- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The amyloid precursor protein (APP) has been under intensive study in recent years, mainly due to its critical role in the pathogenesis of Alzheimer's disease (AD). β-Amyloid
(Aβ) peptides generated from APP proteolytic cleavage can aggregate, leading to plaque formation in human AD brains. Point mutations of APP affecting Aβ production are found to be causal for
hereditary early onset familial AD. It is very likely that elucidating the physiological properties of APP will greatly facilitate the understanding of its role in AD pathogenesis. A number
of APP loss- and gain-of-function models have been established in model organisms including _Caenorhabditis elegans_, _Drosophila_, zebrafish and mouse. These _in vivo_ models provide us
valuable insights into APP physiological functions. In addition, several knock-in mouse models expressing mutant APP at a physiological level are available to allow us to study AD
pathogenesis without APP overexpression. This article will review the current physiological and pathophysiological animal models of APP. SIMILAR CONTENT BEING VIEWED BY OTHERS AN _APP_
KNOCK-IN RAT MODEL FOR ALZHEIMER’S DISEASE EXHIBITING AΒ AND TAU PATHOLOGIES, NEURONAL DEATH AND COGNITIVE IMPAIRMENTS Article Open access 17 November 2021 INTEGRATIVE ANALYSIS REVEALS A
CONSERVED ROLE FOR THE AMYLOID PRECURSOR PROTEIN IN PROTEOSTASIS DURING AGING Article Open access 03 November 2023 DELTA-SECRETASE TRIGGERS ALZHEIMER’S DISEASE PATHOLOGIES IN WILD-TYPE
HAPP/HMAPT DOUBLE TRANSGENIC MICE Article Open access 12 December 2020 INTRODUCTION The amyloid precursor protein (APP) is one of the most intensively studied molecules since it was first
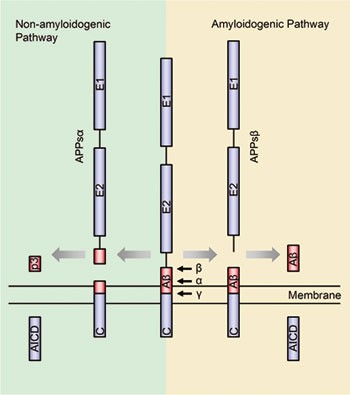
cloned more than 20 years ago1,2,3. A notable feature of APP is that it undergoes finely regulated secretase cleavage processing (Figure 1). In general, the α-secretase or β-secretase
cleaves the APP extracellular domain generating the soluble N-terminal fragments APPsα or APPsβ and the membrane-associated C-terminal fragments CTFα and CTFβ, respectively. CTFα or CTFβ
will further be cleaved by the γ-secretase within the transmembrane domain releasing the p3 peptide or β-amyloid (Aβ) peptide. Following γ-secretase cleavage, both CTFs will also release the
APP intracellular domain (AICD) into the cytoplasm. α-Secretase-mediated APP cleavage prevents Aβ production, and thus is called the non-amyloidogenic pathway, whereas the
β-secretase-mediated APP cleavage generates Aβ peptides and is called the amyloidogenic pathway. However, recent studies showed that APP proteolytic cleavage could be more complicated than
above (reviewed by Chow _et al_.4). For instance, besides the γ-site cleavage of the CTFs by γ-secretase, CTFs may also undergo 'ɛ-site' cleavage near the cytoplasmic membrane
boundary of APP5. The normal physiological functions of APP have raised curiosity, since it belongs to an evolutionarily conserved protein family (Figure 2) and combined deficiency of APP
and its homologs leads to early postnatal lethality in mammals6,7. A number of APP loss-of-function and gain-of-function _in vivo_ models have been established and characterized in different
model organisms, including _Caenorhabditis elegans_, _Drosophila_, zebrafish and mouse. Studies from different species indicate conservation of function between APP family members,
including roles in cell adhesion, neuron migration and synaptogenesis. Enormous scientific efforts have been put into APP-related studies mainly because of its vital pathophysiological
functions in Alzheimer's disease (AD). AD is the leading cause of dementia in the aged population. There are three pathological hallmarks of AD: extracellular amyloid plaques,
intracellular neurofibrillary tangles and neuronal degeneration including synapse loss. Amyloid plaques are extracellular deposits primarily composed of Aβ peptides that are derived from
proteolytic cleavage of APP and associated with other components (reviewed by Armstrong _et al_.8). The pathogenic mechanism of AD is very controversial, however, the Aβ cascade hypothesis
is currently most favored. This hypothesis states that the excessive Aβ peptides generated from mis-regulated cleavage of APP or impaired clearance play a central role in AD9. While the vast
majority of AD cases occur after 65 years of age, a small percentage of patients (<5%) develop clinical symptoms before the age of 65 years due to autosomal dominant familial AD (FAD)
mutations10. These mutations have been discovered in three different genes: _APP_, _Presenilin 1_ and _2_ (_PSEN1_ and _PSEN2_). Most mutations in the _APP_ and _PSEN_ (or _PS_) genes alter
the production of Aβ cleavage profiles, leading to accelerated amyloid pathology. Several lines of knock-in mice (KI) with _APP_ FAD mutations have been generated in recent years, either by
mutation of _APP_ alone or in combination with _PS1_ mutations. Compared to traditional AD transgenic mice, which have been used extensively in the field, these KI models do not suffer from
the confounding effects of APP overexpression and offer us a more physiologically relevant platform to investigate AD pathogenesis. We believe a review of these _in vivo_ APP animal models
will facilitate the understanding of AD pathogenesis in the context of APP physiology. NON-MAMMALIAN APP ANIMAL MODELS C. ELEGANS The _C. elegans_ homolog of APP, APL-1, is structurally
similar to its mammalian counterpart and shares sequence homology in the N-terminal E1 and E2 domains and the intracellular C-terminal domain, which has the highest sequence conservation
(Figure 2)11. There are no known coding region splice variants detected in APL-1, which most closely resembles the neuronal isoform of APP, APP69511. However, APL-1 does not contain the
amyloid-β sequence, similar to the functionally redundant mammalian APP homologs APLP1 and APLP211,12,13,14. Nematode development includes four larval stages after each of which is a molt
where a new, larger exoskeleton is formed to accommodate the growth of the larvae. In worms, loss of function of the single _apl-1_ gene leads to developmental arrest and lethality during
the first larval stage (L1), likely due to a molting defect15,16. Many of the _apl-1_-null worms arrest at the L1/L2 transition, unable to shed old cuticle and exhibiting massive internal
degradation15,16. RNAi knockdown of _apl-1_ also leads to a molting defect, although less severe than the null mutant, as the worms exhibit loose cuticle primarily around the head beginning
at the L3/L4 molt16,17. In addition to the molting defect, _apl-1_ knockdown leads to hypersensitivity to the acetylcholinesterase inhibitor aldicarb, signifying a defect in cholinergic
neurotransmission16. The neuronal phenotype and the molting defect were found to be independent of one another, suggesting _apl-1_ contributes to multiple functions within the worm. However,
more studies are needed to determine the mechanism of _apl-1_ regulation in each of these functions. Structure-function studies have been performed in an attempt to identify functional
domains of _apl-1_ through rescue of the _apl-1_-null lethality and molting defect. Surprisingly, both phenotypes were found to be rescued by either a C-terminal truncation of _apl-1_ or the
soluble N-terminus, showing that the highly conserved C-terminus is not required for _apl-1_ function in the worm15,16. This differs from the mammalian system in which the APP C-terminus is
essential for viability on a non-redundant background (see discussion under 'KI models expressing distinct _APP_ fragments')18,19. Hornsten _et al_.15 were unable to accomplish
rescue using the C-terminus alone, indicating, together with the other rescue experiments, that the N-terminus is the primary functional domain of APL-1 in the worm. DROSOPHILA APPL is the
APP homolog in _Drosophila_, and, like the worm homolog, does not contain the Aβ sequence (Figure 2). _Appl_-deficient flies are viable and fertile with no obvious phenotypes; however, these
flies do have subtle behavioral defects, such as fast phototaxis impairment. This defect can be partially rescued by transgenic expression of either wild-type fly APPL or human APP, which
demonstrates the functional conservation of APP between different species20. Subsequent loss and gain-of-function studies revealed more specific functions of APPL. APPL plays an important
role in axonal transport, since either _Appl_ deletion or its overexpression will cause axonal transport defects similar to kinesin and dynein mutants21. APPL is required for the development
of neuromuscular junctions (NMJs), since _Appl_ deletion leads to decreased NMJ bouton number, whereas _Appl_ overexpression dramatically increases bouton number22. This activity can be
explained by the formation of a potential complex including APPL, the APPL-binding protein dX11/Mint, and the cell adhesion molecule FasII, which together can regulate synapse formation23.
APPL also plays a role in the development of the _Drosophila_ peripheral nervous system (PNS) as both _Appl_ deletion and _Appl_ RNAi leads to the loss of scutellar mechano-sensory organs
(MSOs). Overexpression of human APP homologs in the fly leads to MSO Notch gain-of-function phenotypes, possibly through the interaction of the APP YENPTY motif with Numb/Pon and Dab24. APPL
promotes post-developmental neurite arborization, which may be involved in brain injury response and repair. This function was revealed in a study showing that overexpression of human APP
and APPL can induce post-developmental axonal arborization. This phenotype is dependent on the APP C-terminal domain and its interaction with the Abelson tyrosine kinase. Interestingly,
similar to mammals, APPL is strongly upregulated after traumatic brain injury (TBI) in flies upon which APPL-deficient flies suffer a higher mortality rate compared to controls25. Overall,
_Appl_ loss and gain-of-function studies in _Drosophila_ models offer us important insights into the physiological functions of APP and some of these findings have already been validated in
mouse models. ZEBRAFISH Zebrafish have two APP homologs, APPa and APPb. APPa is a 738-aa protein and contains the Kunitz protease inhibitor domain in the N-terminus, which resembles the
human APP770 isoform. APPb is a 694-aa protein, which is more similar to the human APP695 isoform26. The study of a transgenic zebrafish model expressing GFP under the _appb_ promoter
control reveals that the GFP is mainly expressed in subregions of brain, spinal cord and the developing vasculature of zebrafish embryos. The GFP expression level increases during
development, and in adult transgenic zebrafish, GFP was abundantly expressed in the brain27. Though _appa_ knockdown in zebrafish does not show any significant defects in development, _appb_
knockdown animals show several early developmental deficits, including a shortened body axis, a short and curly tail and mild synopthalmia, which can be explained by defective
convergent-extension movements26. These defects can be largely rescued by human APP695 mRNA, partially rescued by human APPsα mRNA, but cannot be rescued by APP bearing the Swedish mutation,
which leads to FAD in humans. It is interesting that this study indicates APP FAD mutations may not only contribute to AD pathogenesis during aging but also affect early development due to
APP loss-of-function. Though there are not many APP physiological function studies employing zebrafish, as a well-established model organism for vertebrate development, we expect to see more
novel findings from the zebrafish system in the near future. APP MODELS IN MICE To understand the physiological functions of APP and its family members, individual and combined mouse
mutants of _APP_, _APLP1_ and _APLP2_ have been generated. For _APP_, there are two knockout (KO) alleles28,29, one hypomorphic allele30, two conditional alleles31,32, four defined
truncation KI alleles18,19,33 and two C-terminal point mutation KI alleles reported34,35. APP, APLP1 AND APLP2 SINGLE KO MICE _APP_ KO mice are viable and fertile but suffer from various
defects and abnormalities, such as reduced body and brain weight, decreased size of forebrain commissures, increased frequency and severity of corpus callosum agenesis28,30, reactive
gliosis28, increased sensitivity to kainate-induced seizures36, increased copper and iron levels in the cerebral cortex and liver37,38, upregulated cholesterol and sphingomyelin levels in
the brain39 and decreased plasma glucose levels as well as hyperinsulinemia40. Behavioral studies revealed that _APP_ KO mice have decreased locomotor activity, reduced forelimb grip
strength and deficits in the Morris water maze task as well as passive avoidance learning41,42,43. Electrophysiology studies show that these mice are also defective in long-term potentiation
(LTP), which is associated with attenuated paired-pulse depression of GABA-mediated inhibitory postsynaptic currents44. Recently, APP was found to be involved in the regulation of L-type
calcium channel function45. Loss of APP leads to increased levels of the L-type calcium channel subunit Cav1.2 in the striatum, which is associated with reduced GABAergic paired pulse
inhibition and increased GABAergic post-tetanic potentiation45. However, these behavioral and electrophysiological impairments may not be caused by a gross loss of neurons or synapses
because unbiased stereological quantification failed to detect any significant neuronal or synapse loss in aged _APP_ KO mice42. Also, attempts to examine dendritic spine density in _APP_ KO
mice have revealed mixed results. Bittner _et al_.46 reported that _APP_ deletion leads to a twofold higher density of spines in apical dendrites of layer III and layer V neurons of the
somatosensory cortex at 4-6 months of age, whereas Lee _et al_.47 found a significant decrease in spine density in cortical layers II/III and hippocampal CA1 pyramidal neurons in 1-year-old
_APP_ KO mice compared with wild-type controls. This discrepancy may be due to a region-specific effect, a contribution of aging and/or an adaptive mechanism. Nevertheless, additional study
is warranted in this area. The phenotypes of _APLP2_ and _APLP1_ single KO mice are also mild6,7. _APLP2_ KO mice are viable, fertile and normal in size. No abnormalities were detected in
tissues examined by histochemical analysis. _APLP2_ KO mice showed no difference from controls in forelimb strength as well as learning and memory performance in the Morris water maze and
conditioned avoidance tasks. Similarly, _APLP1_ single KO mice are viable, fertile, but have a postnatal growth deficit. However, these mice show normal locomotor activity and forelimb grip
strength6,7. MICE WITH COMBINED KO OF APP FAMILY MEMBERS Though the phenotypes of _APP_ family member single KOs are all relatively subtle, _APP/APLP2_ or _APLP1/APLP2_ double KO (dKO) mice
exhibit early postnatal lethality. This strongly indicates that functional redundancy exists among APP family members. Intriguingly, the _APP/APLP1_ dKO mice are viable7, suggesting a unique
property specific to _APLP2_ that is required when _APP_ or _APLP1_ is absent. In the PNS, APP and APLP2 play an essential yet redundant role in the formation and function of neuromuscular
synapses. _APP/APLP2_ dKO mice have poorly formed NMJs, with excessive nerve growth and reduced apposition of pre- and postsynaptic elements. The number of synaptic vesicles at the
presynaptic terminals is also reduced in parallel with impaired neurotransmitter release48. Moreover, study of the ultrastructure of the submandibular synapse also revealed significant
reductions in synaptic vesicle density, active zone size and docked vesicle number per active zone in _APP/APLP2_ dKO mice49. Mice lacking all three APP family members survive through
embryonic development but die shortly after birth50. Cranial abnormalities were seen in 81% of the triple KO mice while 68% showed cortical dysplasia resembling human type II lissencephaly.
Additionally, the mutant mice showed partial loss of cortical Cajal Retzius cells at E18.5, suggesting a role for APP family members in neuronal migration and/or adhesion50. A very similar
cortical dysplasia phenotype was also observed in mice that are double deficient for Fe65 and Fe65L1, a family of proteins that have been well known to interact with the APP cytoplasmic
domain51, suggesting an APP/Fe65 signaling complex mediating neuronal migration. The role of APP in neuronal migration/adhesion was further supported by the finding that acute knockdown of
_APP_ in rats using shRNA electroporation resulted in defective cortical plate entry and migration of neuronal precursor cells in the embryonic cortex52. APP CONDITIONAL KO To counter
postnatal lethality and further dissect APP physiological functions, two conditional alleles of _APP_ have been generated31,32. Tissue-specific deletion of _APP_ in either neurons or muscle
in the _APLP2_ KO background resulted in neuromuscular defects similar to phenotypes seen in _APP/APLP2_ dKO mice, suggesting that APP expression is required in both motor neurons and muscle
cells for the proper formation and function of neuromuscular synapses31. The authors propose that the trans-synaptic homotypic interaction of APP promotes neuromuscular synapse development.
This was further supported by the synaptogenic properties of APP revealed by hippocampal and HEK293 mixed culture experiments31. Interestingly, muscle APP expression is required for the
proper pre-synaptic localization of CHT (high-affinity choline transporter) and synaptic transmission, suggesting that trans-synaptic APP interaction is necessary for recruiting the
presynaptic APP/CHT complex. Consistent with the role of APP in synaptic plasticity and synaptic function, a recent study that attempted to identify APP-interacting proteins using a tagged
APP transgenic approach revealed a list of synaptic proteins including bassoon and neurexin53. Whether the trans-synaptic interaction of APP is involved in the recruitment of these
presynaptic molecules and how it coordinates with other synaptic adhesion complexes such as neuroligin/neurexin are exciting questions awaiting further investigation. KNOCK-IN MODELS
EXPRESSING DISTINCT APP FRAGMENTS AND SINGLE AMINO ACID MUTATIONS As mentioned earlier, sequential proteolytic processing of APP generates three fragments: a large soluble ectodomain (APPsα
or APPsβ), a short peptide such as P3 or Aβ and the AICD. To investigate physiological functions exerted by these different APP fragments, two research groups have generated four different
gene-targeted _APP_ mutant KI alleles, in which Aβ and AICD regions, are missing or mutated (Figure 3). In order to determine the role of APPsα and APP C-terminal domain, the Müller group
created two KI mouse lines33. The first expresses APPsα (_APPsα_-ki) and the second expresses a C-terminal truncation of APP removing the highly conserved YENPTY motif (_APPΔCT15-ki_) by
deletion of the last 15 aa residues (Figure 3B and 3C). Both of the lines have an overall reduction of mutant APP expression in both mRNA and protein levels, which is attributed to an effect
of SV40 polyA cassette. In APPΔCT15-ki brains, even with a reduction of mutant APP expression of more than 50%, there is a relatively higher percentage of cell surface APP and less Aβ
production compared to controls. This is consistent with the previous _in vitro_ studies showing that the C-terminal region was involved in APP endocytosis and processing54,55. In comparison
with _APP_ KO mice, the _APPsα_-ki and _APPΔCT15_-ki mice appear to rescue a variety of phenotypes observed in _APP_ KO mice. For instance, the body and brain weight loss phenotype of _APP_
KO animals at different ages is largely rescued. Behaviorally, these KI mice do not exhibit any defects in grip strength test and water maze test. Electrophysiology experiments showed that
LTP deficits observed in _APP_ KO mice at 9-12 months were also corrected in both KI lines. Taken together, these results suggest that, similar to what was observed with APL-1 in the _C.
elegans_ model15, the C-terminal region of APP may not be required for certain physiological functions of APP. To further dissect the function of different APP domains, our research group
has also generated two distinct KI lines. The first model expresses APPsβ with a FLAG tag at the C-terminus named _APPsβ_-ki18 (Figure 3D). The second model, named _APP/hAβ/mutC_19,
expresses APP with a humanized Aβ region, including three FAD mutations (Swedish, Arctic and London) to facilitate Aβ production, and a frameshift mutation in the C-terminus that deletes the
last 39 aa residues of APP (Figure 3E). Similar to the _APPsα_-ki and _APPΔCT15_-ki lines, _APPsβ_-ki and _APP/hAβ/mutC_-ki mice do not show any obvious growth or anatomical deficits. As
mentioned above, when APP KO mice are bred to the APLP2 KO background, _APP/APLP2_ dKO pups exhibit postnatal lethality. Similarly, when crossed with _APLP2_ KO mice, neither the _APPsβ_-ki
nor _APP/hAβ/mutC_-ki allele is able to rescue the lethality. Moreover, the deficits of NMJs observed in _APP/APLP2_ dKO pups, including aberrant apposition of presynaptic proteins with
postsynaptic endplates and diffuse synaptic band width, also exist in _APPsβ__ki/ki__/APLP2__−/−_ and _APP/hAβ/mutC__ki/ki__/APLP2__−/−_ pups. Considering that neither of the KI alleles
contains a functional APP C-terminal domain, our data clearly demonstrate a critical and indispensable role of the conserved C-terminal region of APP in early development. Though our KI
alleles are not able to rescue lethality, they do rescue certain gene expression changes caused by _APP_ KO. In _APP/APLP2_ dKO mice, the mRNA level of transthyretin (TTR) and Klotho is
reduced18. However, the expression of TTR and Klotho is normal in the _APPsβ__ki/−__/APLP2__−/−_ pups. This finding indicates that soluble APP can modulate the expression of certain genes
through a currently unknown pathway. TTR has been shown to have Aβ binding ability56,57,58 and Klotho has been extensively implicated in the aging process59,60,61. The enhancement of TTR and
Klotho expression by APPsβ offers the fascinating possibility of a self-protective mechanism in the APP processing pathway to counter the production and toxicity of Aβ during aging. When we
crossed the _APP/hAβ/mutC_ KI line onto the _PS1_ M146V (a _PS1_ FAD mutation) KI background to accelerate Aβ deposition, we found massive plaques in the hippocampus and cortex in mice over
1 year of age. This implies that amyloid pathogenesis could progress independently without the conserved C-terminal sequence19. The essential role of the highly conserved C-terminal region
of APP in early development revealed by _APPsβ__ki/__ki__/APLP2__−/−_ and _APP/hAβ/mutC__ki/ki__/APLP2__−/−_ mice was further confirmed by a recent study of Barbagallo _et al_.34. They
generated a KI mouse with a single amino acid mutation at Tyr682 (Y682G), and showed that, in the absence of _APLP2_ functional redundancy, the Y682G mutation resulted in postnatal lethality
and neuromuscular synapse defects similar to _APP/APLP2_-double-deficient mice34,62. The importance of this Tyr682 site is strengthened by a parallel study, which found that a distinct
mutation at a different Thr site, Thr668 (T668A), had no effect on both animal survival and formation of neuromuscular synapses34,35. Tyr682 is an essential residue of the Y682ENPTY687
motif, which is the docking site for numerous cytosolic proteins63. Some APP interactors require phosphorylated Tyr68264,65,66,67, whereas others require non-phosphorylated Tyr68268. This
leads to questions in regards to how the selective phosphorylation state of Tyr682 is regulated and their corresponding functional consequences. In summary, these different KI lines
expressing different segments of _APP_ have revealed that APP is involved in multiple functions. Some of these functions are carried out by APP N-terminal ectodomain alone without the APP
C-terminal intracellular domain. However, the AICD, especially the YENPTY motif, which is well conserved from worms to humans, plays a critical role in developmental regulation in mice.
Finally, Aβ aggregation and amyloid deposition can proceed without the APP C-terminus, implying that APP physiological functions could be genetically uncoupled with AD pathogenesis. APP FAD
MUTANT KI MODELS Many APP transgenic models expressing APP with FAD mutations under the control of a robust exogenous promoter have been generated and have provided us with valuable
knowledge about AD pathogenesis, especially Aβ plaque deposition. However, in order to accelerate plaque development, transgenic mice usually overexpress mutant APP at a very high level. For
instance, a commonly used transgenic line, Tg2576, overexpresses APP to almost sixfold using the hamster prion promoter69. Considering the crucial and various functions of APP in nervous
system development, this provokes the question of whether the memory and behavioral defects observed in AD-transgenic mice are due to Aβ pathology or simply APP overexpression. To avoid the
complications of overexpression and build a more physiologically relevant model, several labs generated different FAD _APP_ KI lines by introducing human FAD mutations and/or humanized Aβ to
the endogenous mouse _APP_ gene. Compared to traditional transgenic models, _APP_ KI mice exhibit a number of advantages. Since the KI allele is under the native _APP_ promoter control, the
amount of APP expression remains at physiological level. For the same reason, the temporal and spatial expression patterns of mutant APP are not affected. In contrast to the transgenic
lines in which the wild-type mouse _APP_ gene is still intact, where mouse APP and Aβ may complicate the phenotype, the KI models have mouse Aβ replaced with the human Aβ sequence. One FAD
KI model was developed by Reaume _et al_.,70 who generated _APP__NLh/NLh_ mice bearing the Swedish FAD mutation (K670N/M671L) with a humanized Aβ sequence expressed at endogenous levels.
Amyloidogenic β-secretase cleavage of this mutant APP is accurate and also enhanced, while the non-amyloidogenic processing is repressed. Human Aβ production is significantly increased in
these mice, a ninefold increase compared to normal human brain tissue70. Though this _APP_ KI model overproduces human Aβ40 and Aβ42, Aβ plaque deposition was not detectable71. Later, the
_APP__NLh/NLh_ strain was crossed with the _PS-1__P264L/P264L_ line (a PS1 FAD mutant KI line) to generate the _APP__NLh/NLh__/PS-1__P264L/P264L_ double KI (DKI) model71.
_APP__NLh/NLh__/PS-1__P264L/P264L_ DKI animals exhibit elevated levels of Aβ42 and initial Aβ deposition begins at 6 months of age. Aβ deposition at 6 months was predominately seen in the
cerebral cortex with the majority of deposits found in the frontal cortex, followed by the parietal cortex71. Microarray analysis was performed to compare the gene expression profile changes
between _APP__NLh/NLh__/PS-1__P264L/P264L_ animals and two transgenic models, Tg2576/PS-1P264L/+ and Tg2576/PS-1P264L/P264L, both before and after detectable Aβ deposition72. Gene
expression difference was analyzed in _APP__NLh/NLh__/PS-1__P264L/P264L_ DKI animals and their wild-type littermates at 2 months of age, when plaques in DKI brains are absent, and at 18
months of age, when the DKI cortical plaque load is 3.2%. Similar experiments were also performed to compare expression changes in Tg2576/PS-1P264L/+, Tg2576/PS-1P264L/P264L and
non-transgenic controls (non-Tg/PS-1+/+) at 2 months of age, when Tg2576/PS-1P264L/+ had no plaques and the cortical plaque load of Tg2576/PS-1P264L/P264L is 0.09%, and at 12 months of age,
when the plaque load of Tg2576/PS-1P264L/+ and Tg2576/PS-1P264L/P264Lwas 10.6% and 25.8%, respectively. In young animals before Aβ deposition, the DKI animals do not share any altered gene
expression when compared with the two transgenic models, which is possibly due to APP overexpression in transgenic models and/or a genetic background difference. However, there are a number
of common changes found in the three models with Aβ deposition and many of these changes are also found in human AD brains72. This finding strongly supports this DKI animal model as a
valuable AD research tool. Chang _et al_.73 performed electrophysiological and behavioral studies with _APP__NLh/NLh__/PS-1__P264L/P264L_ animals. DKI animals showed an age-related decrease
of AMPA receptor-mediated evoked currents and spontaneous miniature currents. The downregulation of synaptic AMPA receptor is confirmed by electron microscopic study. DKI animals also
exhibit age-related deficits in bidirectional plasticity seen during analysis of LTP and LTD in these mice as well as memory flexibility when examined by Morris water maze. Additionally,
long-lasting and selective impairment in adult hippocampal neurogenesis is found in 9-18 months old _APP__NLh/NLh__/PS-1__P264L/P264L_ DKI animals, while the olfactory bulb neurogenic system
is not affected. The number of MCM2-positive neural stem and progenitor cells is decreased by 3-fold compared to controls and doublecortin-positive neuroblasts are decreased by twofold as
well. This neurogenesis impairment phenotype may be explained by Aβ deposition-induced microglia activation and neuroinflammation74. Another similar FAD mutant _APP_ KI line was generated by
Kohler _et al_.75, which includes the humanized Aβ sequence containing both Swedish (K670N/M671L) and London (V717F) mutations. _APP__SL/SL_ single KI animals do not show any Aβ deposits.
Once this strain was crossed to an FAD _PS1_ transgenic strain (_PS1__M146L-tg_), then Aβ deposition occurred in a similar pattern to APP transgenic animals. However, the Aβ deposits in
_APP__SL/SL__/PS1__M146L-tg_ mice began much later and developed more slowly than APP transgenic animals, with no apparent neurodegeneration75. Further characterization revealed that
_APP__SL/SL__/PS1__M146L-tg_ mice have increased levels of Aβ peptides at 10 months of age and amyloid plaques at 14 months of age. Microdialysis showed that the hippocampal extracellular
acetylcholine (ACh) levels of 15–27 months old _APP__SL/SL__/PS1__M146L-tg_ mice are slightly but significantly reduced. However, there were no major changes found in stimulated ACh release
and overall central cholinergic function was not affected76. The KI model attempting to genetically mimic human FAD to the greatest extent was generated by Kawasumi _et al_.77. This strain
contains an _APP_ _V642I_ mutant KI allele without modifying the mouse Aβ sequence, which was then studied in a heterozygous state. _APP V642I/+_ animals appear normal compared to their
wild-type littermates up to 2.5 years of age. However, their long-term spatial memory is significantly impaired and acquisition of spatial memory is slightly affected. Histologically, the
ratio of Aβ42 (43)/Aβ40 is greatly increased in mutants, but there are no plaques or neurofibrillary tangles in the mutant brains. All the mouse models mentioned above are summarized in
Table 1. CONCLUDING REMARKS _In vivo_ models of APP family members provide us with a considerable amount of insight into the physiological function of APP. It is now well known that APP and
its homologs are important for early developmental events, such as mediating cell adhesion, cell migration and synaptogenesis. These models also highlight several intriguing directions for
future study. First, full-length APP is vital for neuromuscular synapse assembly and possibly central synaptogenesis. Second, APPsβ, independent of full-length APP, can regulate the
expression of certain genes like _TTR_ and _Klotho_. The receptor mediating such an event remains to be identified. Third, _APP/APLP2_ germline dKO animals are early postnatal lethal,
whereas animals with a conditional deletion of _APP_ only in the central nervous system (CNS) on an _APLP2_-null background are viable. This finding strongly suggests that APP has vital, yet
undiscovered, functions outside the CNS. Finally, it is known that APP is strongly upregulated in brains that have experienced TBI in both human patients and in mice. TBI-induced APP
upregulation may contribute to higher Aβ production and increased risk of AD in later life. Since studies in _Drosophila_ demonstrated that the mortality rate of _Appl_-deficient flies is
higher than controls after TBI treatment, it will be extremely interesting to know how _APP/APLP2_ conditional KO animals survive TBI and to further dissect the functional role of APP in
post-TBI brain repair. All of the FAD _APP_ KI AD mouse models share mild phenotypes and pathological manifestations. To generate plaque pathology, both an APP KI mutation and an FAD _PS1_
allele are needed. Considering that a heterozygous _APP_ FAD mutation is able to trigger early onset AD in humans, the effects of these mutations in mice are considerably weaker. This
difference may be due to the short-life span of mice and the intrinsic physiological differences between humans and mice. Nevertheless, these _APP_ KI models provide us with an alternative
platform and a more physiologically relevant disease model to study human AD. More comprehensive and sensitive assays will be required for better characterizing _APP_ KI mice and
consequently AD pathogenesis in the future. REFERENCES * Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC . Characterization and chromosomal localization of a cDNA encoding brain
amyloid of Alzheimer's disease. _Science_ 1987; 235:877–880. Article CAS PubMed Google Scholar * Kang J, Lemaire HG, Unterbeck A, _et al_. The precursor of Alzheimer's disease
amyloid A4 protein resembles a cell-surface receptor. _Nature_ 1987; 325:733–736. Article CAS PubMed Google Scholar * Tanzi RE, Gusella JF, Watkins PC, _et al_. Amyloid beta protein
gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. _Science_ 1987; 235:880–884. Article CAS PubMed Google Scholar * Chow VW, Mattson MP, Wong PC, Gleichmann M .
An overview of APP processing enzymes and products. _Neuromolecular Med_ 2010; 12:1–12. Article CAS PubMed PubMed Central Google Scholar * Weidemann A, Eggert S, Reinhard FB, _et al_.
A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. _Biochemistry_ 2002; 41:2825–2835. Article
CAS PubMed Google Scholar * von Koch CS, Zheng H, Chen H, _et al_. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. _Neurobiol Aging_ 1997;
18:661–669. Article CAS PubMed Google Scholar * Heber S, Herms J, Gajic V, _et al_. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid
precursor protein family members. _J Neurosci_ 2000; 20:7951–7963. Article CAS PubMed PubMed Central Google Scholar * Armstrong RA, Lantos PL, Cairns NJ . What determines the molecular
composition of abnormal protein aggregates in neurodegenerative disease? _Neuropathology_ 2008; 28:351–365. Article PubMed Google Scholar * Korczyn AD . The amyloid cascade hypothesis.
_Alzheimers Dement_ 2008; 4:176–178. Article CAS PubMed Google Scholar * Zheng H, Koo EH . The amyloid precursor protein: beyond amyloid. _Mol Neurodegener_ 2006; 1:5. Article PubMed
PubMed Central Google Scholar * Daigle I, Li C . _apl-1_, a _Caenorhabditis elegans_ gene encoding a protein related to the human beta-amyloid protein precursor. _Proc Natl Acad Sci USA_
1993; 90:12045–12049. Article CAS PubMed PubMed Central Google Scholar * Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS . Expression of a ubiquitous, cross-reactive
homologue of the mouse beta-amyloid precursor protein (APP). _J Biol Chem_ 1994; 269:2637–2644. CAS PubMed Google Scholar * Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F
. Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. _Proc Natl Acad Sci USA_ 1992; 89:10758–10762.
Article CAS PubMed PubMed Central Google Scholar * Wasco W, Gurubhagavatula S, Paradis MD, _et al_. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's
associated amyloid beta protein precursor. _Nat Genet_ 1993; 5:95–100. Article CAS PubMed Google Scholar * Hornsten A, Lieberthal J, Fadia S, _et al_. APL-1, a _Caenorhabditis elegans_
protein related to the human beta-amyloid precursor protein, is essential for viability. _Proc Natl Acad Sci USA_ 2007; 104:1971–1976. Epub 2007 Jan 1931. Article CAS PubMed PubMed
Central Google Scholar * Wiese M, Antebi A, Zheng H . Intracellular trafficking and synaptic function of APL-1 in _Caenorhabditis elegans_. _PLoS ONE_ 2010; 5:e 12790. Article Google
Scholar * Hada K, Asahina M, Hasegawa H, Kanaho Y, Slack FJ, Niwa R . The nuclear receptor gene _nhr-25_ plays multiple roles in the _Caenorhabditis elegans_ heterochronic gene network to
control the larva-to-adult transition. _Dev Biol_ 2010; 344:1100–1109. Article CAS PubMed PubMed Central Google Scholar * Li H, Wang B, Wang Z, _et al_. Soluble amyloid precursor
protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. _Proc Natl Acad Sci USA_ 2010; 107:17362–17367. Article CAS PubMed PubMed
Central Google Scholar * Li H, Wang Z, Wang B, _et al_. Genetic dissection of the amyloid precursor protein in developmental function and amyloid pathogenesis. _J Biol Chem_ 2010;
285:30598–30605. Article CAS PubMed PubMed Central Google Scholar * Luo L, Tully T, White K . Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl
gene. _Neuron_ 1992; 9:595–605. Article CAS PubMed Google Scholar * Gunawardena S, Goldstein LS . Disruption of axonal transport and neuronal viability by amyloid precursor protein
mutations in _Drosophila_. _Neuron_ 2001; 32:389–401. Article CAS PubMed Google Scholar * Torroja L, Packard M, Gorczyca M, White K, Budnik V . The Drosophila beta-amyloid precursor
protein homolog promotes synapse differentiation at the neuromuscular junction. _J Neurosci_ 1999; 19:7793–7803. Article CAS PubMed PubMed Central Google Scholar * Ashley J, Packard M,
Ataman B, Budnik V . Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. _J Neurosci_ 2005; 25:5943–5955. Article CAS PubMed
PubMed Central Google Scholar * Merdes G, Soba P, Loewer A, Bilic MV, Beyreuther K, Paro R . Interference of human and _Drosophila_ APP and APP-like proteins with PNS development in
_Drosophila_. _EMBO J_ 2004; 23:4082–4095. Article CAS PubMed PubMed Central Google Scholar * Leyssen M, Ayaz D, Hebert SS, Reeve S, De Strooper B, Hassan BA . Amyloid precursor protein
promotes post-developmental neurite arborization in the _Drosophila_ brain. _EMBO J_ 2005; 24:2944–2955. Article CAS PubMed PubMed Central Google Scholar * Joshi P, Liang JO, DiMonte
K, Sullivan J, Pimplikar SW . Amyloid precursor protein is required for convergent-extension movements during Zebrafish development. _Dev Biol_ 2009; 335:1–11. Article CAS PubMed Google
Scholar * Lee JA, Cole GJ . Generation of transgenic zebrafish expressing green fluorescent protein under control of zebrafish amyloid precursor protein gene regulatory elements.
_Zebrafish_ 2007; 4:277–286. Article CAS PubMed Google Scholar * Zheng H, Jiang M, Trumbauer ME, _et al_. beta-Amyloid precursor protein-deficient mice show reactive gliosis and
decreased locomotor activity. _Cell_ 1995; 81:525–531. Article CAS PubMed Google Scholar * Li ZW, Stark G, Gotz J, _et al_. Generation of mice with a 200-kb amyloid precursor protein
gene deletion by Cre recombinase-mediated site-specific recombination in embryonic stem cells. _Proc Natl Acad Sci USA_ 1996; 93:6158–6162. Article CAS PubMed PubMed Central Google
Scholar * Muller U, Cristina N, Li ZW, _et al_. Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. _Cell_ 1994; 79:755–765. Article
CAS PubMed Google Scholar * Wang Z, Wang B, Yang L, _et al_. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. _J
Neurosci_ 2009; 29:10788–10801. Article CAS PubMed PubMed Central Google Scholar * Mallm JP, Tschape JA, Hick M, Filippov MA, Muller UC . Generation of conditional null alleles for
_APP_ and _APLP2_. _Genesis_ 2010; 48:200–206. CAS PubMed Google Scholar * Ring S, Weyer SW, Kilian SB, _et al_. The secreted beta-amyloid precursor protein ectodomain APPs alpha is
sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. _J Neurosci_ 2007; 27:7817–7826. Article CAS PubMed PubMed Central Google
Scholar * Barbagallo AP, Weldon R, Tamayev R, _et al_. Tyr(682) in the intracellular domain of APP regulates amyloidogenic APP processing _in vivo_. _PLoS One_ 2010; 5:e 15503. Article
Google Scholar * Barbagallo AP, Wang Z, Zheng H, D'Adamio L . The intracellular threonine of amyloid precursor protein that is essential for docking of pin1 is despensable for
developmental function. _PLoS One_ 2011; 6:e 18006. Article CAS Google Scholar * Steinbach JP, Muller U, Leist M, Li ZW, Nicotera P, Aguzzi A . Hypersensitivity to seizures in
beta-amyloid precursor protein deficient mice. _Cell Death Differ_ 1998; 5:858–866. Article CAS PubMed Google Scholar * White AR, Multhaup G, Maher F, _et al_. The Alzheimer's
disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. _J Neurosci_ 1999; 19:9170–9179. Article CAS PubMed PubMed Central
Google Scholar * Duce JA, Tsatsanis A, Cater MA, _et al_. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. _Cell_ 2010;
142:857–867. Article CAS PubMed PubMed Central Google Scholar * Grimm MO, Grimm HS, Patzold AJ, _et al_. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and
presenilin. _Nat Cell Biol_ 2005; 7:1118–1123. Article CAS PubMed Google Scholar * Needham BE, Wlodek ME, Ciccotosto GD, _et al_. Identification of the Alzheimer's disease amyloid
precursor protein (APP) and its homologue APLP2 as essential modulators of glucose and insulin homeostasis and growth. _J Pathol_ 2008; 215:155–163. Article CAS PubMed Google Scholar *
Dawson GR, Seabrook GR, Zheng H, _et al_. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor
protein. _Neuroscience_ 1999; 90:1–13. Article CAS PubMed Google Scholar * Phinney AL, Calhoun ME, Wolfer DP, Lipp HP, Zheng H, Jucker M . No hippocampal neuron or synaptic bouton loss
in learning-impaired aged beta-amyloid precursor protein-null mice. _Neuroscience_ 1999; 90:1207–1216. Article CAS PubMed Google Scholar * Senechal Y, Kelly PH, Dev KK . Amyloid
precursor protein knockout mice show age-dependent deficits in passive avoidance learning. _Behav Brain Res_ 2008; 186:126–132. Article CAS PubMed Google Scholar * Seabrook GR, Smith DW,
Bowery BJ, _et al_. Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. _Neuropharmacology_ 1999; 38:349–359. Article CAS
PubMed Google Scholar * Yang L, Wang Z, Wang B, Justice NJ, Zheng H . Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic
short-term plasticity. _J Neurosci_ 2009; 29:15660–15668. Article CAS PubMed PubMed Central Google Scholar * Bittner T, Fuhrmann M, Burgold S, _et al_. Gamma-secretase inhibition
reduces spine density _in vivo_ via an amyloid precursor protein-dependent pathway. _J Neurosci_ 2009; 29:10405–10409. Article CAS PubMed PubMed Central Google Scholar * Lee KJ, Moussa
CE, Lee Y, _et al_. Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. _Neuroscience_ 2010; 169:344–356. Article CAS PubMed
Google Scholar * Wang P, Yang G, Mosier DR, _et al_. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. _J Neurosci_ 2005;
25:1219–1225. Article CAS PubMed PubMed Central Google Scholar * Yang G, Gong YD, Gong K, _et al_. Reduced synaptic vesicle density and active zone size in mice lacking amyloid
precursor protein (APP) and APP-like protein 2. _Neurosci Lett_ 2005; 384:66–71. Article CAS PubMed Google Scholar * Herms J, Anliker B, Heber S, _et al_. Cortical dysplasia resembling
human type 2 lissencephaly in mice lacking all three APP family members. _EMBO J_ 2004; 23:4106–4115. Article CAS PubMed PubMed Central Google Scholar * Guenette S, Chang Y, Hiesberger
T, _et al_. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. _EMBO J_ 2006; 25:420–431. Article CAS PubMed PubMed Central Google Scholar
* Young-Pearse TL, Bai J, Chang R, Zheng JB, Loturco JJ, Selkoe DJ . A critical function for amyloid precursor protein in neuronal migration revealed by _in utero_ RNA interference. _J
Neurosci_ 2007; 27:14459–14469. Article CAS PubMed PubMed Central Google Scholar * Norstrom EM, Zhang C, Tanzi R, Sisodia SS . Identification of NEEP21 as a ss-amyloid precursor
protein-interacting protein _in vivo_ that modulates amyloidogenic processing _in vitro_. _J Neurosci_ 2010; 30:15677–15685. Article CAS PubMed PubMed Central Google Scholar * Koo EH,
Squazzo SL, Selkoe DJ, Koo CH . Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. _J Cell Sci_
1996; 109:991–998. CAS PubMed Google Scholar * Perez RG, Soriano S, Hayes JD, _et al_. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the
generation of secreted fragments, including Abeta42. _J Biol Chem_ 1999; 274:18851–18856. Article CAS PubMed Google Scholar * Schwarzman AL, Gregori L, Vitek MP, _et al_. Transthyretin
sequesters amyloid beta protein and prevents amyloid formation. _Proc Natl Acad Sci USA_ 1994; 91:8368–8372. Article CAS PubMed PubMed Central Google Scholar * Choi SH, Leight SN, Lee
VM, _et al_. Accelerated Abeta deposition in APPswe/PS1deltaE9 mice with hemizygous deletions of TTR (transthyretin). _J Neurosci_ 2007; 27:7006–7010. Article CAS PubMed PubMed Central
Google Scholar * Buxbaum JN, Ye Z, Reixach N, _et al_. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of Abeta toxicity. _Proc Natl Acad Sci USA_
2008; 105:2681–2686. Article CAS PubMed PubMed Central Google Scholar * Kuro-o M, Matsumura Y, Aizawa H, _et al_. Mutation of the mouse klotho gene leads to a syndrome resembling
ageing. _Nature_ 1997; 390:45–51. Article CAS PubMed Google Scholar * Kurosu H, Yamamoto M, Clark JD, _et al_. Suppression of aging in mice by the hormone Klotho. _Science_ 2005;
309:1829–1833. Article CAS PubMed PubMed Central Google Scholar * Imura A, Tsuji Y, Murata M, _et al_. alpha-Klotho as a regulator of calcium homeostasis. _Science_ 2007; 316:1615–1618.
Article CAS PubMed Google Scholar * Barbagallo AP, Wang Z, Zheng H, D'Adamio L . A single tyrosine residue in the amyloid precursor protein intracellular domain is essential for
developmental function. _J Biol Chem_ 2011; 286:8717–8721. Article CAS PubMed PubMed Central Google Scholar * King GD, Turner SR . Adaptor protein interactions: modulators of amyloid
precursor protein metabolism and Alzheimer's disease risk? _Exp Neurol_ 2004; 185:208–219. Article CAS PubMed Google Scholar * Zhou D, Noviello C, D'Ambrosio C, Scaloni A,
D'Adamio L . Growth factor receptor-bound protein 2 interaction with the tyrosine-phosphorylated tail of amyloid beta precursor protein is mediated by its Src homology 2 domain. _J Biol
Chem_ 2004; 279:25374–25380. Article CAS PubMed Google Scholar * Russo C, Dolcini V, Salis S, _et al_. Signal transduction through tyrosine-phosphorylated C-terminal fragments of
amyloid precursor protein via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer's disease brain. _J Biol Chem_ 2002; 277:35282–35288. Article
CAS PubMed Google Scholar * Tarr PE, Roncarati R, Pelicci G, Pelicci PG, D'Adamio L . Tyrosine phosphorylation of the beta-amyloid precursor protein cytoplasmic tail promotes
interaction with Shc. _J Biol Chem_ 2002; 277:16798–16804. Article CAS PubMed Google Scholar * Tamayev R, Zhou D, D'Adamio L . The interactome of the amyloid beta precursor protein
family members is shaped by phosphorylation of their intracellular domains. _Mol Neurodegener_ 2009; 4:28. Article PubMed PubMed Central Google Scholar * Zhou D, Zambrano N, Russo T,
D'Adamio L . Phosphorylation of a tyrosine in the amyloid-beta protein precursor intracellular domain inhibits Fe65 binding and signaling. _J Alzheimers Dis_ 2009; 16:301–307. Article
CAS PubMed Google Scholar * Hsiao K, Chapman P, Nilsen S, _et al_. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. _Science_ 1996; 274:99–102.
Article CAS PubMed Google Scholar * Reaume AG, Howland DS, Trusko SP, _et al_. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the
Swedish familial Alzheimer's disease mutations and a 'humanized' Abeta sequence. _J Biol Chem_ 1996; 271:23380–23388. Article CAS PubMed Google Scholar * Flood DG, Reaume
AG, Dorfman KS, _et al_. FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. _Neurobiol Aging_ 2002; 23:335–348. Article CAS PubMed
Google Scholar * Wu ZL, Ciallella JR, Flood DG, O'Kane TM, Bozyczko-Coyne D, Savage MJ . Comparative analysis of cortical gene expression in mouse models of Alzheimer's disease.
_Neurobiol Aging_ 2006; 27:377–386. Article CAS PubMed Google Scholar * Chang EH, Savage MJ, Flood DG, _et al_. AMPA receptor downscaling at the onset of Alzheimer's disease
pathology in double knockin mice. _Proc Natl Acad Sci USA_ 2006; 103:3410–3415. Article CAS PubMed PubMed Central Google Scholar * Zhang C, McNeil E, Dressler L, Siman R . Long-lasting
impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer's disease. _Exp Neurol_ 2007; 204:77–87. Article CAS PubMed
Google Scholar * Kohler C, Ebert U, Baumann K, Schroder H . Alzheimer's disease-like neuropathology of gene-targeted APP-SLxPS1mut mice expressing the amyloid precursor protein at
endogenous levels. _Neurobiol Dis_ 2005; 20:528–540. Article PubMed Google Scholar * Hartmann J, Erb C, Ebert U, _et al_. Central cholinergic functions in human amyloid precursor protein
knock-in/presenilin-1 transgenic mice. _Neuroscience_ 2004; 125:1009–1017. Article CAS PubMed Google Scholar * Kawasumi M, Chiba T, Yamada M, _et al_. Targeted introduction of V642I
mutation in amyloid precursor protein gene causes functional abnormality resembling early stage of Alzheimer's disease in aged mice. _Eur J Neurosci_ 2004; 19:2826–2838. Article PubMed
Google Scholar Download references ACKNOWLEDGEMENTS The authors' work cited in this review was supported by grants from NIH (AG020670, AG032051 and AG033467) and the American Health
and Assistance Foundation (A2008-052). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Huffington Center on Aging, Baylor College of Medicine, One Baylor Plaza, BCM:MS230, Houston, 77030, TX,
USA Qinxi Guo, Zilai Wang, Hongmei Li, Mary Wiese & Hui Zheng * Program in Translational Biology and Molecular Medicine, Houston, 77030, TX, USA Qinxi Guo & Hui Zheng * Department of
Molecular and Human Genetics, Baylor College of Medicine, Houston, 77030, TX, USA Mary Wiese & Hui Zheng Authors * Qinxi Guo View author publications You can also search for this author
inPubMed Google Scholar * Zilai Wang View author publications You can also search for this author inPubMed Google Scholar * Hongmei Li View author publications You can also search for this
author inPubMed Google Scholar * Mary Wiese View author publications You can also search for this author inPubMed Google Scholar * Hui Zheng View author publications You can also search for
this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Hui Zheng. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Guo, Q., Wang, Z.,
Li, H. _et al._ APP physiological and pathophysiological functions: insights from animal models. _Cell Res_ 22, 78–89 (2012). https://doi.org/10.1038/cr.2011.116 Download citation *
Published: 19 July 2011 * Issue Date: January 2012 * DOI: https://doi.org/10.1038/cr.2011.116 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS *
Alzheimer's disease * APP * Aβ * knock-in * animal models







