- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Secondary bacterial pneumonia is a significant complication of severe influenza infection and _Staphylococcus aureus_ and _Streptococcus pneumoniae_ are the primary pathogens of
interest. IL-22 promotes _S. aureus_ and _S. pneumoniae_ host defense in the lung through epithelial integrity and induction of antimicrobial peptides and is inhibited by the soluble decoy
receptor IL-22-binding protein (IL-22BP). Little is known about the effect of the IL-22/IL-22BP regulatory pathway on lung infection, and it has not been studied in the setting of
super-infection. We exposed wild-type and IL-22BP−/− mice to influenza A/PR/8/34 for 6 days prior to infection with _S. aureus_ (USA300) _S. pneumoniae_. Super-infected IL-22BP−/− mice had
decreased bacterial burden and improved survival compared to controls. IL-22BP−/− mice exhibited decreased inflammation, increased lipocalin 2 expression, and deletion of IL-22BP was
associated with preserved epithelial barrier function with evidence of improved tight junction stability. Human bronchial epithelial cells treated with IL-22Fc showed evidence of improved
tight junctions compared to untreated cells. This study revealed that IL-22BP−/− mice are protected during influenza, bacterial super-infection, suggesting that IL-22BP has a
pro-inflammatory role and impairs epithelial barrier function likely through interaction with IL-22 You have full access to this article via your institution. Download PDF SIMILAR CONTENT
BEING VIEWED BY OTHERS AIRWAY EPITHELIAL CD47 PLAYS A CRITICAL ROLE IN INDUCING INFLUENZA VIRUS-MEDIATED BACTERIAL SUPER-INFECTION Article Open access 30 April 2024 THE PDZ MOTIF PEPTIDE OF
ZO-1 ATTENUATES _PSEUDOMONAS AERUGINOSA_ LPS-INDUCED AIRWAY INFLAMMATION Article Open access 12 November 2020 INTERLEUKIN-38 AMELIORATES POLY(I:C) INDUCED LUNG INFLAMMATION: THERAPEUTIC
IMPLICATIONS IN RESPIRATORY VIRAL INFECTIONS Article Open access 07 January 2021 INTRODUCTION Lower respiratory infections are the most common infectious cause of death in children and
adults worldwide. Many types of pathogens cause respiratory infection, but viruses including influenza are especially prominent. Influenza causes an acute respiratory illness that occurs
primarily in the setting of seasonal outbreaks, but it can also occur as pandemics when new strains of virus are introduced to the population. Infection generally causes mild to moderate
self-limited disease in otherwise healthy individuals, but severe disease is associated with significant morbidity and mortality. There are 3–5 million cases of severe influenza and
250,000–500,000 influenza-related deaths worldwide each year, with 12,000–56,000 deaths in the United States alone1. Secondary bacterial pneumonia is a widely known complication of severe
influenza infection. It has been described as a significant cause of excess mortality during influenza pandemics including those in 1918 and 20092,3,4, and it has been linked to an increase
in seasonal influenza-related deaths in children5. While _Streptococcus pneumonia_ has historically been the most commonly implicated organism in these super-infections, _Staphylococcus
aureus_ has recently emerged as the leading associated pathogen. This appears to be a result of the increasing prevalence of methicillin-resistant _S. aureus_ (MRSA) in the community5,6,7.
Community-acquired strains of MRSA including the USA300 clonotype have been shown to be associated with increased mortality in young, healthy individuals with influenza8,9. Interleukin-22
(IL-22) is a cytokine in the IL-10 family that is expressed by cells of the innate and adaptive immune system including TH17 cells. It binds to a heterodimeric receptor (IL-22R) consisting
of IL-22Ra1 and IL-10R2, and signaling occurs through phosphorylation of Jak1 and Tyk2 kinases and activation of STAT1 and STAT3 transcription factors10. The IL-22 receptor is present on the
epithelial surfaces of the lungs, which is a significant source of potential infection due to continuous exposure to the external environment11,12. IL-22 promotes immunity in the lungs
through epithelial protective effects that limit bacterial spread. IL-22 functions in combination with the Type 17 cytokine, IL-17 to induce antimicrobial peptide (AMP) production and the
release of chemokines for phagocyte recruitment. In this way, it has been shown to have a role in host defense against the bacterial pathogens _Klebsiella pneumoniae_ and _Streptococcus
pneumoniae_13,14. A soluble binding protein known as IL-22BP (also IL-22Ra2) competitively inhibits the activity of IL-2215,16,17. Single-nucleotide polymorphisms in the IL-22BP gene have
been associated with multiple sclerosis (MS) and Schistosoma liver infection in humans18,19. It has also been shown to impact immunity and microbiome homeostasis within the gastrointestinal
tract20. The role of IL-22BP in host defense of the lung is less clear. A recent study found that mice treated with an IL-22BP neutralizing antibody had reduced susceptibility to infection
and lung damage in a _Pseudomonas aeruginosa_ pneumonia model21. To our knowledge, the role of IL-22BP on super-infection in the lung has not been reported. Our group has previously shown
that IL-22−/− mice have impaired bacterial clearance during _S. aureus_ pneumonia compared to wild-type mice22. During super-infection with influenza A and _S. aureus_, mice demonstrated
reduced production of Type 17 cytokines, including IL-22, in addition to reduced clearance of virus and bacteria22. Similarly, it has been established that IL-22 is protective against
secondary bacterial infection by _S. pneumoniae_ following influenza A infection23,24. However, the specific role of IL-22BP during super-infection of the lung is not known and that is the
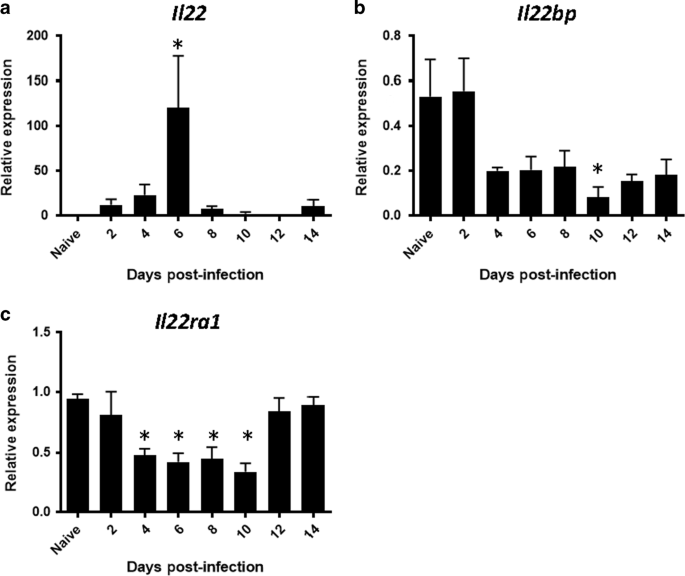
focus of our current work. RESULTS THE KINETICS OF IL22, IL22RA1, AND IL22BP GENE EXPRESSION DURING INFLUENZA INFECTION In order to determine the role of IL-22BP during influenza and
super-infection we interrogated the temporal expression of _Il22_ and its receptors during influenza infection. The super-infection model utilized is initiated on day 6 post-influenza
infection. _Il22_ expression was significantly increased by influenza infection on day 6 (Fig. 1a). Influenza infection significantly decreased expression of IL-22’s signaling (_Il22ra1_)
and decoy (_Il22bp_) receptor (Fig. 1b, c), although expression of both receptors was readily detectable at all time points. These data suggest that day 6 post-influenza is a likely time
point to observe maximal effects of IL-22 and/or IL-22BP in our model. IL-22BP−/− MICE ARE PROTECTED FROM INFLUENZA INFECTION EXACERBATED BY _S. AUREUS_ OR _S. PNEUMONIAE_ Based on the known
protective effects of IL-22, we hypothesized that IL-22BP would have a negative effect on super-infection due to inhibition of IL-22 activity. We tested this hypothesis by infecting
wild-type C57BL/6 and IL-22BP−/− mice with influenza A/PR/8/34 or vehicle for 6 days prior to infection with _S. aureus_ or vehicle for an additional 24 h. Compared to WT mice, IL-22BP−/−
mice were protected against _S. aureus_ infection as demonstrated by decreased bacterial burden in the lung (Fig. 2a). Given the decreased bacterial burden, we set out to determine whether
this phenotype provided a morbidity or survival benefit over WT mice. IL-22BP−/− mice did not display altered weight loss to single influenza infection at days 6 or 7 post-infection or
super-infection at day 7 post-infection compared to WT mice (Fig. 2b). Following a lethal challenge of influenza and _S. aureus_, IL-22BP−/− mice were however protected from
super-infection-induced mortality compared to WT mice although not completely rescued (Fig. 2c). This result indicates that the observed phenotype of the IL-22BP−/− mice is relevant to
disease course and outcomes during super-infection. IL-22 has also been shown to be protective against _S. pneumoniae_ during influenza infection23,24. We looked to see if we could identify
an IL-22BP phenotype in _S. pneumoniae_ secondary bacterial pneumonia that was similar to what was observed with influenza and _S. aureus_. We infected WT and IL-22BP−/− mice with influenza
for 6 days prior to infection with _S. pneumoniae_ for an additional 48 h. IL-22BP−/− mice had decreased bacterial burden compared to WT mice (Fig. 2d). These findings are similar to what
was seen in our model of influenza, _S. aureus_ super-infection, but the differences between the two groups were quantitatively more substantial. Most notably, after 48 h there was nearly
complete clearance of _S. pneumoniae_ in most IL-22BP−/− mice while the bacterial burden in WT mice remained high. We next examined if bacterial dissemination from the lung differed in WT
and IL-22BP−/− mice. We observed little dissemination to other organs in the influenza, _S. aureus_ model (data not shown); however a greater amount was observed in the pneumococcal model.
There were no differences in bacterial dissemination between WT and IL-22BP−/− mice (Fig. 2e). Influenza matrix protein gene expression was then measured as an indicator of viral burden in
IL-22BP−/− and WT mice. We found that there was no reduction in influenza viral burden in IL-22BP−/− mice, but rather, viral burden was increased compared to WT mice in super-infection (Fig.
2f). These findings suggest that the effect of IL-22BP is on super-infection rather than influenza infection alone. IL-22BP−/− MICE HAVE REDUCED AIRWAY INFLAMMATION DURING SUPER-INFECTION
We next evaluated whether IL-22BP−/− mice demonstrated suppression of inflammation during super-infection by either _S. aureus_ or _S. pneumoniae_. Analysis revealed that IL-22BP−/− mice
super-infected with influenza and _S. aureus_ had decreased cellular infiltration into the BAL fluid compared to WT mice (Fig. 3a). A trend towards decreased cellular infiltration into the
BAL fluid was also noted in IL-22BP−/− mice super-infected with influenza and _S. pneumoniae_ (Fig. 3b). Differential cell counts from the BAL fluid showed reduced neutrophil and lymphocyte
accumulation in the lung of IL-22BP−/− mice compared to WT mice. There was a trend towards reduced accumulation of macrophages in IL-22BP−/− mice that did not reach statistical significance
(Fig. 3c). In the influenza, _S. pneumoniae_ model, differential cell counts showed a trend towards reduced neutrophil accumulation in the IL-22BP−/− mice that were not statistically
significant (Fig. 3d). The decreased cellular infiltration in IL-22BP−/− mice was also associated with a reduction in pro-inflammatory cytokines. Quantities of MCP-1, a regulator of the
monocyte inflammatory response, were reduced in IL-22BP−/− mice; as were quantities of MIP-1α, a mediator of neutrophil activation and recruitment (Fig. 3e). Protein levels of IL-1β and
MIP-1β trended towards reduction in IL-22BP−/− mice, while the neutrophil chemoattractant KC was unchanged. These data provide evidence for reduced airway inflammation in super-infected
IL-22BP−/− mice with a specific reduction in neutrophil recruitment and accumulation. DELETION OF IL-22BP DID NOT IMPACT THE FREQUENCY OF MONOCYTES IN THE LUNG DURING SUPER-INFECTION In
order to potentially explain the improved bacterial clearance in WT IL-22BP−/− compared to WT mice, we examined if the cellular identities of the monocytes in the lung were altered.
Different classes of inflammatory monocytes have been shown to drive lung injury and epithelial apoptosis and leak25,26,27,28. We performed flow cytometry analysis to examine the frequency
of alveolar macrophages, inflammatory monocytes, interstitial macrophages, and monocyte-derived dendritic cells in our model. Somewhat surprisingly, there were no differences in these cell
populations in the lung (Fig. 4). There were few alveolar macrophages present at this time point consistent with influenza-induced depletion29. We also examined the total number of
TRAIL-positive monocytes, as TRAIL-mediated epithelial cell death has been implicated in influenza-induced lung injury25,26. No differences were seen in this cellular compartment either.
These data do not exclude potential functional differences in the monocyte compartment as a mechanism for improved bacterial clearance. IL-22BP−/− MICE HAVE LESS HISTOLOGIC EVIDENCE OF
TISSUE INFLAMMATION DURING SUPER-INFECTION Next, we examined the degree of lung injury induced by super-infection in WT and IL-22BP−/− mice. Sections of lung tissue from IL-22BP−/− and WT
mice were compared by light microscopy and scored based on the extent of cellular infiltration and tissue damage in the parenchymal, peribronchial, and perivascular regions of the lung.
Comparisons of lung injury revealed no significant difference between the IL-22BP−/− and WT mice super-infected with influenza, and _S. aureus_ (Fig. 5a). However, damage was noted to be
more intense in WT mice while IL-22BP−/− mice had fewer areas of severe damage (Fig. 5c). IL-22BP−/− mice super-infected with influenza and _S. pneumoniae_ had significantly less lung tissue
damage than WT mice (Fig. 5b). The _S. pneumoniae_ super-infection model resulted in worse lung pathology than _S. aureus_. Notably, WT mice super-infected with influenza and _S.
pneumoniae_ showed evidence of extensive tissue inflammation and hemorrhage above and beyond what was observed in super-infected IL-22BP−/− mice (Fig. 5c). Tissue necrosis was also visible
on gross examination of the harvested lungs of some WT mice (data not shown). These data indicate that IL-22BP−/− mice had ameliorated lung injury during influenza, bacterial
super-infection. IL-22BP−/− MICE HAVE DECREASED EPITHELIAL PERMEABILITY DURING SUPER-INFECTION IL-22BP−/− mice super-infected with influenza and _S. aureus_ had a trend towards decreased
quantity of total protein in bronchoalveolar lavage (BAL) fluid (Fig. 6a). This finding seemed to suggest a change in the airway epithelium towards reduced permeability. No changes were seen
between naïve WT and IL-22BP−/− mice. In order to investigate this further, we injected Evans Blue dye intravenously into the tail vein of WT and IL-22BP−/− mice 30 min prior to lung
harvest and measured the leakage of the dye into the BAL fluid spectrophotometrically. The amount of dye in the BAL fluid of IL-22BP−/− mice was decreased compared to WT mice confirming that
IL-22BP−/− mice have improved epithelial barrier function (Fig. 6b). Protein analysis of BAL fluid during influenza, _S. pneumoniae_ super-infected mice were also obtained. Like in the
influenza and _S. aureus_ model, there was a trend towards decreased total protein in the BAL fluid of IL-22BP−/− mice compared to WT that did not reach statistical significance (Fig. 6c).
Finally, measurement of LDH in BAL fluid revealed no difference in cytotoxic effects between groups (Fig. 6d). In total, these data suggest improved epithelial barrier function in IL-22BP−/−
mice in the influenza, _S. aureus_ model. IMPROVED TIGHT JUNCTION STABILITY IS A LIKELY MECHANISM BEHIND THE IMPROVED EPITHELIAL BARRIER FUNCTION IN IL-22BP−/− MICE We next began to explore
potential mechanisms for the protective barrier phenotype observed in IL-22BP−/− mice. Our group and others have previously shown that IL-22 is critical for lung epithelial repair and
integrity during influenza23,30,31,32. We therefore hypothesized that increased IL-22 activity may improve epithelial barrier function and reduced lung leakage in IL-22BP−/− mice. The tight
junction protein (Tjp) 1 and claudins are proteins involved in tight junction formation and stabilization in many different organ systems33,34. We measured _Tjp1_ and claudin gene expression
in influenza, _S. aureus_ super-infected IL-22BP−/− and WT mice, and found that gene expression of _Cldn1_, _Cldn5_, and _Cldn8_ (trend with _Cldn4_) were significantly increased in the
IL-22BP−/− mice compared to WT (Fig. 7a). No significant changes were seen in epithelial junctional protein expression during influenza infection alone (Fig. 7b). These data combined with
pulmonary leak data suggest that IL-22BP−/− mice are protected by increased epithelial barrier integrity. To examine a potential effect of IL-22 on epithelial barrier integrity, we used an
air–liquid interface epithelial model to evaluate the effects of IL-22Fc treatment on human bronchial epithelial cells (HBEs). HBEs were challenged with _S. aureus_ for 24 h in the presence
or absence of IL-22Fc. Tight junction integrity was measured by transepithelial electrical resistance (TEER). Resistance was maintained at a higher level in the IL-22Fc-treated group
compared to untreated cells providing evidence for superior barrier integrity (Fig. 7c). To examine molecular mechanisms that underlie the improved junctional integrity, we used
immunofluorescence microscopy to observe the association between occludin-1, a protein essential for tight junction stability, to the tight junction-associated protein zonula occludens-1
(ZO-1) in IL-22Fc-treated cells. We found increased localization of ZO-1 with occludin-1 in IL-22Fc-treated cells compared to the untreated cells (Fig. 7d, e). These data confirm a role for
IL-22 in the maintenance of tight junction stability and barrier function. POTENTIAL MECHANISM FOR INCREASED BACTERIAL CLEARANCE IN IL-22BP−/− MICE DURING SUPER-INFECTION First, we examined
whether _Il22bp_ is differentially expressed in super-infection versus single infection. Interestingly, _Il22bp_ expression was significantly lower during influenza or super-infection
compared to _S. aureus_ single infection (Fig. 8a). These data are consistent with the decrease in _Il22bp_ observed over time during influenza infection. A potential mechanism by which
IL-22 may exert protective function in IL-22BP−/− mice would be altered _Il22ra1_ expression. However, _Il22ra1_ expression was not different between WT and IL-22BP−/− mice during single or
super-infection (Fig. 8b). We have previously shown that _Il17_ and _Il22_ expressions are decreased in super-infection compared to _S. aureus_ single infection22. This was later associated
with defective AMP production in super-infection35. IL-10 was shown to limit Type 17 responses and increase bacterial burden in this model36. To determine if IL-22BP deletion impacts these
known pathways we examined each. WT and IL-22BP−/− mice had similar expression of _Il22_ and _Il17_ (Fig. 8c, d). The lack of change in _Il22_ expression in IL-22BP−/− mice does not exclude
the possibility of increased IL-22 in the absence of its decoy receptor. IL-10 protein levels trended to be decreased in the lungs of IL-22BP−/− compared to WT mice (Fig. 8e). We then
measured AMP expression in the lungs of WT and IL-22BP−/− mice during influenza alone or super-infection. Lipocalin 2 expression was higher in IL-22BP−/− compared with WT mice during
super-infection (Fig. 8f). There were no changes in AMP expression between WT and IL-22BP−/− mice during influenza infection alone. We and others have previously shown that lipocalin 2
promotes _S. aureus_ clearance in super-infection models35,37. These data suggest that improved bacterial clearance in IL-22BP−/− mice may be due to increased lipocalin 2 expression.
DISCUSSION Few studies have examined the role of IL-22BP in lung infection and, to our knowledge, this study is the first to identify an IL-22BP effect in the context of bacterial
super-infection of the lungs. Secondary bacterial pneumonia is a major contributor to influenza-related mortality and we have shown that IL-22BP is a critical negative regulator of secondary
infection by both _S. aureus_ and _S. pneumoniae_ following influenza. When IL-22BP is absent, mice are protected from super-infection with decreased bacterial burden, decreased
inflammation, and preservation of epithelial barrier integrity. Together these alterations in the disease characteristics provide a survival benefit to IL-22BP−/− mice during
super-infection. Interestingly, IL-22BP−/− mice did not have a bacterial clearance benefit in _S. aureus_ single infection. This is likely explained by the higher levels of IL-22 present in
this setting compared to super-infection. IL-22BP may not be a critical regulator of IL-22 when IL-22 levels are highly elevated. Alternatively, influenza may create an immune setting, where
IL-22 is more critical to _S. aureus_ clearance and thus IL-22BP’s role is enhanced in super-infection. The ability of IL-22BP deletion to ameliorate super-infection in two bacterial models
is an important consideration. _S. aureus_ does not replicate well in the mouse lung, whereas _S. pneumoniae_ does. IL-22BP−/− mice had improved phenotypes in both high dose bacterial
challenge (_S. aureus_) and low dose infection (_S. pneumoniae_). This finding strengthens the potential role of IL-22BP in disease pathogenesis. First, we examined the effect of influenza
infection on the IL-22 pathway. Influenza infection increased IL-22 production in the lung with a peak at day 6 post-infection. During the same time frame IL-22Ra1 and IL-22BP expression is
decreased below naïve levels (~50%), although expression of each remains readily detectable. This correlates with the timing of heightened susceptibility to secondary bacterial infection.
Deletion of IL-22BP−/− did not impact influenza-induced weight loss or cellular inflammation in the BAL. There was a trend towards an increase in viral burden and a trend towards decreased
BAL protein leak in IL-22BP−/− versus WT mice during single influenza infection. These data suggest that the role of IL-22BP during acute influenza single infection is less robust than
during super-infection. We have previously shown that IL-22 plays a critical role in lung repair during influenza infection with the effect observed at time points later than those studies
herein32. It is likely that IL-22BP has an impact at later time points during influenza infection resolution. We did observe increased viral burden in IL-22BP−/− mice during super-infection.
This may be the result of decreased epithelial cell death and thus higher viral replication; however, we do not know the mechanism for this result. IL-22BP is a soluble decoy receptor with
a high affinity for IL-22. Binding by IL-22BP neutralizes the effect of IL-22 by preventing it from binding to its cell surface receptor. This inhibitory role of IL-22BP has been previously
shown to be important in a variety of inflammatory disease states including those of skin and gastrointestinal tract. Although the effect of IL-22 may be positive or negative depending on
the disease state, in all instances IL-22BP has been associated with close regulation of IL-22 activity15. Investigation into the source of IL-22BP is ongoing with most studies focusing on
cellular sources within the gut. Recent studies have found evidence of IL-22BP expression in the spleen, lymph nodes, and epithelial tissues of the lungs, skin, and intestines. Specifically,
they have shown evidence of IL-22BP expression by subsets of dendritic cells38,39 as well as CD4+ T cells and eosinophils in the small intestine and lymph nodes38. In the lung IL-22BP
appears to exacerbate the highly inflammatory response to super-infection. When absent there is less inflammation visualized microscopically and reduced influx of both pro-inflammatory
cytokines and neutrophils across the epithelial barrier. The result is a stark contrast of uncontrolled inflammation and infection in WT mice and a more regulated host response in IL-22BP−/−
mice. Unlike the influenza, _S. pneumoniae_ model, we did not observe a significant decrease in lung pathology in the influenza, _S. aureus_ model. This may be due to the high inoculum of
bacteria in that model. Overall, these results are consistent with previous studies that associate uncontrolled or atypical inflammatory responses with increased, morbidity and mortality
from influenza and secondary bacterial pneumonia40,41. It is therefore no surprise that super-infected WT mice exhibit higher disease burden compared to IL-22BP−/− mice during
super-infection. The association we observed between IL-22BP and increased inflammation is in agreement with previous studies from other organ systems as well. These include a number of
studies focusing on IL-22BP in the setting of inflammatory diseases of the intestines and liver. A recent study found that IL-22BP is produced by CD4+ T cells in a mouse model of
inflammatory bowel disease (IBD) and appears to be essential for the development of the inflammation that defines the disease. This study also revealed that treatment of mice with IBD with
an anti-TNF-α agent reduced IL-22BP expression suggesting that some of the anti-inflammatory effects of the treatment may be a result of a down-regulating effect on IL-22BP42. We did not
observe a difference in TNF-α protein levels in WT and IL-22BP−/− super-infected mice (data not shown). A separate study examined the effect of IL-22BP injections into the proximal colon in
a mouse model of ulcerative colitis to evaluate the effect on disease process. Inflammation in the area of injection was notably increased in IL-22BP-injected mice compared to those that
received a mock injection43. In the liver, chronic inflammation is a primary driver for the development of fibrosis and cirrhosis, and IL-22BP has been implicated in the regulation of
fibrosis and cirrhosis in chronic hepatitis C disease based on an association with IL-22BP gene variants19. However, a study on acute liver injury by ischemic reperfusion and acetaminophen
administration showed contrasting results. It identified a protective effect of IL-22BP with evidence of increased inflammatory monocyte infiltration in IL-22BP-deficient mice44. This may
suggest an effect of IL-22BP in the liver that is in opposition to what has been described in respiratory and intestinal inflammatory diseases. This may be explained in part by the dual role
of IL-22 depending on the disease context and the overall need for tightly regulated IL-22 activity45. Similar effects of IL-22BP have been observed in inflammatory disorders of the nervous
system. IL-22BP has been identified as a risk gene for MS and has been studied in the experimental autoimmune encephalomyelitis (EAE) mouse model, where it was found to have a
pro-inflammatory role in the central nervous system. The study found that IL-22BP−/− mice had less severe disease with reduced spinal cord inflammation consistent with what we observed in
the super-infected lungs of IL-22BP−/− mice. In contrast to our study, they found no differences in neutrophil accumulation between the IL-22BP−/− and control groups18. These differences are
likely justified by the distinctions between autoimmune inflammatory diseases and infectious disease processes. Psoriasis is a disease where the pro-inflammatory role of IL-22 has been
well-described as a contributor to epithelial remodeling and inflammation. Two recent studies have identified an inflammation-modulating role for IL-22BP in psoriasis, albeit an
anti-inflammatory effect given the opposing role of IL-22 in the disease. The first examined a rat model of imiquimod-induced skin disease and identified increased inflammatory changes on
clinical and histologic examination in IL-22BP−/− rats. In addition, IL-22BP−/− rats had increased expression of pro-inflammatory cytokines compared to those with preserved IL-22BP46. The
second study examined a similar imiquimod-induced skin disease in mice as well as skin biopsies of patients with and without psoriatic skin disease. They noted reduced expression of IL-22BP
in mouse-model as well as in patients with psoriasis. They concluded that regulation of IL-22BP is a key step in the development of skin inflammation47. While the effect of IL-22BP is
reversed in skin disease, its ability to regulate inflammation through action on IL-22 is consistent with our findings. Delving into the potential mechanisms of protection from mortality in
super-infected IL-22BP−/− mice revealed a notable change in barrier function of the airway mucosal layer. A fundamental aspect of host defense in this region is the ability to provide a
barrier against pathogens from the external environment. Impairment raises the infection risk and can potentially lead to increased disease severity. Our study revealed that the absence of
IL-22BP was associated with reduced permeability of the airway epithelium and markedly improved barrier function. The results were consistent between our influenza, _S. aureus_ and
influenza, _S. pneumoniae_ models of super-infection that both demonstrated decreased passage of inflammatory cells and protein into the BAL fluid of IL-22BP−/− mice. This superior barrier
integrity may be beneficial for host defense against bacterial pathogens. First, it may limit bacterial spread from the airway lumen into the lung parenchyma therefore allowing the infection
to be better contained. We did not observe a significant amount of bacterial dissemination from the lungs to other organs in the influenza, _S. aureus_ model and dissemination was not
altered in IL-22BP−/− compared with WT mice in the pneumococcal model. These data indicate that bacterial dissemination to other organs does not likely contribute to mortality in these
models. We cannot exclude the possibility that epithelial barrier function dictates bacterial localization in the lung itself, which is likely. Further, IL-22BP−/− mice may have altered
bacterial dissemination in models where bacterial escape from the lung is more common. Second, barrier integrity may limit the degree of uncontrolled inflammation that occurs through the
influx of inflammatory mediators that would otherwise tend to exacerbate disease course. Influx of recruited monocytes to the lung was not different in WT and IL-22BP−/− mice, although the
localization of these cells was not determined. It has been shown previously that in the GI tract, at least some of IL-22BP’s effect appears to be via inhibition of the epithelial protective
effects of IL-22 on the intestinal lining42. This could explain our findings based on the hypothesis that in the lungs IL-22BP regulates IL-22 in a similar fashion. In addition to
alterations in epithelial protective effects, we have shown evidence in our influenza, _S. aureus_ super-infection model that the improvement in barrier function in IL-22BP−/− mice is
related to the maintenance of tight junctions. Specifically, the absence of IL-22BP is associated with increased gene expression of claudins 1, 4, 5, and 8, barrier forming tight junction
proteins. Claudins are the largest family of tight junction proteins and are expressed by multiple cell types throughout the airways33,34. Influenza infection has been shown to decrease
expression of claudins 1 and 4 in human epithelial cell culture models resulting in decreased tight junctions48,49. Increased claudin expression in IL-22BP−/− mice may contribute to the
reduced passage of substances across the airway epithelium during super-infection. This is supported by our observations that pro-inflammatory cytokines and neutrophil accumulation are both
reduced in the BAL fluid of IL-22BP−/− mice. Further study is necessary to determine whether the effect of IL-22BP on tight junction function is related to IL-22 activity. IL-22 is also
known to contribute to barrier function and has been shown to be involved in the maintenance and repair during influenza and bacterial infection30,31,32,50. Our results suggest that the
mechanism of barrier preservation by IL-22 may be an effect on tight junctions similar to what was observed in the super-infected IL-22BP−/− mice. HBECs treated with IL-22Fc had increased
TEER compared to untreated cells and this resistance is maintained primarily by the integrity of tight junctions. This is indicative of enhanced function of tight junctions with IL-22Fc
treatment. Immunofluorescence staining furthermore showed that IL-22Fc treatment increased the localization of ZO-1 to occludin at tight junctions, a necessary step to assure tight junction
stability. IL-22Fc has recently been shown to be an effective treatment in a mouse model of influenza, _S_. pneumoniae super-infection. Treatment with IL-22Fc was associated with increased
epithelial-adhesion protein, extracellular matrix, and epithelial cell proliferation gene expression that was suggestive of improved barrier function. Treated mice also had decreased lung
pathology and reduced dissemination of _S. pneumoniae_ to the spleen23. These findings of improved barrier function with IL-22 supplementation are consistent with what we observed in vitro.
The concept of regulation of tight junction proteins by IL-22 is consistent with previous studies evaluating non-respiratory organ systems. Within the gastrointestinal tract, IL-22 has been
shown to induce gene expression of claudin-2, a pore forming tight junction protein that, among other functions, increases intestinal permeability in IBD51. The role of Type 17 immunity in
bacterial clearance of _S. aureus_ in the context of influenza super-infection has been demonstrated52. We have previously shown that preceding influenza attenuates IL-17 and IL-22 and
subsequent AMP production in response to _S. aureus_ infection22,35. Further IL-10−/− mice displayed increased Type 17 immune activation during super-infection indicating its detrimental
role in this model36. We first examined whether IL-22BP deletion impacts IL-17 or IL-22 gene expression. We did not see significant changes in either cytokine in comparison with WT mice.
However, this finding does not exclude the possibility that IL-22 has enhanced activity in the lungs of IL-22BP−/− mice. This potential effect is supported by increased expression of
lipocalin 2 in IL-22BP−/− compared to WT mice during super-infection, but not during single influenza infection, where lipocalin 2 expression was lower than super-infected mice. Our group
and others have shown that small magnitude changes in lipocalin 2 expression is associated with improved bacterial control in influenza, super-infection35,37. In that study, we showed that
while IL-22 treatment of mouse airway epithelial cells did not induce lipocalin 2, IL-22 did further enhance IL-17 and TNFα-induced lipocalin 2 expression. It is possible that this increase
in lipocalin 2 is responsible for the improved bacterial clearance observed, likely through regulation of iron availability. In conclusion, this study has demonstrated the detrimental,
pro-inflammatory role of IL-22BP during influenza, bacterial super-infection of the lungs. This expands upon the current body of literature that has identified similar roles for IL-22BP in
other inflammatory diseases. Furthermore, we have identified a novel protective effect of IL-22BP deletion on super-infection. Improved tight junction function and lipocalin 2 production and
reduced membrane permeability may represent possible mechanisms behind this protection and warrant additional study to evaluate IL-22BP as a therapeutic target. In humans, there are three
IL-22BP isoforms present with only IL-22BPi2 likely to have IL-22-binding activity in the context of infection53. Further study is required to determine the potential efficacy of IL-22BP
antagonism in disease. METHODS MICE Six to eight-week-old male C57BL/6 mice were purchased from Taconic Farms (Germantown, NY). IL-22BP−/− mice backcrossed 9 times onto the C57BL/6
background were obtained from Genentech (San Francisco, CA)38. Mice were maintained under pathogen-free conditions and studies were performed on age and sex-matched mice. _S. AUREUS_
INFECTION MRSA (USA 300) was propagated as previously described54. Mice were inoculated with MRSA by oropharyngeal aspiration. Mice received doses of either 2.5 × 108 or 5 × 107
colony-forming units of MRSA (in 50 μL sterile phosphate-buffered saline (PBS)) during the survival study and all other studies, respectively. _S. PNEUMONIAE_ INFECTION One milliliter of a
stock of _S. pneumoniae_ serotype 3 (ATCC 6303) was grown in 5 mL of Todd-Hewitt broth for 6 h at 37 °C, 5% CO2. A 100 μL aliquot of the solution was then inoculated into 100 mL of
Todd-Hewitt broth and grown for an additional 12 h. The concentration was determined by measuring the absorbance at 600 nm with an optical density of 0.5 corresponding to a concentration of
1 × 108CFU/mL. Mice were inoculated with 104 CFU/mL (in 50 μL sterile PBS) by oropharyngeal aspiration. The stock was serially diluted and plated on trypticase soy agar with 5% sheep blood
plates to confirm the concentration given. INFLUENZA A AND SUPER-INFECTION Influenza A PR/8/34 H1N1 was propagated as previously described55. Mice were inoculated with 100 plaque-forming
units (PFU) of influenza A PR/8/34 H1N1 (in 40 μL sterile PBS) by oropharyngeal aspiration. Six days later mice were infected with bacteria or PBS vehicle as described and were harvested 24
or 48 h later. For time course studies, mice were harvested every 2 days following infection for 2 weeks. Weight loss induced by influenza or super-infection was tracked by the percentage of
loss compared to original starting weight of each mouse. EPITHELIAL PERMEABILITY Mice were injected with 20 mg/kg of Evans Blue dye (Sigma, St. Louis, MO) via the dorsal tail vein 30 min
prior to lung harvesting and BAL fluid was obtained as described below. BAL fluid was centrifuged and the absorbance of 100 μL undiluted supernatant was measured at 630 nm using a linear
standard curve. ANALYSIS OF LUNG INFECTION Mouse lungs were lavaged with 1 mL sterile PBS for inflammatory cell counts. Cytospin preparations of BAL fluid were stained using Diff-Quick
(Sigma, St. Louis, MO) and differential cell counts were conducted based on cell morphology. The superior lobe of the right lung was homogenized in sterile PBS (1 mL) by mechanical grinding.
The lung homogenate was use for bacterial colony counting cytokine analysis by Bio-plex Multiplex immunoassay (BioRad, Hercules, CA). Middle and inferior lobes of the right lung were
snap-frozen and homogenized under liquid nitrogen for RNA extraction using an RNA isolation kit (Agilent Technologies, Santa Clara, CA). RNA analysis was performed by RT-PCR using Assay on
Demand TaqMan probes and primers (Applied Biosystems, Foster City, CA). For bacterial dissemination studies, the liver, spleen, and kidneys were homogenized in PBS and plated for colony
counts. Influenza burden was determined by RT-PCR for matrix protein expression as previously described56. HISTOLOGY SCORING A cold, 10% neutral buffered formalin solution (Sigma, St. Louis,
MO) was used for fixation of the left lungs. Lungs were inflated and immersed in the formalin solution prior to storage. Slides were then prepared by Histo-Scientific Research Laboratories
(Mount Jackson, VA) and blindly reviewed and scored using a validated histology scoring system that has been previously described57. Each slide received three scores from 1 to 4 based on the
degree of cellular infiltration and tissue damage in the parenchymal, peribronchial, and perivascular regions. IN VITRO CELL CULTURE TECHNIQUE The role of IL-22 in barrier integrity was
studied in human airway epithelial cells (16HBE cells). The derivation and characterization of these cells have been described in detail58. Airway epithelial cells between passages 45 and 55
were grown and polarized in an air–liquid interface culture at 37 °C for 6–9 days, as described previously59. TRANSEPITHELIAL ELECTRICAL RESISTANCE TEER was measured on air–liquid
differentiated HBEs (16HBE) at 0 through 24 h after inoculation with _S. aureus_ (MRSA; USA 300) and treatment with IL-22Fc or vehicle, as described previously59. IMMUNOFLUORESCENCE STAINING
AND MICROSCOPY Confocal microscopy was performed, as described previously60 to visualize the localization of junctional proteins in response to IL-22 Fc treatment, polarized 16HBE cells
were treated basolaterally with IL-22 Fc for 24 h at 37 °C. Cells were washed with PBS and fixed with 4% paraformaldehyde, followed by permeabilization with 0.25% Triton X-100 in PBS and
incubation with the indicated primary antibodies for 1 h at room temperature. The cells were then washed, incubated with secondary antibodies for 30 min at room temperature, washed, and
mounted with ProLong Gold (Invitrogen). Images were captured using an FV1000 confocal laser scanning microscope (Olympus) and merged and colocalization measured using Nikon Elements
software. Antibodies or other reagents for fluorescence microscopy were as follows: rabbit anti-ZO-1 (mid-region; Invitrogen), mouse anti-occluding-1 (Invitrogen). Experiments were repeated
three times, with six fields imaged for each experiment. FLOW CYTOMETRY To obtain a single-cell suspension, lungs were mechanically dissected, and then incubated shaking at 37 °C for 30 min
in DMEM containing 10% FBS and 1 mg/mL collagenase. After collagenase treatment, tissue was forced through a 70 µM filter and treated with ACK buffer to lyse erythrocytes. The resulting
single-cell suspension was pre-treated with anti-CD16/32 for 5 min to block Fc receptor binding before incubating with fixable viability dye and fluorochrome-conjugated anti-surface marker
monoclonal antibodies for 30 min at 4 °C. The following antibodies were used: anti-CD45, anti-CD11b, anti-CD11c, anti-CD64, anti-SiglecF, anti-Ly6g, and anti-MHCII. LIVE/DEAD Fixable Aqua
Stain (Life Technologies, Carlsbad, CA) was used to determine cell viability. Samples were collected using a LSRFortessa flow cytometer (BD Biosciences, San Jose, CA) and analyzed using
FlowJo software (vX.0.7, Tree Star, Ashland, OR). Flow gating began with doublet exclusion by comparing forward light scatter area versus height, then debris exclusion by comparing forward
versus side light scatter. Dead cells were excluded based on viability dye staining. Within the CD45 positive, Ly6g negative gate inflammatory monocytes were defined as SiglecF− MHCII−
CD11b+ CD11c−; interstitial macrophages as SiglecF− MHCII− CD11b− CD11c+; monocyte-derived dendritic cells as SiglecF− MHCII+ CD11b+. Alveolar macrophages were defined as CD45+ SiglecF+
CD64+ cells. STATISTICS All of the data are presented as the mean ± SEM. Significance was tested by the unpaired _t_-test (for two means) or one-way ANOVA followed by Tukey test (for
multiple data groups). The Gehan–Brislow–Wilcoxon test was used for survival data analysis. All data were analyzed using GraphPad Prism software. STUDY APPROVAL All animal experiments were
conducted with approval from the University of Pittsburgh Institutional Animal Care and Use Committee. REFERENCES * Rolfes, M. A. et al. Effects of influenza vaccination in the United States
during the 2017–2018 influenza season. _Clin. Infect. Dis_. (2019). https://doi.org/10.1093/cid/ciz075. [Epub ahead of print]. * Centers for Disease Control and Prevention (CDC). Bacterial
coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1)—United States, May–August 2009. _MMWR Morb. Mortal. Wkly. Rep._ 58, 1071–1074 (2009). * Morens, D.
M., Taubenberger, J. K. & Fauci, A. S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. _J. Infect.
Dis._ 198, 962–970 (2008). Article Google Scholar * Shieh, W. J. et al. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. _Am. J.
Pathol._ 177, 166–175 (2010). Article Google Scholar * Finelli, L. et al. Influenza-associated pediatric mortality in the United States: increase of _Staphylococcus aureus_ coinfection.
_Pediatrics_ 122, 805–811 (2008). Article Google Scholar * Rice, T. W. et al. Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. _Crit.
Care Med_ 40, 1487–1498 (2012). Article Google Scholar * Williams, D. J. et al. Influenza coinfection and outcomes in children with complicated pneumonia. _Arch. Pediatr. Adolesc. Med._
165, 506–512 (2011). Article Google Scholar * Hageman, J. C. et al. Severe community-acquired pneumonia due to _Staphylococcus aureus_, 2003–04 influenza season. _Emerg. Infect. Dis._ 12,
894–899 (2006). Article Google Scholar * Kallen, A. J. et al. _Staphylococcus aureus_ community-acquired pneumonia during the 2006 to 2007 influenza season. _Ann. Emerg. Med._ 53, 358–365
(2009). Article Google Scholar * Lejeune, D. et al. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are
shared with and distinct from IL-10. _J. Biol. Chem._ 277, 33676–33682 (2002). Article CAS Google Scholar * Liang, S. C. et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells
and cooperatively enhance expression of antimicrobial peptides. _J. Exp. Med._ 203, 2271–2279 (2006). Article CAS Google Scholar * Wolk, K. et al. IL-22 increases the innate immunity of
tissues. _Immunity_ 21, 241–254 (2004). Article CAS Google Scholar * Aujla, S. J. et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. _Nat. Med._ 14,
275–281 (2008). Article CAS Google Scholar * Trevejo-Nunez, G., Elsegeiny, W., Conboy, P., Chen, K. & Kolls, J. K. Critical role of IL-22/IL22-RA1 signaling in Pneumococcal pneumonia.
_J. Immunol._ 197, 1877–1883 (2016). Article CAS Google Scholar * Dumoutier, L., Lejeune, D., Colau, D. & Renauld, J. C. Cloning and characterization of IL-22 binding protein, a
natural antagonist of IL-10-related T cell-derived inducible factor/IL-22. _J. Immunol._ 166, 7090–7095 (2001). Article CAS Google Scholar * Kotenko, S. V. et al. Identification, cloning,
and characterization of a novel soluble receptor that binds IL-22 and neutralizes its activity. _J. Immunol._ 166, 7096–7103 (2001). Article CAS Google Scholar * Xu, W. et al. A soluble
class II cytokine receptor, IL-22RA2, is a naturally occurring IL-22 antagonist. _Proc. Natl Acad. Sci. USA_ 98, 9511–9516 (2001). Article CAS Google Scholar * Laaksonen, H. et al. The
multiple sclerosis risk gene IL22RA2 contributes to a more severe murine autoimmune neuroinflammation. _Genes Immun._ 15, 457–465 (2014). Article CAS Google Scholar * Sertorio, M. et al.
IL-22 and IL-22 binding protein (IL-22BP) regulate fibrosis and cirrhosis in hepatitis C virus and schistosome infections. _Hepatology_ 61, 1321–1331 (2015). Article CAS Google Scholar *
Ratsimandresy, R. A., Indramohan, M., Dorfleutner, A. & Stehlik, C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. _Cell.
Mol. Immunol._ 14, 127–142 (2017). Article CAS Google Scholar * Broquet, A. et al. Interleukin-22 level is negatively correlated with neutrophil recruitment in the lungs in a _Pseudomonas
aeruginosa_ pneumonia model. _Sci. Rep._ 7, 11010 (2017). Article Google Scholar * Kudva, A. et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice.
_J. Immunol._ 186, 1666–1674 (2011). Article CAS Google Scholar * Barthelemy, A. et al. Interleukin-22 immunotherapy during severe influenza enhances lung tissue integrity and reduces
secondary bacterial systemic invasion. _Infect. Immun_. 86, pii: e00706–17 (2018). * Ivanov, S. et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and
protects against secondary bacterial infection. _J. Virol._ 87, 6911–6924 (2013). Article CAS Google Scholar * Herold, S. et al. Lung epithelial apoptosis in influenza virus pneumonia:
the role of macrophage-expressed TNF-related apoptosis-inducing ligand. _J. Exp. Med._ 205, 3065–3077 (2008). Article CAS Google Scholar * Peteranderl, C. et al. Macrophage-epithelial
paracrine crosstalk inhibits lung edema clearance during influenza infection. _J. Clin. Invest._ 126, 1566–1580 (2016). Article Google Scholar * Dawson, T. C., Beck, M. A., Kuziel, W. A.,
Henderson, F. & Maeda, N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. _Am. J. Pathol._ 156, 1951–1959 (2000). Article
CAS Google Scholar * Lin, K. L., Suzuki, Y., Nakano, H., Ramsburg, E. & Gunn, M. D. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary
immune pathology and mortality. _J. Immunol._ 180, 2562–2572 (2008). Article CAS Google Scholar * Ghoneim, H. E., Thomas, P. G. & McCullers, J. A. Depletion of alveolar macrophages
during influenza infection facilitates bacterial superinfections. _J. Immunol._ 191, 1250–1259 (2013). Article CAS Google Scholar * Kumar, P., Thakar, M. S., Ouyang, W. & Malarkannan,
S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. _Mucosal Immunol._ 6, 69–82 (2013). Article CAS Google Scholar *
Paget, C. et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. _J.
Biol. Chem._ 287, 8816–8829 (2012). Article CAS Google Scholar * Pociask, D. A. et al. IL-22 is essential for lung epithelial repair following influenza infection. _Am. J. Pathol._ 182,
1286–1296 (2013). Article CAS Google Scholar * Gunzel, D. & Yu, A. S. Claudins and the modulation of tight junction permeability. _Physiol. Rev._ 93, 525–569 (2013). Article Google
Scholar * Wittekindt, O. H. Tight junctions in pulmonary epithelia during lung inflammation. _Pflug. Arch._ 469, 135–147 (2017). Article CAS Google Scholar * Robinson, K. M. et al.
Influenza A virus exacerbates _Staphylococcus aureus_ pneumonia in mice by attenuating antimicrobial peptide production. _J. Infect. Dis._ 209, 865–875 (2014). Article CAS Google Scholar
* Robinson, K. M. et al. The role of IL-27 in susceptibility to post-influenza _Staphylococcus aureus_ pneumonia. _Respir. Res._ 16, 10 (2015). Article CAS Google Scholar * Planet, P. J.
et al. Lambda interferon restructures the nasal microbiome and increases susceptibility to _Staphylococcus aureus_ superinfection. _mBio_ 7, e01939–01915 (2016). Article CAS Google Scholar
* Huber, S., Gagliani, N., Zenewicz, L. A., Huber, F. J., Bosurgi, L. & Hu, B. et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. _Nature_
491, 259–263 (2012). Article CAS Google Scholar * Martin, J. C. et al. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and
is strongly induced by retinoic acid. _Mucosal Immunol._ 7, 101–113 (2014). Article CAS Google Scholar * Kash, J. C. et al. Genomic analysis of increased host immune and cell death
responses induced by 1918 influenza virus. _Nature_ 443, 578–581 (2006). Article CAS Google Scholar * Kobasa, D. et al. Aberrant innate immune response in lethal infection of macaques
with the 1918 influenza virus. _Nature_ 445, 319–323 (2007). Article CAS Google Scholar * Pelczar, P. et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease.
_Science_ 354, 358–362 (2016). Article CAS Google Scholar * Sugimoto, K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. _J. Clin. Invest._ 118,
534–544 (2008). CAS PubMed PubMed Central Google Scholar * Kleinschmidt, D. et al. A protective function of IL-22BP in ischemia reperfusion and acetaminophen-induced liver injury. _J.
Immunol._ 199, 4078–4090 (2017). Article CAS Google Scholar * Munoz, M. et al. Interleukin (IL)-23 mediates _Toxoplasma gondii_-induced immunopathology in the gut via
matrixmetalloproteinase-2 and IL-22 but independent of IL-17. _J. Exp. Med._ 206, 3047–3059 (2009). Article CAS Google Scholar * Martin, J. C. et al. Limited presence of IL-22 binding
protein, a natural IL-22 inhibitor, strengthens psoriatic skin inflammation. _J. Immunol._ 198, 3671–3678 (2017). Article CAS Google Scholar * Voglis, S. et al. Regulation of IL-22BP in
psoriasis. _Sci. Rep._ 8, 5085 (2018). Article Google Scholar * Short, K. R. et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. _Eur.
Respir. J._ 47, 954–966 (2016). Article CAS Google Scholar * Tian, T. et al. H3N2 influenza virus infection enhances oncostatin M expression in human nasal epithelium. _Exp. Cell Res._
371, 322–329 (2018). Article CAS Google Scholar * Ahn, D., Wickersham, M., Riquelme, S. & Prince, A. The effects of IFN-lambda on epithelial barrier function contribute to _Klebsiella
pneumoniae_ ST258 pneumonia. _Am. J. Respir. Cell Mol. Biol._ 60, 158–166 (2019). Article CAS Google Scholar * Wang, Y., Mumm, J. B., Herbst, R., Kolbeck, R. & Wang, Y. IL-22
increases permeability of intestinal epithelial tight junctions by enhancing claudin-2 expression. _J. Immunol._ 199, 3316–3325 (2017). Article CAS Google Scholar * Robinson, K. M.,
Kolls, J. K. & Alcorn, J. F. The immunology of influenza virus-associated bacterial pneumonia. _Curr. Opin. Immunol._ 34, 59–67 (2015). Article CAS Google Scholar * Lim, C., Hong, M.
& Savan, R. Human IL-22 binding protein isoforms act as a rheostat for IL-22 signaling. _Sci. Signal._ 9, ra95 (2016). Article Google Scholar * Gopal, R. et al. STAT2 signaling
regulates macrophage phenotype during influenza and bacterial super-infection. _Front. Immunol._ 9, 2151 (2018). Article Google Scholar * Lee, B. et al. STAT1 is required for suppression
of type 17 immunity during influenza and bacterial superinfection. _Immunohorizons_ 1, 81–91 (2017). Article CAS Google Scholar * Van der Velden, J. et al. Differential requirement for
c-Jun N-terminal kinase 1 in lung inflammation and host defense. _PLoS ONE_ 7, e34638 (2012). Article Google Scholar * Manni, M. L. et al. Bromodomain and extra-terminal protein inhibition
attenuates neutrophil-dominant allergic airway disease. _Sci. Rep._ 7, 43139 (2017). Article CAS Google Scholar * Cozens, A. L. et al. CFTR expression and chloride secretion in polarized
immortal human bronchial epithelial cells. _Am. J. Respir. Cell Mol. Biol._ 10, 38–47 (1994). Article CAS Google Scholar * Hendricks, M. R. et al. Respiratory syncytial virus infection
enhances _Pseudomonas aeruginosa_ biofilm growth through dysregulation of nutritional immunity. _Proc. Natl Acad. Sci. USA_ 113, 1642–1647 (2016). Article CAS Google Scholar * Bomberger,
J. M. et al. Long-distance delivery of bacterial virulence factors by _Pseudomonas aeruginosa_ outer membrane vesicles. _PLoS Pathog._ 5, e1000382 (2009). Article Google Scholar Download
references ACKNOWLEDGEMENTS This work was supported by NIH NHLBI R01HL107380 (J.F.A.), Pennsylvania Department of Health (J.F.A.), NIDDK P30DK072506 (J.F.A., J.M.B.), and T32HL129949
(R.N.A.). FUNDING This work was supported by NIH NHLBI R01HL107380 (J.F.A.), Pennsylvania Department of Health (J.F.A.), NIDDK P30DK072506 (J.F.A., J.M.B.), and T32HL129949 (R.N.A.). AUTHOR
INFORMATION AUTHORS AND AFFILIATIONS * Department of Pediatrics, UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA, USA Robert N. Abood, Kevin J. McHugh, Helen E. Rich, Marianna A.
Ortiz, Joshua M. Tobin, Krishnaveni Ramanan, Michelle L. Manni & John F. Alcorn * Department of Medicine, University of Pittsburgh Medical Center, Pittsburgh, PA, USA Keven M. Robinson *
Department of Microbiology and Molecular Genetics, University of Pittsburgh, Pittsburgh, PA, USA Jennifer M. Bomberger * Center for Translational Research in Infection and Inflammation,
Tulane University School of Medicine, New Orleans, LA, USA Jay K. Kolls * Department of Pulmonary Critical Care and Environmental Medicine, Tulane University School of Medicine, New Orleans,
LA, USA Derek A. Pociask Authors * Robert N. Abood View author publications You can also search for this author inPubMed Google Scholar * Kevin J. McHugh View author publications You can
also search for this author inPubMed Google Scholar * Helen E. Rich View author publications You can also search for this author inPubMed Google Scholar * Marianna A. Ortiz View author
publications You can also search for this author inPubMed Google Scholar * Joshua M. Tobin View author publications You can also search for this author inPubMed Google Scholar * Krishnaveni
Ramanan View author publications You can also search for this author inPubMed Google Scholar * Keven M. Robinson View author publications You can also search for this author inPubMed Google
Scholar * Jennifer M. Bomberger View author publications You can also search for this author inPubMed Google Scholar * Jay K. Kolls View author publications You can also search for this
author inPubMed Google Scholar * Michelle L. Manni View author publications You can also search for this author inPubMed Google Scholar * Derek A. Pociask View author publications You can
also search for this author inPubMed Google Scholar * John F. Alcorn View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.F.A., J.M.B.,
J.K.K., D.A.P., K.M.R. conceived the studies. R.N.A., K.J.M., H.E.R., K.R., J.M.B., M.L.M., M.A.O., J.M.T. performed the studies. R.N.A., J.M.B., J.F.A. wrote the manuscript. CORRESPONDING
AUTHOR Correspondence to John F. Alcorn. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Abood,
R.N., McHugh, K.J., Rich, H.E. _et al._ IL-22-binding protein exacerbates influenza, bacterial super-infection. _Mucosal Immunol_ 12, 1231–1243 (2019).
https://doi.org/10.1038/s41385-019-0188-7 Download citation * Received: 08 June 2018 * Revised: 15 June 2019 * Accepted: 24 June 2019 * Published: 11 July 2019 * Issue Date: September 2019 *
DOI: https://doi.org/10.1038/s41385-019-0188-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is
not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative