- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Most anoxic environments are populated by small (<10 μm) heterotrophic eukaryotes that prey on different microbial community members. How predatory eukaryotes engage in
beneficial interactions with other microbes has rarely been investigated so far. Here, we studied an example of such an interaction by cultivating the anerobic marine flagellate,
_Carpediemonas frisia_ sp. nov. (supergroup Excavata), with parts of its naturally associated microbiome. This microbiome consisted of so far uncultivated members of the Deltaproteobacteria,
Bacteroidetes, Firmicutes, Verrucomicrobia and Nanoarchaeota. Using genome and transcriptome informed metabolic network modeling, we showed that _Carpediemonas_ stimulated prokaryotic
growth through the release of predigested biomolecules such as proteins, sugars, organic acids and hydrogen. Transcriptional gene activities suggested niche separation between biopolymer
degrading Bacteroidetes, monomer utilizing Firmicutes and Nanoarchaeota and hydrogen oxidizing Deltaproteobacteria. An efficient metabolite exchange between the different community members
appeared to be promoted by the formation of multispecies aggregates. Physiological experiments showed that _Carpediemonas_ could also benefit from an association to these aggregates, as it
facilitated the removal of inhibiting metabolites and increased the availability of prey bacteria. Taken together, our results provide a framework to understand how predatory microbial
eukaryotes engage, across trophic levels, in beneficial interactions with specific prokaryotic populations. SIMILAR CONTENT BEING VIEWED BY OTHERS PHYTOPLANKTON CONSORTIA AS A BLUEPRINT FOR
MUTUALLY BENEFICIAL EUKARYOTE-BACTERIA ECOSYSTEMS BASED ON THE BIOCOENOSIS OF _BOTRYOCOCCUS_ CONSORTIA Article Open access 18 January 2021 THE ABILITY OF _PHAEOBACTER INHIBENS_ TO PRODUCE
TROPODITHIETIC ACID INFLUENCES THE COMMUNITY DYNAMICS OF A MICROALGAL MICROBIOME Article Open access 03 November 2022 MICROBIOMES OF BLOOM-FORMING _PHAEOCYSTIS_ ALGAE ARE STABLE AND
CONSISTENTLY RECRUITED, WITH BOTH SYMBIOTIC AND OPPORTUNISTIC MODES Article 28 June 2022 INTRODUCTION Small heterotrophic eukaryotes (<10 μm) perform essential functions in marine
ecosystems (Pernthaler, 2005; Edgcomb, 2016). For example, these eukaryotes capture and digest similar-sized microbes and release a mixture of nutrients, dissolved organic carbon and
detritus to the environment, stimulating growth of prokaryotes. At the same time, microbial eukaryotes can become prey for the marine mesofauna and so link prokaryotic primary production to
higher trophic levels (Sherr and Sherr, 2002). In environments with high influxes of organic carbon, microbial activity often leads to a depletion of oxygen (Glud, 2008). Most eukaryotes
cope with anoxia by switching from oxygen respiration to fermentation (Müller et al., 2012; Stairs et al., 2015). Just like in prokaryotes, fermentation in eukaryotes proceeds via glycolysis
followed by the decarboxylation of pyruvate (Müller et al., 2012). In strictly anerobic microbial eukaryotes, pyruvate decarboxylation often takes place in mitochondria that lost their
capability to respire oxygen (Boxma et al., 2005). These mitochondria recycle reducing equivalents by transferring electrons to organic metabolites or protons (H+). Depending on the
fermentation pathway used, this leads to the production of molecular hydrogen, fatty acids, alcohols and amino acids (Müller et al., 2012). In marine sediments, fermentation reactions may be
syntrophically linked into a cascade of downstream metabolism, including respiration and further fermentation reactions (Fenchel and Jørgensen, 1977). This mediates a direct consumption of
fermentation products and keeps their steady-state concentrations low. It is well established that such syntrophic interactions provide important bioenergetic advantages to prokaryotes
(Sieber et al., 2012). From the thermodynamic perspective, consumption of fermentation products enables fermentation reactions, which require low product concentrations. At low product
concentrations, fermentative prokaryotes can maximize their growth efficiency and colonize environments with scarce energy resources (McInerney et al. 2007). Among eukaryotes, similar
nutritional interactions are found in ciliates or flagellates that harbor hydrogen-scavenging symbionts (van Hoek et al., 2000; Boxma et al., 2005; Ohkuma et al., 2015). Parts of these
symbionts might also be digested by their host, providing it with an additional carbon source. Yet, their main function appears to be the creation of a biochemical environment that favors
fermentation reactions in the host’s cytosol (Ohkuma et al., 2015; Hamann et al., 2016). Even though symbiont-free eukaryotes are vital components of anerobic microbial communities (Edgcomb
et al., 2002; Wylezich and Jürgens, 2011), it has remained largely unaddressed how their physiological activity is affected by syntrophic interactions with free-living prokaryotes and vice
versa. This prompted us to enrich the marine flagellate _Carpediemonas frisia_ sp. nov. (Fornicata, supergroup Excavata), with part of its naturally associated bacterial and archaeal
community. For this enrichment culture, we applied an experimental approach that combined physiological experiments with metabolic network modeling informed by genomics and transcriptomics.
Our analysis focused on two main questions: (i) how do prokaryotes take advantage from the predatory behavior and metabolic activity of _C. frisia_? (ii) How does the biochemical activity of
prokaryotes affect the fitness of _C. frisia_? MATERIALS AND METHODS ENRICHMENT STRATEGY For the initial enrichment of _C. frisia_, we sampled sediment from the tidal flat ‘Janssand’
located in the German North Sea (53.73585N 7.69905 E, September 3, 2012). The samples were taken from a sulfidic sediment layer (located at 6 cm depth), which is known to host a rich
diversity of sulfate-reducing and fermenting microorganisms (Gittel et al., 2008; Dyksma et al., 2016). The sediment was incubated in anoxic seawater medium (34 g l−1, 1 mmol l−1 HEPES,
pH=8, Red Sea Deutschland, Germany) and supplemented with prey bacteria (109 cells ml−1) as carbon source and sulfate (28 mm) as electron acceptor for bacterial respiration. As prey we used
_Alteromonas macleodii_ (strain ATCC 27126), a small, strictly aerobic bacterium that represents an abundant genus in the oxygenated water column _in situ_ (García-Martínez et al., 2002).
Growth of flagellates was monitored by regular microscopic inspection and by measuring the turbidity of the cultures. After an enrichment of flagellates was observed, a subsample of liquid
culture was removed and grown in a sediment-free culture. This sediment-free culture was always transferred into fresh medium as soon as a depletion of bacterial prey was observed (within 4
to 5 days). Growth experiments and DNA/RNA sequencing were conducted after 26 days (after five culture transfers). MICROSCOPY For transmission electron microscopy, the cells were collected
with a table top centrifuge at 2000 r.p.m. (Stat Spin Microprep 2). After centrifugation, the pelleted sample was vitrified in a high-pressure freezer (BAL-TEC HPM-010). Substitution was
carried out at −90 °C with 0.1% tannic acid in anhydrous acetone for 24 h, and for an additional 8 h in 2% OsO4 in anhydrous acetone. After a further incubation over 20 h at −2 °C, the
samples were warmed up to +4 °C and washed with anhydrous acetone. The samples were embedded at room temperature in Agar 100 (Epon 812 equivalent) at 60 °C for 24 h. After thin sectioning
(60 nm), the sections were counter-stained with lead citrate. The samples were analyzed with a Philips CM 120 transmission electron microscope (Philips Inc., Eindhoven, The Netherlands).
Images were taken with a TemCam F416 CMOS camera (TVIPS, Gauting, Germany). For scanning electron microscopy, the cells were centrifuged at 400 r.p.m. for 6 min, collected and placed on
teflon slides for 5 min. The cells were then fixed with 2% glutaraldehyde solution (in 34 g l−1 sterile seawater, 1 mm HEPES, pH=8) for 60 min at room temperature. Fixation was followed by a
washing step in deionized water and ethanol dehydration in 30%, 50%, 70%, 90% and 100% ethanol (20 min each). Before microscopic evaluation, the specimens were subjected to critical point
drying with CO2 (Leica EM CPD300, Leica, Wetzlar, Germany). Imaging was performed with a nova NanoLab 600 scanning electron microscope (FEI Company, Eindhoven, The Netherlands). For
fluorescence microscopy, the cells were fixed in 1.8% formaldehyde solution (2 h) and stained with the DNA specific stain 4',6-diamidino-2-phenylindole (DAPI 1 μg ml−1, 3 min, at 37
°C). Pictures were taken with an epifluorescence microscope (Leila DM: Osram centra mercury-vapor lamp) connected to an AxioCam MRm camera (Carl Zeiss, Jena, Germany). Mitochondria-related
organelles were stained with MitoTracker (Red CMXRos) according to the manufacturer's protocol (Thermo Fisher Scientific GmbH, Dreieich, Germany). NEXT-GENERATION SEQUENCING, BINNING
AND QUALITY CONTROL The samples for DNA and RNA were retrieved from an exponentially growing culture in mid-exponential phase and subjected to next-generation sequencing. For this, biomass
was harvested from an enrichment culture that had been transferred five times. DNA and RNA extraction was performed as previously described (Smith et al., 2007). For DNA sequencing, a
Nextera mate pair library and a PCR-free shotgun library were constructed from genomic DNA and sequenced on an Illumina MiSeq Sequencing System (Illumina, San Diego, CA, USA). This yielded
11.8 million mate pair reads and 16.8 million paired-end reads after quality trimming. Both trimmed read sets had an average read length of 250 bp. In addition, RNA was prepared for
sequencing in two separate approaches. One library was prepared according to the Illumina TruSeq RNA Sample Preparation v2 Guide, using poly-T oligo-attached magnetic beads to enrich
eukaryotic mRNA. A second library was prepared by depletion of prokaryotic rRNA (Ribo-Zero Kit for Bacteria, epicenter) without enrichment of eukaryotic mRNA. Both mRNA libraries were
sequenced on a MiSeq instrument yielding 3.6 million poly-A tail enriched paired-end reads and 24.7 million non-enriched paired-end reads after quality trimming. Both trimmed read sets had
an average read length of 75 bp. Genomic reads were quality trimmed and filtered using nesoni (https://github.com/Victorian-Bioinformatics-Consortium/nesoni) and NextClip (Leggett et al.,
2014). Assembly of the quality filtered reads was done with SPAdes (Bankevich et al. 2012). Genomes were binned based on % GC, tetranucleotide composition and sequence coverage using
Metawatt (Strous et al., 2012). Genome completeness for prokaryotic bins was evaluated with CheckM (Parks et al., 2015) using the implemented conserved reference gene sets. Genome
completeness for the eukaryotic bin was estimated by searching for the presence of 169 conserved eukaryotic genes as previously described (Hamann et al., 2016). All sequence data for this
project are publicly available at the National Center for Biotechnology Information with the accession numbers SRX2012078-SRX2012079, SRR4448896-SRR4448897 (high-throughput DNA and RNA
sequence read archive). The Sanger-sequenced eukaryotic SSU rDNA gene was submitted to GenBank (accession no. KY031954). GENE ANNOTATION AND PREDICTION OF SYNTROPHIC LINKAGES Gene
predictions and functional annotations for the prokaryotic populations were done with the RAST online annotation pipeline (Glass et al., 2010). For the eukaryotic genome, structural
annotations and gene predictions were performed with the MAKER pipeline (Cantarel et al., 2008). After an initial _ab initio_ gene prediction with GeneMark-ES (Ter-Hovhannisyan et al.,
2008), we refined the obtained gene models with evidence-driven gene predictions using snap (Korf, 2004). As evidence, we used 14 190 assembled transcripts (5.5 Mb) of a poly-A-tail enriched
transcriptome (see above). Repeat identification, annotation and masking were done with RepeatMasker (Smit et al., 2013). Genes for the core metabolism were manually identified by blastx
and blastp homology searches against the UniProtKB/Swiss-Prot protein database. Translocation of gene products to mitochondria-related organelles was predicted based on the presence of
N-terminal target peptides using TargetP (Emanuelsson et al., 2007). Protein architectures and conserved protein domains were identified using the Pfam (Finn et al., 2014) and the SMART
(Letunic et al., 2015) protein domain detection tools. Metabolic linkages between different microbial populations were predicted based on transcriptional activities of each gene of each
population. Genes involved in energy and carbon metabolism were identified based on the RAST annotations and ranked according to their transcriptional activity. Transcriptional gene
activities were estimated by calculating the sum of all mapped mRNA reads per gene, divided by the length of each gene. Mapping and read counting was performed using bbmap
(http://sourceforge.net/projects/bbmap/) and samtools (http://samtools.sourceforge.net/). For the prokaryotic populations, only metabolic pathways that had transcription levels above the
per-population average gene activity were included into the metabolic network. Syntrophic linkages between different populations were inferred by identifying metabolites that were predicted
to be a substrate for one population and a metabolic end product of another population. PHYLOGENETIC ANALYSIS To evaluate the phylogeny of the enriched microbial populations each genomic bin
was screened for the presence of SSU rRNA sequences using HMMER (Finn et al., 2015). The retrieved sequences were then blasted to the NCBI database and closely related sequences were
downloaded for phylogenetic tree calculation. Gene alignments were calculated with MAFFT (Katoh and Standley, 2013). Phylogenetic trees were constructed with RAxML (Stamatakis, 2014) using
the GTR+GAMMA model with 400 rapid bootstrap iterations followed by a search for the best scoring maximum-likelihood tree. For the flagellate, we calculated a separate tree based on a
multigene alignment consisting of six universal eukaryotic genes (Takishita et al., 2012). For this tree, the amino acid partitions were calculated with the WAG replacement matrix with
maximum-likelihood estimated base frequencies at 400 rapid bootstrap iterations. PHYSIOLOGICAL EXPERIMENTS AND WET LAB CHEMISTRY To determine the influence of interspecies hydrogen transfer
on the fitness of _C. frisia_, four cultures were grown in parallel until they reached the early exponential phase. The cultures were then supplied with 13C-labeled _Alteromonas_ to monitor
organic carbon remineralization rates (production of 13CO2 from 13C biomass). Shortly afterwards, the sulfate-reducing activity in two cultures was inhibited with 2 mm of sodium molybdate, a
competitive inhibitor of sulfate reduction (Oremland and Capone, 1988). 13CO2 production, hydrogen concentration and eukaryotic cell numbers were recorded (in time steps of 6 min) to
monitor the immediate effect of hydrogen accumulation on the microbial activity. For the determination of protist abundance, 50 μl subsamples of liquid culture was removed from the bottles,
mixed with formaldehyde solution to a final concentration of 0.02% to immobilize the swimming cells. The cell abundance was determined by manually counting with an improved Neubauer counting
chamber (BRAND counting chamber, Neubauer improved, 0.1 mm depth). To monitor the respiration rates (production of 13CO2 from 13C-labeled biomass), liquid samples (1 ml) were filled into
exetainers and 0.2% zinc acetate solution was added to abolish microbial activity. The isotopic component of DIC was determined after acidifying with 1% (final) hypophosphoric acid on a gas
chromatography isotope ratio monitoring mass spectrometer (Optima Micromass, Manchester, UK). Volatile fatty acids were measured with a Syham HPLC system (Fürstenfeldbruck, Germany) equipped
with an Aminex HPX-87 H HPLC column (300 × 7.8 mm) and 5 mm H2SO4 as eluent. Separation was performed in isothermal mode at 40 °C and the eluted compounds were simultaneously detected with
an ultraviolet and a refractive index detector at a detection limit of 0.1 mm. As calibration standard, a mixture of the fatty acids succinate, lactate, formate, acetate, propionate and
butyrate was measured at different concentrations. RESULTS ENRICHMENT OF _C. FRISIA_ AND ITS ASSOCIATED MICROBIOTA We stimulated growth of predatory flagellates and prokaryotes by
anerobically incubating marine sediment with prey bacteria as the only carbon source and sulfate as the only electron acceptor. Light microscopy indicated growth of flagellates and
indigenous bacteria after 3 days of incubation. We cultivated a subsample of the enrichment culture in sediment-free medium and repeatedly transferred it every 4 to 5 days. After four
transfers, a morphologically uniform population of flagellates was observed. PCR amplification and Sanger sequencing of 18S rRNA genes confirmed the presence of a single eukaryotic
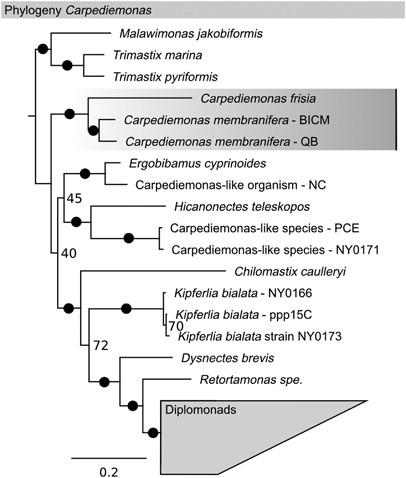
phylotype. Phylogenetic analysis placed this phylotype in a well-supported clade within Fornicata (supergroup Excavata; Figure 1). _Carpediemonas membranifera_ was its closest cultivated
relative (94% sequence identity of the 18S rRNA gene). Consistently, electron microscopy confirmed the presence of all morphological features typical for excavate flagellates (Figure
2,Supplementary Figure 1). We designated the flagellate as the novel species _Carpediemonas frisia_ (see below for a formal description). _C. frisia_ is a biflagellated protist that preys on
suspended bacteria by using a beating anterior flagellum that directs prey bacteria towards the cell. Ingestion of bacteria into membrane-enclosed digestive vacuoles took place at the
ventral feeding groove. In these vacuoles, bacteria appeared to be only incompletely digested as shown by the accumulation of membrane remnants and other high molecular weight material
(Figure 2d, Supplementary Figures 1a–c). Transmission electron microscopy also revealed the presence of mitochondria-related organelles with slight membrane folding (Figure 2f). Staining
with mitotracker, a dye retained in the presence of an active membrane potential, showed positive signals, indicating physiological activity of these organelles (Figure 2e). Microscopic
observation further indicated that a morphologically diverse prokaryotic community was co-enriched with _C. frisia_. Prokaryotes were often associated with aggregates. Aggregation appeared
to be supported by the formation of a network of pili and extracellular material (Figure 2a). _C. frisia_ itself was frequently found associated to these aggregates. MICROBIAL COMMUNITY
STRUCTURE AND COMMUNAL METABOLISM After we confirmed that _C. frisia_ was the only eukaryotic species in the culture, we determined the ecological functions of the individual community
members. For this, DNA and RNA was extracted from the exponentially growing culture and subjected to next-generation sequencing. The genomic reads were assembled and binned, resulting in
provisional whole genome sequences for all abundant microbial populations (Figure 3). For prokaryotes, the genome completeness and quality was estimated with CheckM (Parks et al., 2015). For
_C. frisia_ genome completeness was estimated by searching for the presence of conserved eukaryotic marker genes (Hamann et al., 2016). The results are shown in Table 1. The genomes
belonged to _C. frisia_ itself and members of Deltaproteobacteria, Bacteroidetes, Firmicutes, Verrucomicrobia and Archaea. Based on the annotated genomes and transcriptional activities, we
reconstructed the core metabolism of these populations. The genomes of the subpopulations of Deltaproteobacteria, Bacteroidetes and Firmicutes encoded for very similar metabolic
capabilities. Therefore, we treated them as single ecological guilds in our analysis. The predicted metabolic interactions of _C. frisia_ with these guilds are shown in Figure 4. For _C.
frisia_, we recovered a 12.38 MB provisional genome with an estimated completeness of 93%. We predicted 5695 protein-coding genes, covering 59% of the genome. The free-living, predatory
lifestyle of _C. frisia_ was well reflected in its genome. _C. frisia_ encoded typical features for actin-based phagotrophy, motility and the ability to use hydrolysis products from ingested
bacteria. _C. frisia_ also encoded genes for an exocytosis machinery, supporting the excretion of undigested particles and other substances through secretory vesicles. Consistent with the
known physiological capabilities of the related genera _Trichomonas_ and _Giardia_ (Müller et al., 2012), _C. frisia_ lacked the capability for autotrophy, oxygen respiration and
proton-driven adenosine triphophate (ATP) synthesis (Figure 4b). Instead, it was inferred to depend on the use of carbohydrates and amino acids to drive a strictly fermentative metabolism.
Transcriptional gene activities indicated that ATP production was coupled to glycolysis followed by the decarboxylation of pyruvate. Pyruvate decarboxylation appeared to be catalyzed by the
subsequent activity of pyruvate:ferredoxin oxidoreductase and acetyl-CoA synthetase. Further, we detected transcriptional activity of a Fe-hydrogenase and the NADH oxidizing subunits
(NouE/NuoF) of respiratory Complex I (Supplementary Figure 2). The combined activity of these enzymes is known to catalyze the proton dependent, simultaneous (‘confurcating’) re-oxidation of
NADH and reduced ferredoxin, leading to the production of hydrogen (‘H2’). In addition, _C. frisia_ was inferred to be capable of the re-oxidation of NADH through the activity of lactate
and/or alcohol dehydrogenases. We also checked for the presence of N-terminal target peptides that support translocation of gene products to the mitochondria-related organelles. None of the
predicted genes involved in _C. frisia's_ energy metabolism contained such target peptides. Thus, energy conservation in _C. frisia_ appears to take place entirely in the cytosol.
Bacteroidetes was the most abundant group of bacteria co-cultured with _C. frisia_. Bacteroidetes were represented by two different populations. One population was affiliated to the genus
_Marinifilum_ (7.8 MB, 96% complete) and another was most closely related to a group of deeply branching, so far uncultivated Prolixibacteraceae (4.3 MB, 98% complete). The latter appeared
to be most abundant, with a relative abundance of 33% as estimated by normalized read counts. (Figure 3c). From previous studies, it is known that Bacteroidetes are specialized consumers of
high molecular weight compounds such as proteins and polysaccharides (Fernández-Gómez et al., 2013). To break down these compounds Bacteroidetes make use of a variety of specialized
transporters and enzymes with hydrolytic activity (Bauer et al., 2006). The Prolixibacteraceae population detected here, also displayed high activity of these enzymes. Specifically, we
recorded high transcriptional activity of genes encoding for proteases, glycoside hydrolases, and glycosyltransferases (Supplementary Figure 3). In addition, activity of TonB-dependent and
ABC-like biopolymer transport systems indicated the consumption of macromolecular organic material (Supplementary Figure 5). Energy appeared to be conserved by fermentation as shown by high
transcriptional activity of genes involved in glycolysis. In addition, genes encoding for acetyl-CoA synthetase, acetate kinase and Fe-hydrogenases showed high transcriptional activity.
Taken together, both Bacteroidetes populations were inferred to belong to the same ecological guild that performed biopolymer degradation and fermentation with hydrogen and acetate as main
metabolic end products. In addition to Bacteroidetes, we also recovered genomes for two major populations of Firmicutes belonging to the genera _Fusibacter_ (14.7 MB, 100% complete) and
_Bacillus_ (19.4 MB, 97% complete). Both populations consisted of several subpopulations exhibiting different degrees of genetic heterogeneity, as indicated by the detection of multiple
single copy marker genes and the large bin sizes (Table 1). Together, all Firmicutes were estimated to make up about 12% of the total microbial community. They were inferred to consume a
broad range of biomolecules including amino acids, sugars and other low molecular weight organic acids. This was indicated by high transcriptional activity of genes encoding for
oligopeptide, nucleoside and sugar ABC-like transporters as well as TRAP-like C4-dicarboxylate transporters (Supplementary Figures 3 and 4). These substrates were predicted to be fermented
to butyrate, acetate, formate, ethanol and hydrogen. Hydrogen production in Firmicutes appeared to proceed via the same mechanism as in _C. frisia,_ through the simultaneous oxidation of
NADH and reduced ferredoxin. This was indicated by transcriptional activity of genes encoding for Fe-hydrogenases and the NADH oxidizing subunits of respiratory Complex I (Supplementary
Figure 2). These genes were encoded in a single operon. In sum, all Firmicutes were inferred to perform very similar functions by metabolizing low molecular weight biomolecules in a
fermentative catabolism. The remainder of the bacterial community mainly consisted of Deltaproteobacteria. Phylogenetic analyses showed that Deltaproteobacteria were present as three main
populations belonging to the genera _Desulfotalea_ (4.3 MB, 100% complete), _Desulfovibrio_ (3.8 MB, 97% complete) and _Desulfofaba_ (8.6 MB, 96% complete). Together, these populations made
up approximately 30% of the total microbial community. The encoded core metabolisms of these populations indicated that they were mainly involved in the consumption of hydrogen and other
fermentation products. Specifically, we found high activities of genes supporting the uptake of short chain fatty acids and amino acids, as well as the oxidation of hydrogen. Hydrogen
consumption was catalyzed by a high-affinity Ni/Fe-hydrogenase and coupled to a membrane-bound respiratory chain that used sulfate as terminal electron acceptor. In addition, all
Deltaproteobacteria populations transcribed genes encoding for type IV pilus assembly proteins and the fimbria forming protein pilin. This was in agreement with observations made via
scanning electron microscopy, which suggested the presence of pili-like structures aiding in aggregation of prokaryotes (compare Figure 2). Apart from the major bacterial populations, we
also detected a small population of Verrucomicrobia and Archaea. 16S rDNA gene phylogenies showed that the detected population of Verrucomicrobia was related to the genus _Fucuphilus_.
Similar to the Firmicutes, this population was involved in the uptake and fermentation of sugar monomers and amino acids. The archael population was related to a clade of so far uncultivated
species from mesophilic and thermophilic marine environments. The closest isolated and described relative was _Nanoarchaeum equitans_. This species is known for its reduced genome size,
limited metabolic capabilities and an epibiotic association with the archaeum _Ignicoccus_ (Waters et al., 2003). The archaeal genome recovered here was also small (1.75 Mb) and encoded only
basic metabolic capabilities. The most basic requirements to sustain life appeared to be present given the detection of genes for ATP synthesis, DNA replication, transcription and
translation. Based on its encoded core catabolism, the archaeon appeared to be dependent on a fermentative metabolism. Transcriptional activities of genes encoding for a sugar ABC-like
transporter, a pyruvate kinase and an acetate kinase indicated an involvement in the fermentation of sugar monomers. In addition, activity of genes encoding for a methyl-accepting chemotaxis
protein and archaeal flagellin were detected. Genes supporting methanogenesis, hydrogen oxidation, sulfur oxidation or ammonium oxidation, were not detected. RELEVANCE OF SYNTROPHY FOR THE
FITNESS OF _C. FRISIA_ Our metagenomic and transcriptomic analyses suggested that fermentation in _C. frisia_ was facilitated by interspecies hydrogen transport from _C. frisia_ to
sulfate-reducing _Desulfotalea, Desulfovibrio and Desulfofaba_. To confirm this inference experimentally, molybdate, a competitive inhibitor of sulfate reduction, was added to the
exponentially growing _C. frisia_ enrichment culture (Figure 5). For this experiment, we used 13C-labeled prey bacteria and monitored their consumption and conversion by the analysis of
produced 13CO2. Immediately after inhibition of sulfate reduction, we observed a rapid accumulation of hydrogen in the culture (Figure 5c). In addition, we observed an accumulation of
butyrate, formate and acetate (Supplementary Figure 6b). Growth of _C. frisia_ completely stopped shortly after the onset of hydrogen accumulation (Figure 5a). Concomitantly, 13CO2
production rates leveled off compared with the control. In addition, we incubated _C. frisia_ in medium containing only limiting amounts of sulfate (200 μm). This cultivation condition
favored the growth of fermenting species but did not support hydrogen oxidation coupled to sulfate reduction. Also, for this treatment, we observed a significantly reduced growth rate and
growth yield of _C. frisia_ (Supplementary Figures 6c and d). DISCUSSION In this study, we showed that anoxic incubation of a marine sediment supplemented with prey bacteria as carbon source
and sulfate as electron acceptor led to the enrichment of the predatory protist _C. frisia_ and co-enrichment of a prokaryotic community. After five subsequent culture transfers in
sediment-free medium, this community consisted of distinct populations affiliated with Bacteroidetes, Firmicutes, Deltaproteobacteria, Verrucomicrobia and Nanoarchaeota. Metabolic
interactions were inferred from the analysis of metagenomic and transcriptomic sequence data. _C. frisia_ appeared to provide two major nutritional opportunities for prokaryotes: First, the
degradation of incompletely digested macromolecular organic material and second, the oxidation of sugars, organic acids and hydrogen with sulfate. Physiological experiments confirmed that
the fitness of _C. frisia_ was directly impacted by the hydrogen oxidizing activity of Deltaproteobacteria. Our genomic and transcriptomic analysis suggested that _C. frisia_ produced
hydrogen through the simultaneous (confurcating) oxidation of NAD(P)H and ferredoxin using an enzyme complex that consisted of the catalytic subunits of respiratory Complex I (NouE/ NuoF)
and a Fe-hydrogenase. This reaction is very common among fermentative bacteria (Schut and Adams, 2009) and has also been predicted to be used by a variety of eukaryotic species (Stairs et
al. 2015). However, the available free energy released during NAD(P)H-dependent hydrogen production decreases exponentially with increasing product concentration. For the synergistic
production of molecular hydrogen from NAD(P)H and ferredoxin to proceed, the maximum hydrogen concentration is 0.1 mm (at NADH/NAD+=10; Stams and Plugge, 2009). Many fermenting eukaryotes
have evolved symbiotic relationships with hydrogen-scavenging prokaryotes, which allow them to maintain intracellular hydrogen levels below this threshold. For example, ciliates and amoeba
often harbor hydrogen-consuming methanogens inside their cytosol (van Bruggen et al., 1985; van Hoek et al., 2000; Boxma et al., 2005). Another example is the breviate _Lenisia limosa_,
which forms epibiotic associations with hydrogen oxidizing _Arcobacter_ (Hamann et al., 2016). In this association, the maximum hydrogen concentration has been reported to be 20 times lower
than predicted for _C. frisia_. This can be explained by the observation that _L. limosa_ mainly uses NAD(P)H for hydrogen production, a reaction that is more prone to product inhibition
than the simultaneous oxidation of NAD(P)H and ferredoxin. This may also explain why _C. frisia_ can sustain its metabolism without the tight symbiotic association observed for _L. limosa_
and other hydrogen-producing eukaryotes. In any case, if the maximum hydrogen concentration is exceeded, NAD(P)H needs to be re-oxidized by reduction of organic metabolites such as pyruvate
or succinate. Less energy is conserved in this metabolic bypass, leading to a reduced growth efficiency. In addition, NAD(P)H-dependent pyruvate oxidation may lead to the production of
ethanol and acetaldehyde (Müller et al., 2012), which are growth inhibitors at elevated concentrations (Maiorella et al., 1983). The removal of inhibitory metabolic byproducts by
Deltaproteobacteria therefore provides an important fitness benefit to _C. frisia_. The second major metabolic function performed by the bacteria co-enriched with _C. frisia_ appeared to be
the degradation of incompletely digested, macromolecular organic material. Microscopic imaging showed that this material originated from fecal pellets excreted by _C. frisia_. As we did not
provide any organic carbon other than prey bacteria to our enrichment, this material likely represented a major carbon source for bacterial growth. This also explains the high abundance of
bacterial species in our enrichment that are adapted to use this type of material. Bacteroidetes are known as consumers of macromolecules (Fernández-Gómez et al., 2013) and were inferred to
use detritus as their substrates. The presence and activity of the two populations of Firmicutes also suggested the availability of smaller organic molecules such as oligopeptides, amino
acids and sugars. These might be released either directly by _C. frisia_, or during the hydrolysis of proteins and polysaccharides by Bacteroidetes. Similar niche partitioning between
Bacteroidetes and Firmicutes is known for many other organic carbon-rich environments (for example, the digestive tract of animals (Lozupone et al., 2012) or marine oxygen minimum zones
(Wright et al., 2012)). The processing of organic waste products by Bacteroidetes and Firmicutes is, therefore, in good agreement with the known physiological capabilities of these species.
A typical characteristic for syntrophic bacterial communities is the formation of multispecies aggregates. Aggregate formation can enhance the exchange of metabolites between different
populations, thereby accelerating fermentative reactions and degradation of organic material (Schink and Thauer, 1988). In our enrichment, aggregates most likely arose from bacterial
colonization of detritus excreted by _C. frisia_. As _C. frisia_ was often directly associated to these aggregates, it could likely benefit from the high turnover of hydrogen and other
metabolites within these aggregates. In addition, _C. frisia_ may benefit from an increased availability of prey bacteria in aggregates. This way, excretion of detritus by _C. frisia_ and
the subsequent bacterial colonization of this material, can be interpreted as a primitive form of microbial agriculture. It will be an interesting research avenue, to address how widespread
similar bi-directional metabolic interactions between predatory flagellates and bacteria are in the environment. To date, there are many reported cases of specific protists-bacterial
co-occurrences, which may be a result of metabolic interdependencies. For example, community fingerprinting by 16S rDNA amplicon sequencing showed that a bloom of _Carpediemonas_ and related
_Diplomonads_ was accompanied by a specific enrichment for Bacteroidetes, Firmicutes and known sulfate-reducing Deltaproteobacteria (Holmes et al., 2013). Another study based on single-cell
sequencing of flow-sorted planktonic eukaryotes showed that Firmicutes and Deltaproteobacteria are preferentially associated with heterotrophic protists (Martinez-Garcia et al., 2012). So
far, a common assumption is that an enrichment of specific prokaryotic phenotypes stimulates the growth of flagellates that are adapted to prey on these species (Pernthaler, 2005; Holmes et
al., 2013). The results presented here, however, show that for anoxic environments, the opposite provides an equally compelling explanation—predatory microbial eukaryotes can also stimulate
the growth of specific prokaryotic populations, which are adapted to consume their metabolic waste products. In conclusion, we have shown that _C. frisia_ is a newly identified anerobic
flagellate, that likely engages in a hydrogen- and organic carbon-based metabolic syntrophy with sulfate-reducing Deltaproteobacteria. This syntrophy appears to benefit _C. frisia_ by
removing inhibitory metabolites and by creating a biochemical environment that promotes fermentative hydrogen production. In return, _C. frisia_ appears to provide its associated microbiota
with predigested organic macromolecules, sugars, organic acids and hydrogen. The described metabolic interdependences provide a framework to predict the ecological importance of specific
co-occurrences between heterotrophic microbial eukaryotes and prokaryotic populations. In addition, the results show that the concept of metabolic syntrophy, as a form of microbial
mutualism, can also apply for inter-kingdom interactions between anaerobic flagellates and free-living prokaryotes. GENUS _CARPEDIEMONAS_ EKEBOM _ET AL._ 1996 _CARPEDIEMONAS FRISIA_ SP. NOV.
HAMANN _ET AL._ 2017 _Description._ _Carpediemonas frisia_ is a flagellated marine protist with characteristics of the genus (Simpson and Patterson, 1999). The cells are typically around 5
μm long, pear-shaped, slightly compressed laterally, with two flagella of unequal length. Starving cells can be as small as 3 μm. Flagella emerge from the anterior end of the ventral groove.
The anterior flagellum is about 5 μm long. The posterior flagellum is two to three times longer and has a thickened, shuffle-like morphology near its origin. The anterior flagellum performs
a sweeping motion, bending its tip backwards. In swimming cells, the longer posterior flagellum beats and provides additional motive force. The cells preferentially swim along surfaces,
holding the anterior side in contact with the substrate. Small filamentous pseudopodia emerge from all parts of the cell. No cysts are observed. _C. frisia_ is differentiated from
_Carpediemonas membranifera_ by the presence of mitochondria-related organelles with slight membrane folding. _Habitat._ This species was isolated from marine tidal flat sediment collected
from a sulfidic sediment layer. The sampling site is commonly known as ‘Janssand’ and located in the German Wadden Sea south of the island Spiekeroog (53.73585 N, 7.69905 E). _Etymology._
The species name frisia is Latin and refers to the coastal region of the southeastern North Sea ‘Friesland’, which is the historical settlement area of the Friesians. _Gene sequence data._
The nearly complete SSU rRNA gene of this isolate (strain S16) is deposited in GenBank under accession no. KY031954. REFERENCES * Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M,
Kulikov AS _et al_. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. _J Comp Biol_ 9: 455–477. Article Google Scholar * Bauer M, Kube M,
Teeling H, Richter M, Lombardot T, Allers E _et al_. (2006). Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii” reveals adaptations to degradation of polymeric organic
matter. _Environ Microbiol_ 8: 2201–2213. Article CAS Google Scholar * Boxma B, de Graaf R, Staay GWM, Alen TA, Ricard G, Gabaldon T _et al_. (2005). An anaerobic mitochondrion that
produces hydrogen. _Nature_ 434: 72–74. Article Google Scholar * van Bruggen JJA, Stumm CK, Zwart KB, Vogels GD . (1985). Endosymbiotic methanogenic bacteria of the sapropelic amoeba
Mastigella. _FEMS Microbiol Ecol_ 1: 187–192. Google Scholar * Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B _et al_. (2008). MAKER: an easy-to-use annotation pipeline designed
for emerging model organism genomes. _Genome Res_ 18: 188–196. Article CAS Google Scholar * Dyksma S, Bischof K, Fuchs BM, Hoffmann K, Meier D, Meyerdierks A _et al_. (2016). Ubiquitous
Gammaproteobacteria dominate dark carbon fixation in coastal sediments. _ISME J_ 10: 1939–1953. Article CAS Google Scholar * Edgcomb VP . (2016). Marine protist associations and
environmental impacts across trophic levels in the twilight zone and below. _Curr Opin Microbiol_ 31: 169–175. Article CAS Google Scholar * Edgcomb VP, Kysela DT, Teske A, de Vera Gomez
A, Sogin ML . (2002). Benthic eukaryotic diversity in the Guaymas Basin hydrothermal vent environment. _Proc Natl Acad Sci USA_ 99: 7658–7662. Article CAS Google Scholar * Emanuelsson O,
Brunak S, vonHeijne G, Nielsen H . (2007). Locating proteins in the cell using TargetP, SignalP, and related tools. _Nat Protoc_ 2: 953–971. Article CAS Google Scholar * Fernández-Gómez
B, Richter M, Schüler M, Pinhassi J, Acinas SG, González JM _et al_. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. _ISME J_ 7: 1026–1037. Article Google Scholar
* Fenchel TM, Jørgensen BB . (1977) Detritus food chains of aquatic ecosystems: the role of bacteria. In: Alexander M (ed), _Advances in Microbial Ecology_. Plenum Press: New York, NY,
USA, pp 1–58. Book Google Scholar * Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR _et al_. (2014). Pfam: the protein families database. _Nucleic Acids Res_ 42:
D222–D230. Article CAS Google Scholar * Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F _et al_. (2015). HMMER web server: 2015 update. _Nucleic Acids Res_ 43: W30–W38.
Article CAS Google Scholar * García-Martínez J, Acinas SG, Massana R, Rodríguez-Valera F . (2002). Prevalence and microdiversity of Alteromonas macleodii-like microorganisms in different
oceanic regions. _Environ Microbiol_ 4: 42–50. Article Google Scholar * Gittel A, Mußmann M, Sass H, Cypionka H, Könneke M . (2008). Identity and abundance of active sulphate-reducing
bacteria in deep tidal flat sediments determined by directed cultivation and CARD-FISH analysis. _Environ Microbiol_ 10: 2645–2658. Article CAS Google Scholar * Glass EM, Wilkening J,
Wilke A, Antonopoulos D, Meyer F . (2010). Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. _Cold Spring Harb Protoc_ 5: e-pub ahead of print;
doi:10.1101/pdb.prot5368. Article Google Scholar * Glud RN . (2008). Oxygen dynamics of marine sediments. _Mar Biol Res_ 4: 243–289. Article Google Scholar * Hamann E, Gruber-Vodicka H,
Kleiner M, Tegetmeyer HE, Riedel D, Littmann S _et al_. (2016). Environmental Breviatea harbour mutualistic Arcobacter epibionts. _Nature_ 534: 254–258. Article CAS Google Scholar * van
Hoek AH, van Alen TA, Sprakel VS, Leunissen JA, Brigge T, Vogels GD _et al_. (2000). Multiple acquisition of methanogenic archaeal symbionts by anaerobic ciliates. _Mol Biol Evol_ 17:
251–258. Article CAS Google Scholar * Holmes DE, Giloteaux L, Williams KH, Wrighton KC, Wilkins MJ, Thompson CA _et al_. (2013). Enrichment of specific protozoan populations during _in
situ_ bioremediation of uranium-contaminated groundwater. _ISME J_ 7: 1286–1298. Article CAS Google Scholar * Katoh K, Standley DM . (2013). MAFFT multiple sequence alignment software
version 7: improvements in performance and usability. _Mol Biol Evol_ 30: 772–780. Article CAS Google Scholar * Korf I . (2004). Gene finding in novel genomes. _BMC Bioinformatics_ 5: 59.
Article Google Scholar * Leggett RM, Clavijo BJ, Clissold L, Clark MD, Caccamo M . (2014). Next clip: an analysis and read preparation tool for nextera long mate pair libraries.
_Bioinformatics_ 30: 566–568. Article CAS Google Scholar * Letunic I, Doerks T, Bork P . (2015). SMART: Recent updates, new developments and status in 2015. _Nucleic Acids Res_ 43:
D257–D260. Article CAS Google Scholar * Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R . (2012). Diversity, stability and resilience of the human gut microbiota. _Nature_ 489:
220–230. Article CAS Google Scholar * Maiorella B, Blanch HW, Wilke CR . (1983). By-product inhibition effects on ethanolic fermentation by Saccharomyces cerevisiae. _Biotechnol Bioeng_
25: 103–121. Article CAS Google Scholar * Martinez-Garcia M, Brazel D, Poulton NJ, Swan BK, Gomez ML, Masland D _et al_. (2012). Unveiling _in situ_ interactions between marine protists
and bacteria through single cell sequencing. _ISME J_ 6: 703. Article CAS Google Scholar * McInerney MJ, Rohlin L, Mouttaki H, Kim U, Krupp RS, Rios-Hernandez L _et al_. (2007). The
genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. _Proc Natl Acad Sci USA_ 104: 7600–7605. Article Google Scholar * Müller M, Mentel M, van
Hellemond JJ, Henze K, Woehle C, Gould SB _et al_. (2012). Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. _Microbiol Mol Biol Rev_ 76: 444–495. Article Google
Scholar * Ohkuma M, Noda S, Hattori S, Iida T, Yuki M, Starns D _et al_. (2015). Acetogenesis from H2 plus CO2 and nitrogen fixation by an endosymbiotic spirochete of a termite-gut
cellulolytic protist. _Proc Natl Acad Sci USA_ 698: 201423979. Google Scholar * Oremland RS, Capone DG . (1988). Use of specific inhibitors in biogeochemistry and microbial ecology. _Adv
Microb Ecol_ 10: 285–383. Article CAS Google Scholar * Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW . (2015). CheckM: assessing the quality of microbial genomes recovered
from isolates, single cells, and metagenomes. _Genome Res_ 25: 1043–1055. Article CAS Google Scholar * Pernthaler J . (2005). Predation on prokaryotes in the water column and its
ecological implications. _Nat Rev Microbiol_ 3: 537–546. Article CAS Google Scholar * Schink B, Thauer RK . (1988). Energetics of syntrophic methane formation and the influence of
aggregation. In: Lettinga G, Zehnder AJB, Grotenhuis JTC, Hulshoff LW (eds), Granular anaerobic sludge: microbiology and technology. Pudoc:: Wageningen, The Netherlands, pp 5–17. * Schut GJ,
Adams MWW . (2009). The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. _J Bacteriol_ 191:
4451–4457. Article CAS Google Scholar * Sherr EB, Sherr BF . (2002). Significance of predation by protists in aquatic microbial food webs. _Antonie Van Leeuwenhoek_ 81: 293–308. Article
CAS Google Scholar * Sieber JR, McInerney MJ, Gunsalus RP . (2012). Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. _Annu Rev Microbiol_ 66: 429–452.
Article CAS Google Scholar * Simpson AGB, Patterson DJ . (1999). The ultrastructure of Carpediemonas membranifera (Eukaryota) with reference to the ‘excavate hypothesis’. _Eur J
Protistol_ 35: 353–370. Article Google Scholar * Smit A, Hubley R, Green P . (2013), RepeatMasker Open-4.0. 2013–2015. Available at http://www.repeatmasker.org. * Smith CJ, Nedwell DB,
Dong LF, Osborn AM . (2007). Diversity and abundance of nitrate reductase genes (narG and napA), nitrite reductase genes (nirS and nrfA), and their transcripts in estuarine sediments. _Appl
Environ Microbiol_ 73: 3612–3622. Article CAS Google Scholar * Stairs CW, Leger MM, Roger AJ . (2015). Diversity and origins of anaerobic metabolism in mitochondria and related
organelles. _Philos Trans R Soc B Lond B Biol Sci_ 370: 20140326. Article Google Scholar * Stamatakis A . (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of
large phylogenies. _Bioinformatics_ 30: 1312–1313. Article CAS Google Scholar * Stams AJM, Plugge CM . (2009). Electron transfer in syntrophic communities of anaerobic bacteria and
archaea. _Nat Rev Microbiol_ 7: 568–577. Article CAS Google Scholar * Strous M, Kraft B, Bisdorf R, Tegetmeyer HE . (2012). The binning of metagenomic contigs for microbial physiology of
mixed cultures. _Front Microbiol_ 3: 410. Article Google Scholar * Takishita K, Kolisko M, Komatsuzaki H, Yabuki A, Inagaki Y, Cepicka I _et al_. (2012). Multigene phylogenies of diverse
carpediemonas-like organisms identify the closest relatives of ‘Amitochondriate” Diplomonads and Retortamonads. _Protist_ 163: 344–355. Article Google Scholar * Ter-Hovhannisyan V,
Lomsadze A, Chernoff YO, Borodovsky M . (2008). Gene prediction in novel fungal genomes using an _ab initio_ algorithm with unsupervised training. _Genome Res_ 18: 1979–1990. Article CAS
Google Scholar * Waters E, Hohn MJ, Ahel I, Graham DE, Adams MD, Barnstead M _et al_. (2003). The genome of Nanoarchaeum equitans: insights into early archaeal evolution and derived
parasitism. _Proc Natl Acad Sci USA_ 100: 12984–12988. Article CAS Google Scholar * Wright JJ, Konwar KM, Hallam SJ . (2012). Microbial ecology of expanding oxygen minimum zones. _Nat Rev
Microbiol_ 10: 381–394. Article CAS Google Scholar * Wylezich C, Jürgens K . (2011). Protist diversity in suboxic and sulfidic waters of the Black Sea. _Environ Microbiol_ 13: 2939–2956.
Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Theresa Hargesheimer, Lulu Liu, Gabi Klockgether, Ramona Appel and Ines Kattelmann for technical assistance,
Xiaoli Dong for her help with metagenomics and Alastair Simpson, Ger Strous and Sergio for comments on electron micrographs. We thank Kai-Uwe Hinrichs and Nicole Dubilier for discussions and
advice. This study was supported by the European Research Council (ERC) starting grant MASEM 242635 (to MS and EH), the Campus Alberta Innovation Chair Program (to MS and EH), the Canadian
Foundation for Innovation (to MS), the German Federal State Nordrhein-Westfalen (to MS), the Max Planck Society and NSERC for a Discovery Grant to MS. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Department of Geoscience, University of Calgary, Calgary, AB, Canada Emmo Hamann, Jianwei Chen & Marc Strous * Max Planck Institute for Marine Microbiology, Bremen,
Germany Emmo Hamann, Halina E Tegetmeyer, Sten Littmann, Soeren Ahmerkamp, Jianwei Chen, Philipp F Hach & Marc Strous * Institute for Genome Research and Systems Biology, Center for
Biotechnology, Bielefeld University, Bielefeld, Germany Halina E Tegetmeyer & Marc Strous * Max Planck Institute for Biophysical Chemistry, Göttingen, Germany Dietmar Riedel Authors *
Emmo Hamann View author publications You can also search for this author inPubMed Google Scholar * Halina E Tegetmeyer View author publications You can also search for this author inPubMed
Google Scholar * Dietmar Riedel View author publications You can also search for this author inPubMed Google Scholar * Sten Littmann View author publications You can also search for this
author inPubMed Google Scholar * Soeren Ahmerkamp View author publications You can also search for this author inPubMed Google Scholar * Jianwei Chen View author publications You can also
search for this author inPubMed Google Scholar * Philipp F Hach View author publications You can also search for this author inPubMed Google Scholar * Marc Strous View author publications
You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHORS Correspondence to Emmo Hamann or Marc Strous. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
conflict of interest. ADDITIONAL INFORMATION Supplementary Information accompanies this paper on The ISME Journal website SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION (PDF 3990 KB)
RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article
are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to
obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Hamann, E., Tegetmeyer, H., Riedel, D. _et al._ Syntrophic linkage between predatory _Carpediemonas_ and specific prokaryotic populations. _ISME J_ 11,
1205–1217 (2017). https://doi.org/10.1038/ismej.2016.197 Download citation * Received: 02 August 2016 * Revised: 28 October 2016 * Accepted: 07 December 2016 * Published: 17 February 2017 *
Issue Date: May 2017 * DOI: https://doi.org/10.1038/ismej.2016.197 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative




:max_bytes(150000):strip_icc():focal(742x607:744x609)/Hiker-Bright-Angel-Trail-01-052623-8e87927a10dd42048a1167885a367680.jpg)