2022-PUMCH-A-086, 2022-PUMCH-B-062), and the National Scientific Data Sharing
Platform for the Population and Health (NCMI‐YF01N‐201906). AUTHOR INFORMATION Author notes * These authors jointly supervised this work: Shuangni Yu, Jie Chen. AUTHORS AND AFFILIATIONS *
Department of Pathology, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Science, Beijing, 100730, China Heng Ma, Xianlong Chen, Shengwei
Mo, Yue Zhang, Xinxin Mao, Jingci Chen, Yilin Liu, Zhaohui Lu, Shuangni Yu & Jie Chen * Department of Pathology, Institute of Basic Medical Sciences, Peking Union Medical College and
Chinese Academy of Medical Science, Beijing, 100730, China Wei-Min Tong Authors * Heng Ma View author publications You can also search for this author inPubMed Google Scholar * Xianlong Chen
View author publications You can also search for this author inPubMed Google Scholar * Shengwei Mo View author publications You can also search for this author inPubMed Google Scholar * Yue
Zhang View author publications You can also search for this author inPubMed Google Scholar * Xinxin Mao View author publications You can also search for this author inPubMed Google Scholar
* Jingci Chen View author publications You can also search for this author inPubMed Google Scholar * Yilin Liu View author publications You can also search for this author inPubMed Google
Scholar * Wei-Min Tong View author publications You can also search for this author inPubMed Google Scholar * Zhaohui Lu View author publications You can also search for this author inPubMed
Google Scholar * Shuangni Yu View author publications You can also search for this author inPubMed Google Scholar * Jie Chen View author publications You can also search for this author
inPubMed Google Scholar CONTRIBUTIONS Jie Chen and SY supervised and administrated this study. HM conceived the project, conducted most experiments, and wrote the manuscript. XC, SM, and YZ
contributed to the clinical sample collection and data preparation. XM, Jingci Chen., and YL responded to pathology. WT provided the facilities for animal experiments and kind suggestions.
All authors have read and approved the final manuscript. CORRESPONDING AUTHORS Correspondence to Shuangni Yu or Jie Chen. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
competing interests. ETHICS APPROVAL All clinical samples and clinicopathological data used in this study were approved by the Institutional Review Board of Peking Union Medical College
Hospital (approval number: S-K1593; date: April 2, 2021). All animal experimental protocols were approved by the Institutional Animal Care Use and Welfare Committee (IACUC-A02-2021-036).
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION
SUPPLEMENTARY FIGURE 1 SUPPLEMENTARY FIGURE 2 SUPPLEMENTARY FIGURE 3 SUPPLEMENTARY FIGURE 4 SUPPLEMENTARY FIGURE 5 SUPPLEMENTARY FIGURE 6 SUPPLEMENTARY FIGURE 7 SUPPLEMENTARY FIGURE 8
SUPPLEMENTARY FIGURE 9 SUPPLEMENTARY TABLE 1 SUPPLEMENTARY TABLE 2 SUPPLEMENTARY TABLE 3 SUPPLEMENTARY TABLE 4 SUPPLEMENTARY TABLE 5 SUPPLEMENTARY FIGURE AND TABLE LEGENDS SUPPLEMENTARY
MATERIALS AND METHODS ORIGINAL FILE OF WESTERN BOLTS RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ma, H., Chen, X., Mo, S. _et al._ Targeting _N_-glycosylation of 4F2hc mediated by glycosyltransferase B3GNT3 sensitizes ferroptosis of
pancreatic ductal adenocarcinoma. _Cell Death Differ_ 30, 1988–2004 (2023). https://doi.org/10.1038/s41418-023-01188-z Download citation * Received: 08 July 2022 * Revised: 09 June 2023 *
Accepted: 26 June 2023 * Published: 21 July 2023 * Issue Date: August 2023 * DOI: https://doi.org/10.1038/s41418-023-01188-z SHARE THIS ARTICLE Anyone you share the following link with will
be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative