- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The emergence of resistance to imatinib mediated by mutations in the BCR-ABL has become a major challenge in the treatment of chronic myeloid leukemia (CML). Alternative therapeutic
strategies to override imatinib-resistant CML are urgently needed. In this study, we investigated the effect of AKI603, a novel small molecule inhibitor of Aurora kinase A (AurA) to
overcome resistance mediated by BCR-ABL-T315I mutation. Our results showed that AKI603 exhibited strong anti-proliferative activity in leukemic cells. AKI603 inhibited cell proliferation and
colony formation capacities in imatinib-resistant CML cells by inducing cell cycle arrest with polyploidy accumulation. Surprisingly, inhibition of AurA by AKI603 induced leukemia cell
senescence in both BCR-ABL wild type and T315I mutation cells. Furthermore, the induction of senescence was associated with enhancing reactive oxygen species (ROS) level. Moreover, the
anti-tumor effect of AKI603 was proved in the BALB/c nude mice KBM5-T315I xenograft model. Taken together, our data demonstrate that the small molecule AurA inhibitor AKI603 may be used to
overcome drug resistance induced by BCR-ABL-T315I mutation in CML. SIMILAR CONTENT BEING VIEWED BY OTHERS INTRACELLULAR ANGIOPOIETIN-1 PROMOTES TKI-RESISTANCE VIA ACTIVATION OF JAK/STAT5
PATHWAY IN CHRONIC MYELOID LEUKEMIA Article 17 November 2022 CDK8/19 INHIBITION ATTENUATES G1 ARREST INDUCED BY BCR-ABL ANTAGONISTS AND ACCELERATES DEATH OF CHRONIC MYELOGENOUS LEUKEMIA
CELLS Article Open access 15 February 2025 PRECLINICAL EVALUATION OF A REGIMEN COMBINING CHIDAMIDE AND ABT-199 IN ACUTE MYELOID LEUKEMIA Article Open access 18 September 2020 INTRODUCTION
Chronic myeloid leukemia (CML) is a myeloproliferative disorder that accounts for 15% of adult leukemia1. This disease is characterized by Philadelphia chromosome, the t (9; 22) (q34; q11)
reciprocal translocation, resulting in the expression of a fusion protein BCR-ABL2,3. BCR-ABL plays a central role in the pathogenesis of CML by activating multiple signal pathways4,5,6.
Thus, BCR-ABL has been an important target for CML therapeutics. Although the development of imatinib, a tyrosine kinase inhibitor (TKI) has redefined the management of CML7, the resistance
to imatinib occurs in 20~30% of CML patients and is commonly attributable to point mutations in the BCR-ABL kinase domain8,9. In more than 100 mutations of BCR-ABL, T315I mutation is one of
the most common mutations, and accounts for about 20% of imatinib-resistant cases10. However, T315I mutation confers resistance to multiple TKIs11. Hence, novel compounds or strategies to
override this challenging problem are urgently required for CML treatment. The discovery that AurA was abnormally expressed in malignancies including leukemia prompted the development of
agents that inhibited kinase activity12. Small molecule kinase inhibitors of AurA have attracted a great interest. For example, MK-0457 (VX-680), PHA-739358 and MLN8237 are being
investigated in clinical trials12,13,14,15. MK-0457 effectively inhibited proliferation and growth of multiple tumor cell types including HL-60 cells14,16. Our and other studies suggested
that AurA kinase activity was responsible for chemo-resistance and tumorigenic ability16,17. MLN8237, MK-0457 and related compound VE-465 exhibited promising results against leukemia cells
expressing T315I mutant form of BCR-ABL _in vitro_, _in vivo_ and in patients18,19,20. Those studies indicate that AurA inhibitors exhibit a desirable therapeutic index in resistance of CML
to imatinib caused by the T315I mutation. The aim of this study was to investigate the antineoplastic effects of the novel AurA small molecule inhibitor AKI603 in CML cells. AKI603 inhibited
cell proliferation and induced senescence both in BCR-ABL wild-type and BCR-ABL-T315I mutant CML cells as well as in nude mouse xenograft models. The results revealed that AKI603 could
efficiently overcome imatinib resistance of CML _in vitro_ and _in vivo_. RESULTS AKI603 EXTENSIVELY INHIBITS PROLIFERATION OF LEUKEMIA CELLS We recently reported that AKI603 could inhibit
the proliferation of breast cancer cells21. To evaluate the effect of AKI603 on proliferation of leukemic cells, six leukemic cell lines (AML (acute myelocytic leukemia): U937, HL-60 and
NB4; CML: KBM5 and K562; ALL (acute lymphoblastic leukemia): Jurkat) were treated with various concentrations of AKI603 for 48 h, and the cell proliferation was determined by cell counting
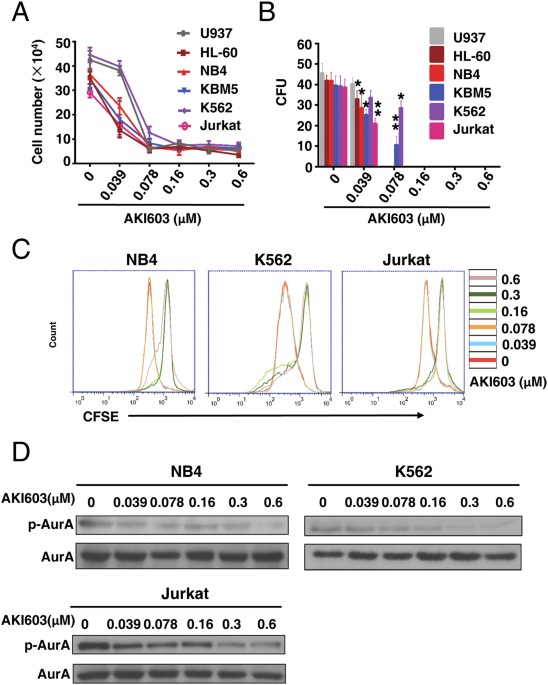
assay and CFSE (carboxyfluorescein diacetate succinimidyl ester) staining assay. As shown in Fig. 1A,C and Supplementary Fig. 1A, all the tested cell lines were inhibited by AKI603
treatment. We performed a colony formation assay in methylcellulose to test the long-term effect of AKI603 on leukemic cells. As shown in Fig. 1B, AKI603 potently decreased the number of
colony units at concentration of 0.16 μM. Next, we assessed the ability of AKI603 to inhibit the kinase activity of AurA in leukemic cells, by testing the phosphorylation of AurA Thr288
(p-AurA). As shown in Fig. 1D, AKI603 significantly inhibited the phosphorylation of AurA in NB4, K562, and Jurkat cell lines in a dose-dependent manner while the level of total AurA protein
was not changed. AKI603 INHIBITS THE PROLIFERATION AND COLONY FORMATION OF IMATINIB RESISTANT CML CELLS K562, KBM5 are sensitive to imatinib treatment, whereas K562/G22 and KBM5-T315I23
cells are resistant to imatinib. To test the effect of AKI603 on proliferation of imatinib resistant CML cells, K562/G and KBM5-T315I cells were incubated for 48 h with different
concentrations of AKI603. As shown in Fig. 2A and Supplementary Fig. 1B, all cell types were inhibited by 0.078 μM AKI603. To further validate this finding, we established a pair of 32D
(32Dcl3) murine cell lines stably expressing wild-type (p210) or T315I-mutant (p210-T315I, T315I) BCR-ABL. 32D-p210 cells were sensitive to imatinib whereas 32D-T315I cells were resistant to
imatinib (Supplementary Fig. S2A). Then we treated these two cells with various concentrations of AKI603 and found that the growth of cells (32D-p210 and 32D-T315I) was significantly
inhibited, with IC50 values of 0.032 μM and 0.039 μM, respectively (Fig. 2A). In colony formation assay, the colony units of imatinib-resistant cells were inhibited in a dose-dependent
manner (Fig. 2B). In addition, we established a pair of BM (bone marrow) cells stably expressing p210 or p210-T315I of BCR-ABL. After 7 days of culture without cytokines, the survival cells
of transfected BM was tested by Western blot for BCR-ABL expression (Supplementary Fig. S2B). Then we tested the sensitivity of those cells to imatinib and found that BM-p210 cells were
sensitive whereas BM-T315I cells were resistant to imatinib (Supplementary Fig. S2C). The proliferation of both cells were significantly inhibited by AKI603 (Fig. 2C) and the colony unit was
completely inhibited at the concentration of 0.3 μM (Fig. 2D). Due to the crucial role of AurA in mitosis, the blocking effect of cell cycle by AKI603 was examined. AKI603 significantly
induced polyploidization in K562, K562/G, 32D-p210 and 32D-T315I cells (Fig. 2E and Supplementary Fig. S3A). These results suggested that the proliferative inhibition induced by AKI603 could
be associated with cell cycle blockage. INHIBITION OF AURA KINASE BY AKI603 RESULTS IN CELLULAR SENESCENCE AKI603 could induce cell cycle arrest with polyploidy accumulation in K562,
K562/G, 32D-p210 and 32D-T315I cells (Fig. 2E), but did not result in obvious apoptosis in 32D-p210 and 32D-T315I cells (Supplementary Fig. S3B). As apoptosis could not account for the
significant reduction in cell number in T315I-mutant or wild-type BCR-ABL cell lines, we predicted that other processes were responsible for reduced cancer cell proliferation in response to
AKI603 treatment. Indeed, after 96 h of treatment, we observed that the cellular size was greatly enlarged (Supplementary Fig. S3C), which is a characteristic of senescence. The
morphological change we observed in leukemia cells was also consistent with the senescence phenotype described in AurA- or AurB (Aurora kinase B)-knockdown cells of solid tumors as well as
leukemia cells24,25. To assess enlarged cellular size induced by AKI603 is associated with senescence, β-galactosidase activity was examined using SA-β-gal (senescence-associated
β-galactosidase) assay. Results showed that β-galactosidase activity was enhanced in drug-treated KMB5, KBM5-T315I, 32D-p210 and 32D-T315I cells (Fig. 3A,B). After 96 h of treatment, the
percentage of SA-β-gal positive cells of KBM5, KBM5-T315I, 32D-p210 and 32D-T315I at 0.3 μM was 68.8% ± 4.4%, 83.6% ± 5.6%, 78.7% ± 5.8% and 81.4% ± 6.2%, respectively (Fig. 3C,D). To
investigate the mechanism of this drug-induced senescence, we tested the expression of p21 in AKI603-treated cells by western blot. In response to drug treatment for 96 h, p21 was induced in
p53 wt (32D-p210 and 32D-T315I)26 and p53-mutant (KBM5 and KBM5-T315I)27 cells (Fig. 3E). However, p53 was induced in 32D-p210 and 32D-T315I cells, but not in KBM5 and KBM5-T315I cells
(Fig. 3E). These results suggested that p21 might be the essential regulator of AKI603-induced senescence independent of p53. INDUCTION SENESCENCE OF AKI603 IS PARTIALLY VIA ENHANCING ROS
GENERATION Our results demonstrated that AKI603 could induce the production of polyploidy (Supplementary Fig. S3A). We and other previously reported that the level of glycolytic metabolism
and the ROS level were significantly increased in the polyploidy cells16,28,29. ROS is considered to be one of the important inducers of senescence30,31, thus we examined whether ROS
production was involved in the occurrence of senescence in KBM5 and KBM5-T315I cells. First, we measured total ROS levels using DCFH (2′,7′-Dichlorofluorescin diacetate) or DHE
(Dihydroethidium) fluorescence assay in AKI603-exposed KBM5 and KBM5-T315I cells. As shown in Fig. 4A, after 0.3 μM AKI603 treatment for 96 h, the levels of total ROS fluorescence increased
significantly in treated groups of KBM5 and KBM5-T315I cells compared to the control groups. Indeed, AurA knockdown by shRNA could increase the ROS level in KBM5 and K562 cells
(Supplementary Fig. S4). Next, we used the ROS scavenger NAC (N-acetyl-L-cysteine) to eliminate cellular ROS28. As shown in Fig. 4B, treatment with NAC blocked AKI603-induced ROS
accumulation in KBM5 cells (AKI603 + NAC vs AKI603: 838.7 ± 85.4 vs 1378.8 ± 127.1, _p_ = 0.007) and KBM5-T315I (AKI603 + NAC vs AKI603: 946.4 ± 92.5 vs 1657.8 ± 105.3, _p_ = 0.0035). Then
we analyzed whether ROS accumulation was responsible for AKI603-induced senescence. As shown in Fig. 4C, treatment with NAC significantly reduced the number of SA-β-gal positive cells
following AKI603 treatment in both KBM5 cells (AKI603 + NAC vs AKI603: 50.8% ± 4.4% vs 68.1% ± 7%, _p_ = 0.0227) and KBM5-T315I (AKI603 + NAC vs AKI603: 60.8% ± 2.6% vs 80.1% ± 4.3%, _p_ =
0.0026). NAC application also partially attenuated AKI603-induced reduction in cell numbers of KBM5 (AKI603 + NAC vs AKI603: 16.38 ± 2.93 × 104 vs 5.86 ± 1.65 × 104, _p_ = 0.0008) and
KBM5-T315I (AKI603 + NAC vs AKI603: 17.19 ± 4.02 × 104 vs 7.94 ± 1.20 × 104, _p_ = 0.0044), and in colony formation units of KBM5 (AKI603 + NAC vs AKI603: 8.0 ± 3.0 vs 0, _p_ = 0.009) and
KBM5-T315I (AKI603 + NAC vs AKI603: 5.7 ± 1.5 vs 0, _p_ = 0.003)(Fig. 4D,E). Taken together, these data indicated that AKI603-induced senescence in CML cells was partially through enhancing
the ROS generation. AKI603 SYNERGISTICALLY ENHANCES THE EFFECTS OF IMATINIB IN BCR-ABL-T315I MUTANT CELLS Anti-tumor therapy with high doses of therapeutic reagents often causes side
effects. Therefore, new strategies with lower dose therapeutic reagents are urgently needed. Synergistic analysis was performed to evaluate the combined effects between AKI603 and imatinib
in BCR-ABL-T315I mutant cells. The experimental setting of AKI603 and imatinib treatment was combined in a fixed ratio (1:1). The results showed that the combination therapy resulted in a
greater growth inhibition of KBM5-T315I and 32D-T315I cells than that was achieved with either AKI603 or imatinib alone (Fig. 5A) and the combination of the two drugs had synergy effect
(Supplementary Tables S1 and S2). To confirm these results, a drug concentration of 0.078 μM was used by a colony formation assay. As shown in Fig. 5B, 0.078 μM imatinib did not obviously
decrease the number of colony formation in KBM5-T515I (34.7 ± 5.5, _p_ = 0.995) and 32D-T315I (40.0 ± 2.0, _p_ = 0.78) compared with the controls (KBM5-T315I: 37.3 ± 4.5; 32D-T315I: 41 ±
5.6). AKI603 could inhibit colony formation (KBM5-T315I: 19.0 ± 5.3, _p_ = 0.0113; 32D-T315I: 5.0 ± 4.0, _p_ = 0.0024) compared with the control groups, whereas the combination (10.7 ± 3.0,
_p_ = 0.0013 in KBM5-T315I; 0, _p_ = 0.000 in 32D-T315I) substantially suppressed colony formation compared with the control groups (Fig. 5B). In addition, the size of the colony formation
was measured. The sizes of colony formation did not obviously be decreased at 0.078 μM of imatinib (KBM5-T315I: 203.0 ± 65.2 μm, _p_ = 0.1193; 32D-T315I: 243.3 ± 118.9 μm, _p_ = 0.997)
compared with the control groups (KBM5-T315I: 258.2 ± 77.5 μm; 32D-T315I: 253.6 ± 117.0 μm) (Fig. 5C). 0.078 μM AKI603 decreased the size of colony formation (KBM5-T315I: 162.0 ± 61.6 μm,
_p_ = 0.0013; 32D-T315I: 136.9 ± 52.7 μm, _p_ = 0.0004), whereas the combination significantly suppressed the colony sizes (KBM5-T315I: 95.2 ± 43.5 μm, _P_ = 0.00016; 32D-T315I: 0, _p_ =
0.000001). These results suggested that the inhibition of AurA by AKI603 provided a potential strategy that overcomes the imatinib resistance in BCR-ABL T315I-mutant cells. AKI603 ABROGATES
THE GROWTH OF XENOGRAFTED KBM5-T315I CELLS IN NUDE MICE We further examined the _in vivo_ effect of AKI603 on KBM5-T315I cells using the nude mouse xenograft model. As shown in Fig. 6A, the
tumor sizes in the AKI603-treated groups (12.5 mg/kg: 699.3 ± 281.2 mm3, _p_ = 0.00005; 25 mg/kg: 493.2 ± 65.5 mm3, _p_ = 0.000014) were smaller than that in the vehicle-treated group
(2877.3 ± 754.7 mm3), indicating that the growth of xenograft tumors was significantly inhibited by AKI603. There was no obvious inhibited effect in imatinib-treated group (2206.5 ± 496.8
mm3, _p_ = 0.222) compared with the vehicle-treated group. The mean weights of tumors in the treated groups were significantly lower than that in the vehicle-treated group (12.5 mg/kg vs
Control: 496.0 ± 145.7 mg vs 1745.2 ± 818.7 mg, _p_ = 0.0019; 25 mg/kg vs Control: 234.7 ± 86.5 mg vs 1745.2 ± 818.7 mg, _p_ = 0.0003) (Fig. 6B,C). The body weights of the mice were stable,
with no significant differences between AKI603 or imatinib-treated and vehicle-treated mice (Fig. 6D). Motor activity and feeding behavior were all normal. The level of phosphorylation of
AurA was significantly decreased in tumor tissues from mice treated with AKI603 than in vehicle treatment, whereas the level of p21 was significantly increased in AKI603-treated groups
compared with the vehicle-treated group (Fig. 6E). H&E staining (Hematoxylin-Eosin staining) of tumor slides revealed that cells in the AKI603-treated KBM5-T315I xenograft exhibited
greatly enlarged cellular size and these cells were often multinucleated (Fig. 6F). Cell proliferation was reduced in AKI603-treated tumors compared to vehicle-treated tumors by Ki-67
staining (Fig. 6F). Together, the results demonstrated that AKI603 inhibited xenografted KBM5-T315I cells _in vivo_. DISCUSSION Although imatinib has revolutionized the management of CML
therapy, drug resistance remains a challenge. Moreover, the prognosis of the patients with imatinib-resistant CML is poor32. Extensive efforts have been made to overcome imatinib-resistance.
Several groups have developed new generation ATP-competitive BCR-ABL kinase inhibitors, such as nilotinib, dasatinib33,34. These modified drugs have stronger affinities for the ATP-binding
site than imatinib, and thus are more effective for imatinib-resistant patients to some extent. Among many mechanisms proposed to explain the resistance to imatinib, mutation of BCR-ABL
(especially T315I mutation) is believed to be the predominant10,32,35. Although these novel inhibitors can effectively inhibit the kinase activity of the mutated BCR-ABL such as E255K and
M351T, they had little effect on T315I mutation36. We aimed to identify effective therapy against leukemic cells carrying the notorious BCR-ABL-T315I mutation. We recently reported a novel
AurA inhibitor AKI603 that against leukemia cells including BCR-ABL wild-type and T315I mutation. Our findings showed that AKI603 was highly effective in inhibiting the proliferation of
leukemic cell lines including CML, AML and ALL (Figs 1 and 2). AKI603 potently inhibited the proliferation and colony formation in CML cells with the T315I mutation (Fig. 2). Interestingly,
we found that inhibition of AurA by AKI603 induced senescence partially via enhancing ROS level (Figs 3 and 4). AKI603 and imatinib synergistically inhibited proliferation of cells with
BCR-ABL-T315I mutation (Fig. 5). Moreover, AKI603 inhibited the growth of KBM5-T315I cells in nude mouse xenografts (Fig. 6). Thus, AKI603 is effective against CML cells, including those
with the T315I mutation _in vitro_ and _in vivo_. Many kinase inhibitors directly bind to the ATP pocket of kinase domain to inhibit the kinase activity37. VX-680, is a potent pan-Aurora
kinase inhibitor and also inhibits BCR-ABL kinase activity38. Different doses of VX-680 have different effects on BCR-ABL. At high doses, VX-680 could inhibit kinase activity of both wild
type and T315I mutation BCR-ABL _in vitro_ and _in vivo_18,39. BCR-ABL oncoprotein could suppress cell apoptosis by interactions with the majority of proteins involved in the oncogenic
pathways40. Some reports showed that low doses of VX-680 and other AurA inhibitor such as MLN8237 inhibited growth of CML cell lines without affecting BCR-ABL activity39,41,42,43. Our
results demonstrated that low doses of AKI603 did not directly inhibit BCR-ABL kinase activity (Fig. 6) and not significantly induce the apoptosis23. These results indicated that inhibition
of AurA kinase but not BCR-ABL activity by AKI603 might contribute to the inhibition of tumor growth. Senescence is an irreversible terminal growth arrest that occurs as a result of cellular
stress or DNA damage. A wide variety of anticancer agents have been shown to induce accelerated senescence in tumor cell44,45. Senescent cells generally display the enlarged and flattened
morphology with increased activity of SA-β-galactosidase. Other features of senescence include increased nucleus, vacuolated cellular morphology, high levels of p53, p21, p27 and p16, the
DNA damage response (DDR), as well as the senescence-associated secretory phenotype (SASP)24. In our study, after treatment with AKI603, CML cells with BCR-ABL-T315I mutation displayed a
serial of senescent morphological and functional changes such as enlarged and flattened morphology, increased levels of p21 protein and enhanced SA-β-gal staining as well as imatinib
sensitive CML cells (Fig. 3). These results suggested that senescence was a mainly terminal outcome of AurA inhibition in some tumor types. The role of p53 in the induction of senescence is
somewhat controversial. Some studies reported that p53 was necessary for senescence and the onset of senescence was associated with increased levels of p5346. Induction of senescence by p53
was associated with the regulation of p53-dependent genes that could participate in cell cycle arrest47. However, other evidences showed that p53 could also function to inhibit senescence
while promoting cell cycle arrest48. In addition, Liu _et al._ recently reported that senescence resulted from inhibition of Aurora kinases was independent of p5324. The role of p53 in
senescence of different cells responded to different stimulations was different. Our data showed that inhibition AurA with AKI603 induced senescence in both p53 wild type and mutant cells.
The level of p21 increased independent of p53 (Fig. 3). This data suggested that p53 was not absolutely required for AKI603-induced senescence. We and others reported that inhibition AurA
kinase by small molecular inhibitors could induce the polyploidization14,16,18. In our study, after treatment with AKI603, the percentage of polyploidy cells was significantly increased. Our
previous study showed that the level of glycolytic metabolism was significantly increased in the polyploidy cells induced by AurA inhibitors16. Recent study reported that polyploidy cells
contained higher levels of ROS due to the higher mitochondrial contents28. ROS played an important role in the cellular senescence30,31. Report also showed that MLN8237 could induce the
generation of ROS49. We found that the level of ROS was higher in AKI603-treated cells than in control cells. Moreover, knockdown of AurA by shRNA could induce the generation of ROS. These
results suggested that AurA inhibited the generation of ROS. Consistent with prior reports24, we observed that decreased ROS production and senescence, increased cell viability and cell
colony formation after prior treatment of NAC. These results demonstrated that induction of senescence by AKI603 was partially via enhancing ROS level at least. Combination treatment with a
BCR-ABL kinase inhibitor and the other kinase inhibitor could possibly prevent resistance caused by mutations in CML50,51. Combination with VX-680 or S1451, a novel and highly specific AurA
inhibitor, and imatinib caused significantly more cell death than imatinib treatment alone. And combination treatment suppressed development of acquired resistance in KCL-22 cells upon
imatinib treatment41. Another AurA inhibitor MLN8237 could inhibit growth of CML cells harboring BCR-ABL-T315I mutation or Ba/F3 cells artificially introduced with T315I mutational BCR-ABL.
Compared to treatment with either agent alone, the percentage of apoptosis was significantly improved in K562 xenograft tumor treated with nilotinib and MLN823742. In our study, compared to
treatment with either agent alone, combination treatment with AKI603 and imatinib also significantly inhibited the growth of BCR-ABL-T315I mutant cells. BCR-ABL is mainly retained within the
cytoplasm, where it interacts with the majority of proteins involved in the oncogenic pathway. Examples include Ras/MAPK pathway, JAK/STAT pathway and PI3K/AKT pathway40,52. One of the most
important factor involved in BCR-ABL/ABL signaling was Myc53,54. ABL could directly activate Myc by phosphorylation on tyrosine 74,54. In addition, we and others demonstrated that AurA
promoted tumorigenesis and cell survival by overexpression and stabilization of Myc55,56,57. Since Myc is the common downstream protein of both BCR-ABL and AurA, we proposed the
combinational effect of AKI603 and imatinib could be caused by inhibition of Myc. Moreover, in this study, we also investigated the effect of AKI603 on KBM5-T315I cells by using the nude
mouse xenograft model. The nude mouse is a useful model for studying the biology and response to therapies of human tumors _in vivo_. In addition, leukemic xenografts implanted
subcutaneously into nude mice have played a significant role in testing antitumor activity and cytotoxics of drugs _in vivo_58,59,60,61. Our results showed that both 12.5 mg/kg and 25 mg/kg
of AKI603 could significantly inhibit the growth of KBM5-T315I xenografts and not affect motor activity and feeding behavior of the nude mice. This indicated that under this administration
method and doses, AKI603 did not cause harm to the mice. Xenografts of human tumors or tumor cell lines in nude mice could reproduce the histology and be used to evaluate tumor response to
therapy _in vivo_62. Our results demonstrated that AKI603 inhibited the growth of KBM5-T315I cells, decreased the activity of AurA and increased the level of p21 in xenografts as well as
that _in vitro_. These results offered a novel perspective on the potential effects of AKI603 in CML to some extent _in vivo_. In summary, we have shown that targeting AurA by AKI603 in
imatinib resistant CML cells induced senescence and inhibited tumor growth _in vitro_ and _in vivo_. Our results provided new approach for treatment of resistant CML caused by BCR-ABL-T315I
mutation with AKI603 in clinical application. MATERIALS AND METHODS CHEMICALS AKI603, designed and synthesized by our lab21, prepared as a 100 mM stock solution in DMSO and stored at −20 °C;
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), DMSO, Trypan Blue, NAC, CFSE, DCFH and DHE were obtained from Sigma; imatinib (Novartis Pharma Schweiz AG, Switzerland).
CELL CULTURE U937, HL-60, Jurkat and K562 were obtained from the American Type Culture Collection (ATCC). NB4 provided by Shanghai Institute of Hematology, Ruijin Hospital. KBM5 and
KBM5-T315I cell lines were gifts from Dr. Peng Huang (Texas MD Anderson Cancer Center, Houston, USA). K562/G gifted from Dr. Wen-Lin Huang (Cancer Center, Sun Yat-sen University). 32D cells
were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). All cells were cultured under recommended conditions. Culture media and FBS were
from Invitrogen (Carlsbad, CA). All cell lines were authenticated by short tandem repeat profiling. PLASMIDS MSCV-BCR-ABL-p210 (p210) and MSCV-BCR-ABL-p210-T315I (T315I) were kindly provided
by Prof. Justus Duyster (University Medical Center Freiburg, Freiburg, Germany). RETROVIRUS PACKAGING AND INFECTION Stocks of retrovirus were generated by transiently co-transfecting 293-T
cell line with p210 or T315I along with the ecotropic packaging plasmids (Addgene) using Lipofectamine 2000 (Invitrogen). Supernatants collected was used to infect the IL-3-dependent murine
progenitor cell line 32D. After 48 h, cells were washed once with PBS and cultured in media devoid of IL-3. IL-3-independent 32D cells were propagated and used for biochemical and biological
experiments. CELL COUNTING ASSAY About 1 × 105 cells per well were plated in 12-well plates (Corning, Costar, USA). Subsequently, cells were treated with different concentrations AKI603 for
48 h. Then, cell count was determined with Trypan blue exclusion assay. MTT ASSAY 1 × 104 cells were seeded in each well of the 96-well plates. Subsequently, cells were treated with
different drugs at different concentrations for indicated time. After different treatment, MTT solution was added to each well and cells were incubated at 37 °C for 4 h. The absorbance was
finally determined at 490 nm using the microplate reader (BioTek, Vermont, USA). CFSE STAINING ASSAY Cell proliferation determination was conducted by CFSE probe. Briefly, cells (5 × 105)
were seeded and stained with CFSE in 6-well plates according to the manufacturer’s protocol. Then, cells were exposed to a series of concentrations of AKI603 for 48 h. CFSE fluorescence was
detected by flow cytometry (Calibur, BD Biosciences, San Diego, CA, USA) and calculated by FlowJo software (Version X; Treestar, Ashland, OR, USA). COLONY FORMATION ASSAY Cells were cultured
in RPMI 1640 medium supplemented with 0.9% methylcellulose and 10% FBS. The colonies were counted and photographed after 10 days using inverted microscope (Olympus, Tokyo, Japan). CELL
CYCLE ANALYSIS The cells were treated with the indicated concentrations of AKI603 for 48 h, collected, and fixed in 70% ice-cold ethanol. After an overnight incubation at 4 °C, the cells
were collected by centrifugation, and resuspended in PBS containing 100 μg/mL RNaseA, 50 μg/mL PI and 0.2% Triton X-100. After PI staining, the quantification of the cell-cycle distribution
was carried out using a FACS Calibur flow cytometer equipped with FlowJo software. ROS ASSAY ROS generation was analyzed using the fluorescent dyes DCFH and DHE. Briefly, after exposure to
AKI603 for 96 h, cells were washed with RPMI 1640 and incubated with one of the fluorescent dyes (10 μM) in RPMI 1640 for 30 min at 37 °C in the dark. Then, cells were washed and analyzed
using a flow cytometer at an excitation/emission wavelength of 488/525 nm and 488/610 nm, respectively. The experiments were repeated a minimum of three times. SA-Β-GAL ASSAY Cells were
subjected to SA-β-gal staining using the senescence-associated SA-β-gal Staining Kit (Cell Signaling Technology, MA, USA), according to the protocol. Briefly, cells were washed twice with
PBS and fixed with the fixative solution for 15 min at room temperature. Then, cells were washed twice with PBS and incubated overnight at 37 °C with the staining solution before observed
using a microscope (Olympus). More than 300 cells per sample were counted to determine the percentage of senescent cells. WESTERN BLOT ANALYSIS Total cellular proteins were isolated with
lysis buffer. Equal amounts of protein were subjected to SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked and then incubated with p-AurA (Thr288), p-ABL
(Tyr245), p53 and p21(Cell Signaling Technology), AurA (Upstate, NewYork, USA), c-ABL and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies. Subsequently, the membranes were
incubated with an HRP-conjugated secondary antibody (Cell Signaling Technology) at room temperature for 1 h and were visualized using enhanced chemiluminescence reagents (Sigma), according
to the manufacturer’s instruction. BONE MARROW TRANSDUCTION AND TRANSFORMATION For BCR-ABL transformation assays, murine BM cells were collected from C57BL/6 mice, and transduced with
retrovirus, as described63. Transduced cells were plated in 6 well cell culture plates for 7 days without cytokines and then used for further experiments. TUMOR GROWTH IN XENOGRAFTS 3 × 107
of KBM5-T315I cells were injected into the flanks of female BALB/c nude mice. Tumors were measured every other day with use of calipers. Tumor volumes were calculated by the following
formula: _A_ × _B_2/2, where _A_ is the greatest diameter and _B_ is the diameter perpendicular to _A_. Other indicators of general health, such as body weight, feeding behavior, and motor
activity, of each animal were also monitored. 6 days after subcutaneous inoculation, when tumors were palpable (50~150 mm3), mice were randomized to receive treatment with vehicle (50%
PEG300 in 50 mM PBS), AKI603 (12.5 mg/kg or 25 mg/kg, injected intraperitoneally every 2 days) and imatinib (50 mg/kg, treated intragastrically every day) for 2 weeks. The animals were then
euthanized, and tumor xenografts were immediately removed, weighed, stored, and fixed. ETHICS STATEMENT The methods were carried out in accordance with the Guide for the Care and Use of
Laboratory Animals (2011). The study protocol was approved by the Animal Ethical and Welfare Committee (AEWC) of Sun Yat-sen University (The Approved No. is IACUC-F3-14-1201). STATISTICAL
ANALYSIS The data are representative of three independent experiments. Data were presented as mean ± SD. Statistical analysis was performed using Prism 6 (GraphPad Software, Inc.) and SPSS
v. 16.0 (SPSS, Inc.). The unpaired two-tailed Student’s t test was used to perform statistical comparison between two groups. The ANOVA test was used for multiple comparisons. The
Kruskal-Wallis test, followed by Dunn’s Multiple Comparison test, was used to perform statistical comparison for colonies size distribution. _p_ < 0.05 was considered statistically.
ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Wang, L.-X. _et al._ Aurora A Kinase Inhibitor AKI603 Induces Cellular Senescence in Chronic Myeloid Leukemia Cells Harboring T315I Mutation.
_Sci. Rep._ 6, 35533; doi: 10.1038/srep35533 (2016). PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
REFERENCES * NCCN. NCCN clinical practice guidelines in oncology. _NCCN Chronic Myelogenous Leukemia Guidelines_ Vers 1, NCCN (2015). * Faderl, S. et al. The biology of chronic myeloid
leukemia. N Engl J Med. 341, 164–172 (1999). CAS PubMed Google Scholar * Melo, J. V. & Barnes, D. J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev
Cancer. 7, 441–453 (2007). CAS PubMed Google Scholar * Gesbert, F., Sellers, W. R., Signoretti, S., Loda, M. & Griffin, J. D. BCR/ABL regulates expression of the cyclin-dependent
kinase inhibitor p27Kip1 through the phosphatidylinositol 3-Kinase/AKT pathway. J Biol Chem. 275, 39223–39230 (2000). CAS PubMed Google Scholar * Lugo, T. G., Pendergast, A. M., Muller,
A. J. & Witte, O. N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 247, 1079–1082 (1990). ADS CAS PubMed Google Scholar * Danial, N. N.
& Rothman, P. JAK-STAT signaling activated by Abl oncogenes. Oncogene. 19, 2523–2531 (2000). CAS PubMed Google Scholar * Druker, B. J. et al. Efficacy and safety of a specific
inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 344, 1031–1037 (2001). CAS PubMed Google Scholar * O’Hare, T., Zabriskie, M. S., Eiring, A. M. &
Deininger, M. W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 12, 513–526 (2012). PubMed Google Scholar * Zabriskie, M. S. et al. BCR-ABL1 compound
mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer cell. 26, 428–442 (2014). CAS PubMed PubMed Central
Google Scholar * Apperley, J. F. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 8, 1018–1029 (2007). CAS PubMed Google Scholar * O’Hare, T. et
al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer cell. 16, 401–412 (2009). PubMed PubMed
Central Google Scholar * Ikezoe, T. et al. A novel treatment strategy targeting Aurora kinases in acute myelogenous leukemia. Mol Cancer Ther. 6, 1851–1857 (2007). CAS PubMed Google
Scholar * Kelly, K. R. et al. Phase I study of MLN8237–investigational Aurora A kinase inhibitor–in relapsed/refractory multiple myeloma, non-Hodgkin lymphoma and chronic lymphocytic
leukemia. Invest New Drugs. 32, 489–499 (2014). CAS PubMed Google Scholar * Harrington, E. A. et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases,
suppresses tumor growth _in vivo_. Nat Med. 10, 262–267 (2004). CAS PubMed Google Scholar * Goldberg, S. L. et al. An exploratory phase 2 study of investigational Aurora A kinase
inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leuk Res Rep. 3, 58–61 (2014). PubMed PubMed Central Google Scholar * Liu, L. L. et al.
Inhibition of mTOR pathway sensitizes acute myeloid leukemia cells to aurora inhibitors by suppression of glycolytic metabolism. Mol Cancer Res. 11, 1326–1336 (2013). CAS PubMed Google
Scholar * Cammareri, P. et al. Aurora-a is essential for the tumorigenic capacity and chemoresistance of colorectal cancer stem cells. Cancer Res. 70, 4655–4665 (2010). CAS PubMed Google
Scholar * Giles, F. J. et al. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood.
109, 500–502 (2007). CAS PubMed Google Scholar * Akahane, D., Tauchi, T., Okabe, S., Nunoda, K. & Ohyashiki, K. Activity of a novel Aurora kinase inhibitor against the T315I mutant
form of BCR-ABL: _in vitro_ and _in vivo_ studies. Cancer Sci. 99, 1251–1257 (2008). CAS PubMed Google Scholar * Carter, T. A. et al. Inhibition of drug-resistant mutants of ABL, KIT, and
EGF receptor kinases. Proc Natl Acad Sci USA. 102, 11011–11016 (2005). ADS CAS PubMed PubMed Central Google Scholar * Zheng, F. M. et al. A novel small molecule aurora kinase inhibitor
attenuates breast tumor-initiating cells and overcomes drug resistance. Mol Cancer Ther. 13, 1991–2003 (2014). CAS PubMed Google Scholar * Pan, X. N. et al. Inhibition of c-Myc overcomes
cytotoxic drug resistance in acute myeloid leukemia cells by promoting differentiation. Plos One. 9, e105381; 10.1371/journal.pone.0105381 (2014). ADS PubMed PubMed Central Google
Scholar * Long, Z. J. et al. A novel compound against oncogenic Aurora kinase A overcomes imatinib resistance in chronic myeloid leukemia cells. Int J Oncol. 46, 2488–2496 (2015). CAS
PubMed Google Scholar * Liu, Y. et al. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-kappaB impairs this drug-induced senescence.
EMBO Mol Med. 5, 149–166 (2013). ADS CAS PubMed Google Scholar * Kim, H. J., Cho, J. H., Quan, H. & Kim, J. R. Down-regulation of Aurora B kinase induces cellular senescence in human
fibroblasts and endothelial cells through a p53-dependent pathway. FEBS lett. 585, 3569–3576 (2011). CAS PubMed Google Scholar * Brusa, G. et al. p53 loss of function enhances genomic
instability and accelerates clonal evolution of murine myeloid progenitors expressing the p(210)BCR-ABL tyrosine kinase. Haematologica. 88, 622–630 (2003). CAS PubMed Google Scholar *
Sen, S., Takahashi, R., Rani, S., Freireich, E. J. & Stass, S. A. Expression of differentially phosphorylated Rb and mutant p53 proteins in myeloid leukemia cell lines. Leuk Res. 17,
639–647 (1993). CAS PubMed Google Scholar * Roh, M., van der Meer, R. & Abdulkadir, S. A. Tumorigenic polyploid cells contain elevated ROS and ARE selectively targeted by antioxidant
treatment. J Cell Physiol. 227, 801–812 (2012). CAS PubMed PubMed Central Google Scholar * McCrann, D. J., Yang, D., Chen, H., Carroll, S. & Ravid, K. Upregulation of Nox4 in the
aging vasculature and its association with smooth muscle cell polyploidy. Cell Cycle. 8, 902–908 (2009). CAS PubMed Google Scholar * Macip, S. et al. Inhibition of p21-mediated ROS
accumulation can rescue p21-induced senescence. EMBO J. 21, 2180–2188 (2002). CAS PubMed PubMed Central Google Scholar * Campisi, J. & d’Adda di Fagagna, F. Cellular senescence: when
bad things happen to good cells. Nat Rev Mol Cell Biol. 8, 729–740 (2007). CAS PubMed Google Scholar * Kantarjian, H. M., Talpaz, M., Giles, F., O’Brien, S. & Cortes, J. New insights
into the pathophysiology of chronic myeloid leukemia and imatinib resistance. Ann Intern Med. 145, 913–923 (2006). PubMed Google Scholar * Kaur, P. et al. Nilotinib treatment in mouse
models of P190 Bcr/Abl lymphoblastic leukemia. Mol Cancer. 6, 67 (2007). MathSciNet PubMed PubMed Central Google Scholar * Talpaz, M. et al. Dasatinib in imatinib-resistant Philadelphia
chromosome-positive leukemias. N Engl J Med. 354, 2531–2541 (2006). CAS PubMed Google Scholar * Shah, N. P. Loss of response to imatinib: mechanisms and management. _Hematology-Am Soc
Hemat_. 1, 183–187 (2005). * Akard, L. P. Second-generation BCR-ABL kinase inhibitors in CML. N Engl J Med. 363, 1672–1673 (2010). PubMed Google Scholar * Cohen, P. & Alessi, D. R.
Kinase drug discovery–what’s next in the field? ACS Chem Biol. 8, 96–104 (2013). CAS PubMed Google Scholar * Cheetham, G. M., Charlton, P. A., Golec, J. M. & Pollard, J. R. Structural
basis for potent inhibition of the Aurora kinases and a T315I multi-drug resistant mutant form of Abl kinase by VX-680. Cancer Lett. 251, 323–329 (2007). CAS PubMed Google Scholar *
Donato, N. J. et al. Targets and effectors of the cellular response to aurora kinase inhibitor MK-0457 (VX-680) in imatinib sensitive and resistant chronic myelogenous leukemia. Biochem
Pharmacol. 79, 688–697 (2010). CAS PubMed Google Scholar * Cilloni, D. & Saglio, G. Molecular pathways: BCR-ABL. Clin Cancer Res. 18, 930–937 (2012). CAS PubMed Google Scholar *
Yuan, H. et al. Overcoming CML acquired resistance by specific inhibition of Aurora A kinase in the KCL-22 cell model. Carcinogenesis. 33, 285–293 (2012). CAS PubMed Google Scholar *
Kelly, K. R. et al. The novel Aurora A kinase inhibitor MLN8237 is active in resistant chronic myeloid leukaemia and significantly increases the efficacy of nilotinib. J Cell Mol Med. 15,
2057–2070 (2011). ADS CAS PubMed PubMed Central Google Scholar * Fiskus, W. et al. Cotreatment with vorinostat enhances activity of MK-0457 (VX-680) against acute and chronic
myelogenous leukemia cells. Clin Cancer Res. 14, 6106–6115 (2008). CAS PubMed PubMed Central Google Scholar * Roninson, I. B. Tumor cell senescence in cancer treatment. Cancer research.
63, 2705–2715 (2003). CAS PubMed Google Scholar * Chang, B. D. et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to
anticancer agents. Cancer Res. 59, 3761–3767 (1999). CAS PubMed Google Scholar * Huck, J. J. et al. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both
_in vitro_ and _in vivo_. Mol Cancer Res. 8, 373–384 (2010). CAS PubMed Google Scholar * Vigneron, A. & Vousden, K. H. p53, ROS and senescence in the control of aging. Aging (Albany
NY). 2, 471–474 (2010). CAS Google Scholar * Demidenko, Z. N., Korotchkina, L. G., Gudkov, A. V. & Blagosklonny, M. V. Paradoxical suppression of cellular senescence by p53. Proc Natl
Acad Sci USA. 107, 9660–9664 (2010). ADS CAS PubMed PubMed Central Google Scholar * Niu, N. K. et al. Pro-apoptotic and pro-autophagic effects of the Aurora kinase A inhibitor alisertib
(MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the activation of mitochondria-mediated pathway and inhibition of p38 MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Dev Ther.
9, 1555–1584 (2015). CAS Google Scholar * O’Hare, T., Eide, C. A. & Deininger, M. W. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid
leukemia. Blood. 110, 2242–2249 (2007). PubMed Google Scholar * Carter, B. Z. et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells.
Sci Transl Med. 8, 355ra117, doi: 10.1126/scitranslmed.aag1180 (2016). Article CAS PubMed PubMed Central Google Scholar * Melo, J. V. & Deininger, M. W. Biology of chronic
myelogenous leukemia–signaling pathways of initiation and transformation. HematolOncol Clin N. 18, 545–568 (2004). Google Scholar * Sawyers, C. L., Callahan, W. & Witte, O. N. Dominant
negative MYC blocks transformation by ABL oncogenes. Cell. 70, 901–910 (1992). CAS PubMed Google Scholar * Sanchez-Arevalo Lobo, V. J. et al. Dual regulation of Myc by Abl. Oncogene. 32,
5261–5271 (2013). CAS PubMed Google Scholar * Zheng, F. et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun.
7, 10180 (2016). ADS CAS PubMed PubMed Central Google Scholar * Lee, J. K. et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer
Cell. 29, 536–547 (2016). CAS PubMed PubMed Central Google Scholar * Dauch, D. et al. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver
cancer. Nat Med. 22, 744–753 (2016). CAS PubMed Google Scholar * Shi, X. et al. Triptolide inhibits Bcr-Abl transcription and induces apoptosis in STI571-resistant chronic myelogenous
leukemia cells harboring T315I mutation. ClinCancer Res. 15, 1686–1697 (2009). CAS Google Scholar * Wu, Y. et al. Cyclin-dependent kinase 7/9 inhibitor SNS-032 abrogates FIP1-like-1
platelet-derived growth factor receptor alpha and bcr-abl oncogene addiction in malignant hematologic cells. Clin Cancer Res. 18, 1966–1978 (2012). CAS PubMed Google Scholar * Shi, X. et
al. Gambogic acid induces apoptosis in imatinib-resistant chronic myeloid leukemia cells via inducing proteasome inhibition and caspase-dependent Bcr-Abl downregulation. Clin Cancer Res. 20,
151–163 (2014). CAS PubMed Google Scholar * Valsasina, B. et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and
hematologic malignancies. Mol Cancer Ther. 11, 1006–1016 (2012). CAS PubMed Google Scholar * Kerbel, R. S. Human tumor xenografts as predictive preclinical models for anticancer drug
activity in humans - Better than commonly perceived - But they can be improved. Cancer Biol Ther. 2, S134–S139 (2003). CAS PubMed Google Scholar * Miething, C. et al. The Bcr-Abl
mutations T315I and Y253H do not confer a growth advantage in the absence of imatinib. Leukemia. 20, 650–657 doi: 10.1038/sj.leu.2404151 (2006). Article CAS PubMed Google Scholar
Download references ACKNOWLEDGEMENTS We thank the members of Quentin Liu lab and Medical Research Center, The Third Affiliated Hospital, Sun Yat-sen University for their critical comments
and technical support. We thank Dr Wen-fang Li (Sun Yat-sen medical school, Sun Yat-sen University) for reviewing our manuscript. This study was supported by the National High Technology
Research and Development Program of China (863 Program; No. 2015AA020926 to Z.-J. Long), the National Basic Research Program of China (973 Program; No. 2012CB967000 to Q. Liu), the Special
Support Program for Training High Level Talents in Guangdong Province (No. 2014TQ01R138 to Z.-J. Long), the Science and Technology Planning Project of Guangdong Province (No. 2016A020215078
to Z.-J. Long), and the China Postdoctoral Science Foundation (No. 2016M590838 to L.-X. Wang). AUTHOR INFORMATION Author notes * Wang Le-Xun, Wang Jun-Dan and Chen Jia-Jie contributed
equally to this work. AUTHORS AND AFFILIATIONS * Department of Hematology, The Third Affiliated Hospital, Sun Yat-sen University, 600 Tianhe Road, Guangzhou 510630, China; Institute of
Hematology, Sun Yat-sen University, Guangzhou, 510630, China Le-Xun Wang, Jun-Dan Wang, Jia-Jie Chen, Bing Long, Ling-Ling Liu, Yuan Hu, Dong-Jun Lin, Zi-Jie Long & Quentin Liu *
Department of Cardiac Surgery II, The First Affiliated Hospital, Sun Yat-sen University, 58 Zhongshan 2 Road, Guangzhou, 510080, China Le-Xun Wang * Institute of Cancer Stem Cell, Dalian
Medical University, Dalian, 116044, China Xi-Xiang Tu & Quentin Liu * Institute of Medicinal Chemistry, School of Pharmaceutical Sciences, Sun Yat-sen University, 132 Waihuan Road East,
Guangzhou, 510006, China Yu Luo & Gui Lu * Sun Yat-sen University Cancer Center; State Key Laboratory of Oncology in South China; Collaborative Innovation Center of Cancer Medicine,
Guangzhou, 510060, China Quentin Liu Authors * Le-Xun Wang View author publications You can also search for this author inPubMed Google Scholar * Jun-Dan Wang View author publications You
can also search for this author inPubMed Google Scholar * Jia-Jie Chen View author publications You can also search for this author inPubMed Google Scholar * Bing Long View author
publications You can also search for this author inPubMed Google Scholar * Ling-Ling Liu View author publications You can also search for this author inPubMed Google Scholar * Xi-Xiang Tu
View author publications You can also search for this author inPubMed Google Scholar * Yu Luo View author publications You can also search for this author inPubMed Google Scholar * Yuan Hu
View author publications You can also search for this author inPubMed Google Scholar * Dong-Jun Lin View author publications You can also search for this author inPubMed Google Scholar * Gui
Lu View author publications You can also search for this author inPubMed Google Scholar * Zi-Jie Long View author publications You can also search for this author inPubMed Google Scholar *
Quentin Liu View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS L.-X.W., J.-D.W. and J.-J.C. researched data and participated in writing of the
manuscript. B.L. and X.-X.T. contributed to the discussion. Y.L., L.-L.L. and Y.H. participated in editing of the manuscript. D.-J.L. provided oversight for the project. G.L., Z.-J.L. and
Q.L. are the guarantors of this work. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY
INFORMATION RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included
in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain
permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Wang, LX., Wang, JD., Chen, JJ. _et al._ Aurora A Kinase Inhibitor AKI603 Induces Cellular Senescence in Chronic Myeloid Leukemia Cells Harboring T315I Mutation. _Sci Rep_
6, 35533 (2016). https://doi.org/10.1038/srep35533 Download citation * Received: 14 April 2016 * Accepted: 29 September 2016 * Published: 08 November 2016 * DOI:
https://doi.org/10.1038/srep35533 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently
available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative