- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT MicroRNAs regulate a spectrum of developmental and biochemical processes in plants and animals. Thus, knowledge of the entire miRNome is essential to understand the complete
regulatory schema of any organism. The current study attempts to unravel yet undiscovered miRNA genes in rice. Analysis of small RNA libraries from various tissues of drought-tolerant
_‘aus’_ rice variety Nagina 22 (N22) identified 71 novel miRNAs. These were validated based on precursor hairpin structure, small RNA mapping pattern, ‘star’ sequence, conservation and
identification of targets based on degradome data. While some novel miRNAs were conserved in other monocots and dicots, most appear to be lineage-specific. They were segregated into two
different classes based on the closeness to the classical miRNA definition. Interestingly, evidence of a miRNA-like cleavage was found even for miRNAs that lie beyond the classical
definition. Several novel miRNAs displayed tissue-enriched and/or drought responsive expression. Generation and analysis of the degradome data from N22 along with publicly available
degradome identified several high confidence targets implicated in regulation of fundamental processes such as flowering and stress response. Thus, discovery of these novel miRNAs
considerably expands the dimension of the miRNA-mediated regulation in rice. SIMILAR CONTENT BEING VIEWED BY OTHERS A CONSERVED SNP VARIATION IN THE PRE-_MIR396C_ FLANKING REGION IN _ORYZA
SATIVA INDICA_ LANDRACES CORRELATES WITH MATURE MIRNA ABUNDANCE Article Open access 07 February 2023 IDENTIFICATION OF KEY SEQUENCE FEATURES REQUIRED FOR MICRORNA BIOGENESIS IN PLANTS
Article Open access 21 October 2020 GENOME-WIDE IDENTIFICATION OF MICRORNAS INVOLVED IN THE REGULATION OF FRUIT RIPENING AND CLIMACTERIC STAGES IN MELON (_CUCUMIS MELO_) Article Open access
01 July 2020 INTRODUCTION MicroRNAs are one of the major components of molecular networks that regulate several plant developmental processes such as leaf development, flowering time, organ
polarity1 as well as biotic and abiotic stress responses2,3. Thus, discovery of novel miRNA genes that have not been reported in any organism would have a significant impact on our
understanding of the complex molecular regulatory networks. Since discovery of miRNA genes is primarily based on detection of their expression in the small RNA (sRNA) NGS libraries, no
single data set is sufficient to identify miRNA genes. Consequently, diverse tissues, growth conditions and varieties need to be explored in order to identify and characterize miRNA genes.
Response to abiotic stress is perhaps one of the most widely researched topics due to its impact on the productivity of major agro-economically important crops such as rice, a model
monocotyledon. Drought imposes a major threat to its productivity. Water stress during reproductive stage severely affects panicle exsertion and anther dehiscence leading to crop failure4.
India has a rich collection of rice germplasm wherein many cultivars/varieties have a natural tolerance to various stress conditions. In general, upland rice cultivars, like Nagina 22 (N22),
have a better tolerance to drought as compared to rainfed lowland cultivars like Pusa Basmati 1 (PB1). N22 is a deep-rooted, drought- and heat-tolerant5,6,7 rice cultivar. Detailed
molecular characterization of such drought-tolerant cultivars would provide valuable information about the nature of adaptive diversity evolved in these cultivars8,9. N22 offers unique
opportunity as a model for studying stress adaptive diversity as it harbors traits, at both physiological6 and molecular level5,10, to combat drought and heat stress. Nevertheless, only a
couple of studies have characterized and explored the dynamism of miRNA population in N228,9. Currently, 592 rice miRNA precursor entries are available in the miRBase database release 21.
Study with sRNA deep sequenced data in N22 rice further identified a few novel miRNAs9, indicating that several miRNA genes remain undiscovered. In general, deep sequenced sRNA populations
have been widely used for identification of novel miRNAs in different plant species11,12,13,14,15,16. A major challenge in miRNA gene mining is the presence of small interfering RNAs
(siRNAs), which are similar to miRNAs and can only be differentiated by their biogenesis17. siRNAs originate from double stranded (ds) precursors formed by intermolecular hybridization of
two complementary RNA strands while miRNAs are derived from RNA Pol II transcribed single stranded (ss) precursors possessing a hairpin structure due to intramolecular
self-complementarity17. The hairpin precursors are processed by Dicer-Like 1 or 4 (DCL1 or DCL4)18,19 to produce a dsRNA duplex of ~21–24 bp. HASTY transports this duplex out of the
nucleus20, which is then methylated by HUA enhancer (HEN1) at 3′ end21,22. The guide or mature strand is incorporated into the RNA induced silencing complex (RISC) whereby the effector
protein Argonaute (AGO)23 mediates target mRNA identification and cleavage or translational arrest. The other strand of the duplex called the miRNA* (star) is usually degraded. However,
there are reports on the functionality of miRNA* as well, under certain conditions24,25. In order to classify sRNA as a bonafide miRNA, certain criteria based on its secondary structure,
expression, biogenesis and conservation need to be considered26. Along with the primary criteria of secondary structure, the mapping pattern of the sRNA tags on the genome helps in the
identification of the probable miRNA gene locus. A tightly stacked mapping of sRNA tags at the locus is indicative of miRNA processing whereas a dispersed mapping all over the locus is more
suggestive of a siRNA-like miRNA27. The main objective of the current study was to identify miRNA genes that have not been reported so far in rice or any other organism. Deep sequencing of
the sRNA populations from different tissues such as flag leaf, spikelets ‘heading stage’ and spikelets ‘anthesis stage’ and mature root of N22 plants grown under control and drought
conditions was done. Subsequent rigorous analysis, based on well-defined criterion26,28, identified several putative novel miRNA genes loci. To the best of our knowledge, except an isolated
study9, sRNA population from these tissues has not been utilized for identification of novel miRNA genes. qRT-PCR analysis was done to validate their expression wherein several of these
novel miRNAs were found to have drought responsive as well as tissue preferential expression patterns. While some novel miRNAs were found to be conserved in other plants, most were
lineage-specific as they were primarily detected in multiple rice sRNA libraries. The biological activity of these novel miRNAs was confirmed by identifying their target genes and the
cut-site with the help of degradome data. Thus, multiple lines of evidences were gathered to support the existence and activity of these novel miRNA genes. RESULTS IDENTIFICATION OF NOVEL
MIRNAS IN N22 Small RNA data is a major resource for identification of novel miRNA genes. Thus, in order to uncover yet unidentified novel MIR gene loci in rice, five sRNA libraries from
adult tissues (flag leaf control & stress, heading spikelet control & stress and anthesis spikelet control) of the rice cultivar, N22 were deep sequenced to generate >19 million
sRNA tags. This data was used for identification of any unreported miRNA genes as well as to study modulation of miRNome (already known miRNAs) under environmental and temporal cues. While
the data on miRNome modulation is presented elsewhere (Balyan S. _et al._, under-review), the current study summarizes the discovery of novel miRNA genes from the sRNA data. The detailed
analysis pipeline has been described in the ‘materials and methods’ section. In summary, all the sRNA tags were mapped to the N22 genome and genomic loci having a non-phasic mapping of sRNA
tags were identified. Subsequently, genomic sequences (~200 bp) adjoining these sites were extracted and considered as the putative novel pre-miRNA sequence. These putative novel pre-miRNA
sequences were then analyzed for their ability to form a miRNA-like hairpin structure. A total of 2,010 intergenic and 542 intronic putative miRNA loci were initially selected based on
hairpin structures. A second round of screening was done to ensure that the sRNA tags within the putative miRNA precursor region clustered at a particular location i.e. mature sequence
region27. Ultimately, we could identify 71 putative novel miRNA gene loci in N22, details of which are summarized in Supplementary Table S1. Further analysis of these putative novel mature
miRNA sequences identified 18 novel miRNAs that had at least 17 bp sequence similarity to miRNAs reported in published literature (Supplementary Table S2) on rice but not available at
miRBase database. Thirteen of them (n-032, n-042, n-054, n-064, n-069, n-098, n-102, n-107, n-120, n-128, n-131, n-149 and n-184) did not have significant similarity at the precursor
sequence level, while 5 of the putative novel miRNAs (n-020, n-055, n-169, n-050 and n-213) shared appreciable similarity at the precursor level. Only one putative novel miRNA i.e., n-020
was found to be identical with miR2175–0.229 or miR301830, which had been reported separately. In summary, 17 of the putative novel molecules identified in this study appear to be family
members of miRNAs reported in the literature while one was found to be the same molecule. CHARACTERIZATION AND CLASSIFICATION OF NOVEL MIRNAS The putative novel miRNAs were further analyzed
from several aspects in order to validate and characterize them. One of the foremost criteria for defining a miRNA gene is that the precursor transcript should be able to fold back into a
typical hairpin structure that has ≤4 symmetrical mismatches within the duplex and/or only 1–2 asymmetric bulge(s) of up to 2 bases26. Close scrutiny of the secondary structures indicated
that hairpin structures of all the 71 putative novel miRNA candidates conform to the established norms. The secondary structures of the 71 candidates are provided in Supplementary Fig. S1
while their precursor sequences are given in Supplementary Table S1. Further, presence of ‘star’ sequence is another important criteria for identification of miRNA genes. However, due to
unstable nature of the sequence it is difficult to detect the ‘star’ sequence for all the miRNA genes. We were able to detect the ‘star’ sequence for 36 novel miRNA genes (Supplementary
Table S1). Another important characteristic feature of a miRNA gene locus is the manner in which sRNA tags map on the precursor sequence. Since the generation of the mature miRNA population
from the precursor sequence is through a near precise cut made by DCL1 and associated proteins, majority of the mature miRNA tags share almost a common 5′ end (with ±3 bases variation)31.
The 3′ end, however is quite variable. Thus, when sRNA tags are mapped on the precursor sequences, most of them should map as a tight cluster in the region that is proposed to give rise to
the mature miRNA sequence. Scrutiny of the mapping pattern of sRNA reads in the entire precursor sequence indicated that majority of tags mapped in the proposed mature miRNA region. However,
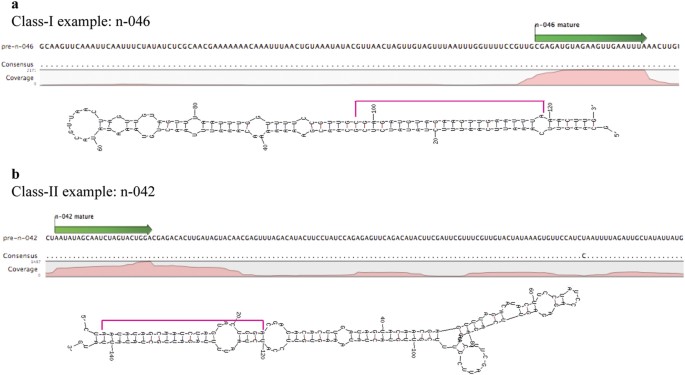
in some cases the mapping of tags was slightly dispersive in nature. Based on the mapping pattern, the putative novel miRNAs were divided into two classes. While both classes had a good
hairpin secondary structure, class-I loci had a consistent mapping pattern (≥40% of total reads mapped to the mature sequence region) while class-II loci had slightly dispersive mapping
pattern (<40% of total reads mapped to the mature sequence region). The percentage of sRNA reads abundance at mature position as compared to the total reads mapped to the precursor is
given in Supplementary Table S1. Examples of each class based on mapping pattern and secondary structure is given in Fig. 1a,b. In summary, there are 40 class-I and 31 class-II novel miRNA
candidates. The putative novel miRNAs were also characterized by assessing their abundance in multiple rice sRNA libraries as well as by detecting the mature miRNA sequence with the help of
qRT-PCR. As expected, all the novel miRNAs were detected in multiple sRNA libraries of N22 and comparison of their normalized tag abundance in these libraries indicated that most of them
express at a moderate level (Fig. 2a). The qRT-PCR based gel profiles of the amplified mature novel miRNA sequences were also generated in order to detect their expression as well as to
confirm their size (Supplementary Fig. S2). The amplified product includes the adapter (~100 nt) as well as the mature miRNA (~20 nt) thereby giving the size of the amplified product in the
range of ~120 bp. Based on mature sequence length, 49 out of 71 are of canonical length (20–23 bp) while 22 are long molecules (24–25 bp). They are further sub-classified as either
intergenic (39) or intronic (32) based on their genomic location. Chromosomal distribution of the 71 novel miRNAs indicates highest abundance at chromosome (chr) 3 followed by chr 1 (Fig.
2b), while size distribution indicates that 22–24 bp novel miRNAs were of highest abundance (Fig. 2c). Further, association of the miRNAs with the RISC complex is an indication of their
functionality, since only functional miRNAs tend to get loaded onto the RISC complex. Thus, publicly available AGO pulldown sRNA libraries published earlier32,33 were analyzed wherein a
significant number (67) of novel miRNAs were found to be associated with the RISC complex (Supplementary Table S3). Tags of 36 out of 67 were found in various AGO1 pulldown libraries while
tags of all the 67 novel miRNAs were present in various AGO4 pulldown libraries (Supplementary Table S3). Moreover, sRNAs with different 5′ starting base of the mature sequence have
differential AGO loading32,33,34,35. The distribution of 5′ starting base in novel and known miRNAs was quite comparative wherein novel miRNAs and known miRNAs (miRBase) had 35.21% and
45.58% with 5′U, 23.94% and 15.01% with 5′G, 33.80% and 11.50% with 5′A and 7.04% and 27.91% with 5′C, respectively (Fig. 2d). Finally, the copy number of all the 71 novel MIR genes was also
checked to ascertain if they originated from multiple loci or discrete genomic locus. The study was done by analyzing the occurrence of precursor as well as the mature novel miRNA sequences
on the rice genome sequence. Based on the analysis of the precursor sequences, almost all the novel MIRs was found to originate from a single genomic locus except n-170, which was present
in 2 copies on the genome. On the other hand, analysis with the mature sequence indicated that, most of the novel miRNAs had <20 genome hits, while 20 of them showed multiple hits
(>20) to the genome (Supplementary Table S4). Similar analysis with known rice miRNAs from published literature revealed that many of them had >20 genome hits as well (Supplementary
Table S4). Further, since recent studies have also indicated that miRNAs may also originate from within transposable elements (TE)36, we explored the rice repeat sequences (RGAP database;
www.rice.plantbiology.msu.edu) for the presence of any novel miRNA precursors. Consequently, four novel miRNA candidates, all belonging to class-II i.e., n-002, n-123, n-128 and n-195 were
found to have 100% sequence similarity with MITE-adh, type D-like repeats ORSgTEMT01700916, ORSgTEMT01700334, ORSgTEMT01701756 and ORSgTEMT01700523, respectively. The secondary structures of
these repeat sequences are shown in Fig. 3a–d. NOVEL MIRNA FAMILY MEMBERS The mature sequences of the novel miRNAs were further analyzed to ascertain if they can be clubbed as miRNA family
members. Based on sequence similarity, 10 novel MIR families containing 27 novel miRNAs were identified. In general, family members share mature sequence similarity and often target related
genes. The 10 novel miRNA families thus identified are MIRn-130, MIRn-050, MIRn-059, MIRn-098, MIRn-099, MIRn-102, MIRn-129, MIRn-134, MIRn-160 and MIRn-195. The multiple alignments of their
mature sequences are shown in Supplementary Fig. S3a–j. Family of MIRn-160 had the highest number of miRNA members namely, n-160, n-100, n-225, n-169 and n-213. Additionally, analysis on
genomic arrangement of novel miRNAs _vis-à-vis_ known miRNAs in N22 genome revealed an interesting fact about a novel miRNA locus. Although present on different chromosomal loci, the
precursor of n-026 (chr 5) is very similar (>80%) to that of osa-miR5788 (chr 3). However, their mature sequences have only 5 bp overlap and thus they cannot be classified as family
members. Nevertheless, there appears to be some kind of evolutionary relationship between the two miRNAs since their precursor sequences share significant similarity. IDENTIFICATION OF
UNREPORTED PUTATIVE MIRNA ORTHOLOGS IN N22 The phylogenetic conservation of miRNA sequences (mature and precursor) as well as the secondary structure facilitates identification of putative
orthologous miRNAs26. Thus, in addition to the aforementioned novel miRNA genes which were not reported in rice or any other organism, analysis was also done to identify rice orthologs of
miRNA genes known in other plant species. Accordingly, as depicted in the analysis pipeline (Supplementary Fig. S4), 22 non-rice known MIR precursors could be aligned with ≥50% sequence
similarity to the N22 genome. Of these, 16 had ≤3 mismatches in the mature region. The pairwise alignments of the parent MIR precursors (miRBase) to that of identified rice putative ortholog
precursors are shown in Supplementary Fig. S5. Subsequently, secondary structure analysis indicated that rice ortholog of 6 i.e. MIR5056, MIR5368, MIR6187, MIR6196, MIR6221 and MIR9774 had
a good hairpin structure while others (MIR6199, MIR5141, MIR9780, MIR6485, MIR6207, MIR6206, MIR6183, MIR2916, MIR7767 and MIR6194) had inconsistent structures, despite a significant
conservation at the precursor sequence level. The details of 16 putative orthologs and their secondary structures are summarized in Table 1 and Supplementary Fig. S6a–p, respectively. NOVEL
MIRNAS ARE LINEAGE-SPECIFIC In order to check the extent of conservation/divergence of novel miRNAs, their abundance was analyzed in other publicly available sRNA libraries of rice and other
plants. A total of 14 sRNA libraries from rice (3), maize (1), wheat (2), barley (3), _Arabidopsis_ (3) and soybean (2) available at NCBI ‘SRA’ database were utilized (Supplementary Table
S5). Tag abundance of 71 miRNAs could be detected in all the 3 rice sRNA libraries, indicating the reliability for their existence and expression in other varieties/stage/tissue of rice.
Further, sRNA tags corresponding to 14 novel miRNAs were detected in other monocots, which include 13 in wheat, 10 in barley and 3 in maize. Heat map of the 71 novel miRNAs in sRNA libraries
of rice, maize, wheat, barley, _Arabidopsis_ and soybean is shown in Fig. 4a. Similarly, sRNA tags for 6 novel miRNAs were detected in dicots including 4 in _Arabidopsis_ and 2 in soybean.
While most of them were detected at low levels, some had >5 tags (Supplementary Table S5). Analysis of the genomic loci in the respective plants revealed that the precursors of 3 novel
miRNAs in barley (n-006, n-107 and n-118) and 3 in wheat (n-024, n-032 and n-107) had acceptable miRNA-like hairpin structures (Supplementary Fig. S7a–f). Similar analysis for known miRNAs
to compare the extent of conservation/divergence indicated that 23.7%, 31.6%, 27.8%, 21.7% and 19.9% of known miRNAs were detected in the sRNA libraries of maize, wheat, barley,
_Arabidopsis_ and soybean, respectively (Fig. 4b, Supplementary Table S5). In summary, most of these novel miRNAs are considerably conserved in rice whereas only few could be detected in
other plants indicating a lineage-specific nature17. IDENTIFICATION OF NOVEL MIRNA TARGETS To ascertain the functionality of these novel miRNAs, ‘degradome sequencing’37 or ‘PARE’ (Parallel
Analysis of RNA Ends)38 was performed to identify and validate target mRNA cleavage. Sequencing of three degradome libraries from anthesis spikelets (ASp), heading spikelets (HSp) and flag
leaf (FL) of N22 generated more than six million reads from each library (Supplementary Table S6). Subsequent analysis of the degradome data by CleaveLand analysis pipeline39 identified
40:105, 20:52 and 30:91 novel miRNA:target pairs in the PARE libraries from ASp, HSp and FL, respectively. CleaveLand analysis was further extended by including five more publicly available
rice PARE libraries (SRR032098, SRR039716-039720, SRR521269, SRR032097 and SRR034102) (Supplementary Table S7). In summary, analyses of data from 8 PARE libraries indicated putative multiple
targets of the novel miRNAs (including low confidence targets). Distribution of miRNA:target pairs based on different confidence parameters (degradome read number, category and P-value) of
CleaveLand pipeline is shown in Supplementary Table S8. Analysis of the top ten targets for each novel miRNAs reveals diverse targets including transcription factors, metabolic enzymes,
signaling components (kinases and phosphatases) that have been implicated in important cellular processes such as plant development and stress (biotic and abiotic) response (Supplementary
Table S9). Of all the putative targets, essentially 33 miRNA:target pairs (25 genes targeted by 19 novel miRNAs) are of high confidence (degradome reads ≥10, category ≤2 and P-value ≤0.05)
(Table 2). A cut-off based on simply the alignment score was not considered as it has been reported that miRNAs could cleave targets even with higher mismatches40. T-plots (target plots) for
selected high confidence and important targets of novel miRNAs are shown in Fig. 5a–i and discussed in subsequent text. Of these 19 novel miRNAs having high confidence target genes, 11 were
of class-I while 8 of class-II. Further, novel miRNAs with multiple loci also had high confidence targets based on the degradome data indicating their biological activity. For example,
novel miRNA n-121 with 397 genomic hits (at mature level) had a target identified with 36 degradome reads (category ‘0’). Similarly, n-118 having 177 genome hits had a target supported by
101 degradome reads (category ‘0’). Subsequently, targets of orthologous miRNAs were also identified from the 8 PARE libraries (Supplementary Table S10). Targeting of 38 genes by 10
orthologous miRNAs are supported by ≥10 degradome reads (Supplementary Fig. S8a) while 11 pairs (4 miRNA:11 target genes) are of ≥10 degradome reads and category ‘0’. Interestingly, two
(osa-miR6485 and osa-miR6207) of the miRNA:target pairs with ‘0’ category do not have a consistent hairpin structures. The targeting of LOC_Os11g15240 (retrotransposon) by osa-miR9774 could
be supported with highest confidence. Representative ‘t-plots’ for some of the orthologous miRNA targets are shown in Supplementary Fig. S8b–m. The degradome libraries were also used to
identify targets of known miRNAs. Consequently, 266:895, 266:925 and 350:3054 miRNA:target pairs were obtained from N22 ASp, HSp and FL, degradome libraries, respectively (Supplementary
Table S6). On further inclusion of 5 more PARE libraries, 551:16,381 miRNA:target pairs were identified (no filters applied on degradome criteria). Out of this, 102:126 miRNA:target pairs
are of high confidence (≥10, category ≤2 and P-value ≤0.05) (Supplementary Table S7). The ratio of high confidence targets among all the targets is quite comparable for both novel and known
miRNAs (Supplementary Tables S9 and S10). EXPRESSIONS OF NOVEL MIRNAS IN RESPONSE TO DEVELOPMENTAL AND ENVIRONMENTAL CUES Expressions of miRNAs are guided by environmental or developmental
cues. Thus, sRNA deep sequenced data from various tissues of N22 (FL, HSp, ASp and MR, i.e. mature root) grown under control and drought conditions was analyzed to assess the tissue-biased
and drought responsive nature of the novel miRNAs. Analysis of the sRNA tags from different tissues of plants grown under control conditions identified tissue preferential expression of
several novel miRNA genes. In summary, 47, 51, 46 and 49 novel miRNAs were found to have ≥10 normalized tag density (RPM) in FL, HSp, ASp and MR of N22. Further, 5 novel miRNA (n-024, n-039,
n-063, n-148 and n-153) were significantly up-regulated (fold change ≥4, P-value ≤0.05) in the FL as compared to both HSp and MR whereas 8 novel miRNAs (n-001, n-019, n-028, n-046, n-050,
n-130, n-140 and n-173) were up-regulated in HSp as compared to both FL and MR (Supplementary Table S11). Incidentally, we did not find any novel miRNA that was significantly up-regulated in
MR as compared to both FL and HSp. Comparison of the spikelet from the two stages of inflorescence development i.e. heading and anthesis identified 28 novel miRNAs that were differentially
regulated even in these closely spaced developmental stages (Supplementary Table 11). While 19 novel miRNAs were up-regulated, 9 were down-regulated in the spikelets during transition from
heading to anthesis. An interesting member of this group is n-001 which is significantly down-regulated (42 folds) in ASp and targets a Casein Kinase-1 protein, “Hd16” (_HEADING DATE 16_) or
“El1” (Early flowering 1)41,42. Further, comparison of the sRNA libraries from control and drought treated N22 plants revealed several drought responsive novel miRNAs. In total, 48 novel
miRNAs were found to be significantly drought stress responsive in the four different tissues of N22. Heat map showing the fold change expression of 71 novel miRNAs in FL, HSp, ASp and MR in
drought stress treated libraries as compared to control libraries are shown in Fig. 6a. Most of the novel miRNAs were down-regulated while only 8, 1, 8 and 1 novel miRNAs were up-regulated
in FL, HSp, ASp and MR, respectively (Supplementary Table S12). Interestingly, four novel miRNAs _viz_., n-016 and n-129 in ASp and n-046 and n-170 in FL had ≤1 (normalized) tag under
control conditions but ≥30 tags (normalized) during drought. The drought-regulated expression of several novel miRNAs which target important genes was validated by qRT-PCR (Fig. 6b–d). Novel
miRNAs n-025, n-032, n-098, n107 and n-200 were found to be down-regulated (Fig. 6b) whereas n-001, n-009, n-016, n-019 and n-130 were up-regulated in the FL during drought (Fig. 6c).
Similarly, except for n-192, miRNAs n-002, n-006, n-024, n-025, n-032, n-063, n-098, n-107, n-137, n-195 and n-200 were down-regulated in ASp during drought (Fig. 6c). Novel miRNAs n-025,
n-032, n-098, n-107 and n-200 were commonly down-regulated in both FL and ASp during drought. We further verified drought responsiveness of the target genes of these drought responsive novel
miRNAs. Indeed, analysis of the transcriptome data (Indica Rice Database; www.genomeindia.org.in/irdb) revealed that targets of many of the novel miRNAs showed anti-correlation in
expression during drought stress (Supplementary Table S13). The sRNA libraries were also used to assess the expression of the putative orthologous miRNAs. The expression profile of 17 mature
putative orthologous miRNAs is depicted by heat map generated from transformed values of sRNA abundance in 8 different N22 sRNA libraries (Supplementary Fig. S8n). Five of them
(osa-miR5368, osa-miR6485, osa-miR6196, osa-miR2916 and osa-miR9774) were constitutively expressed in most of the N22 libraries while expression of osa-miR6187, osa-miR6199, osa-miR6206,
osa-miR6221-5p, osa-miR6221-3p and osa-miR9780 could not be detected in our libraries. DISCUSSION MicroRNAs regulate various metabolic and developmental processes43,44 and thus knowledge of
the complete repertoire of the miRNA genes is critical to understand the complicated regulatory mechanisms in both plants and animals. Discovery of miRNA genes has been revolutionized by
next generation sequencing technologies. In rice, over 700 mature miRNAs (miRBase release 21) have been identified, nevertheless, the list is not saturating. Like any other gene, expression
of miRNAs is under tight control of spatial, temporal and environmental cues. Since identification of miRNA is primarily based on the detection of its expression (e.g. RNA seq), low
expressing miRNAs or those having restricted expression may be missed if appropriate sRNA libraries are not available. In the current study, we have explored sRNA libraries from flag leaves,
spikelets and roots tissues of a drought tolerant _‘aus’_ rice variety Nagina 22 and after rigorous multi-tiered analysis were able to identify 71 novel miRNA genes that have not been
reported earlier in rice or any other organism. Interestingly, 39 of them were intergenic while 32 were localized within the introns of protein coding genes. Four of the novel miRNAs were
found to originate from within the repeat sequences in rice. Earlier studies have also reported miRNAs originating from repetitive regions of genome36. MITEs (Miniature Inverted Transposable
Elements) loci in _Arabidopsis_ and rice have been reported to generate sRNAs45. Moreover, some of these novel genes are probably family members of already known miRNA genes in rice while
others could be grouped as members of novel miRNA families. Family members represent similar/same mature sequence but with distinct MIR precursor genes46. They probably have a common
evolutionary origin and often cleave same/related target genes. In order to confirm the existence of these novel miRNAs, multiple lines of evidences were investigated. These included
acceptable hairpin structure27,28, mapping pattern of sRNA tags on the precursor, qRT-PCR based expression, detection of ‘star’ sequence, conservation in sRNA libraries from rice and other
plants as well as identification of target genes based on degradome data. In general, all the novel miRNAs had hairpin structure similar to a typical miRNA gene and could be detected by both
sRNA NGS data as well as qRT-PCR analysis. Thus, there is a high level of confidence in these novel miRNA genes. Detection of these novel miRNA genes in the AGO pulldown sRNA libraries
further supports their existence and functionality. In-depth characterization of these novel miRNAs indicated that while all had a typical miRNA hairpin structure, the mapping pattern of
sRNA tags on some novel miRNA precursors was not very stacked, indicating an imprecise processing. Thus, we segregated the novel miRNAs based on the mapping patterns wherein class-I genes
can be referred to as typical miRNA genes whereas the class-II genes are more of a siRNA-like miRNA genes27. Several earlier studies have also reported existence of such siRNA-like miRNA
loci27,47. Interestingly, it was possible to identify high confidence targets (with typical miRNA like precise target cleavage) for several class-II novel miRNA genes indicating
functionality of the siRNA-like miRNA genes. Thus, even though class-II genes undergo an imprecise processing, they are able to cleave the target in a typical miRNA like manner. Similarly,
it was possible to identify high confidence targets even for novel miRNA genes that have multiple genome hits (>20). Incidentally, some known miRNA genes (deposited in the miRBase) are
also known to have more than 20 genome hits. For example, miR818 has 472 hits, miR815 has 87 hits in rice genome. Apparently, although the mature sequence may hit the genome multiple times,
however, their corresponding precursor sequences may not be able to fold into a typical miRNA like hairpin. Another important feature of miRNA genes is that most loci are conserved across
organisms. However, non-conserved miRNAs that are weakly expressed and have limited phylogenetic conservation are a universal feature of land plants48. While, almost all the novel miRNAs
could be detected in the publicly available rice (mostly _japonica_) sRNA libraries, only few were conserved in other monocots such as wheat, maize and barley. A couple of novel miRNAs were
even conserved in dicots (_Arabidopsis_ and soybean) as well. Thus, apparently most of the novel miRNAs were lineage-specific (rice) in nature and may have recently evolved17. An interesting
feature of the miRNAs, in general, is the distribution of the first 5′ base of the mature miRNA sequence. In plants, miRNAs and 21 bp siRNAs with 5′ U are primarily directed to AGO1, 24 bp
siRNAs with 5′ A onto AGO4, sRNAs with 5′ A on AGO2 and those with 5′ C are primarily loaded onto AGO532. The distribution of the 5′ base of the novel miRNAs identified in the study was
quite similar to known miRNAs. Further, number of AGO4 associated novel miRNAs was comparatively higher than AGO1 associated miRNAs. Indeed, many of these miRNAs could be defined as long
miRNAs having length ≥24 bp. Studies on these miRNAs would yield interesting results since long miRNAs are known to be involved in regulating DNA methylation33. Besides basic identification
and characterization of the novel miRNA genes, the evidence of the cleavage of the target transcript is one of the most convincing indications of a miRNA activity. Thus, analysis of the
degraded transcripts is essential in order to validate the existence of the novel miRNA genes as well as to assess the biological impact of the novel miRNA activity. Consequently, analysis
of in-house generated degradome libraries as well as several publicly available degradome libraries of rice32,49,50,51 revealed appreciable number (≥10) of degradome tags for more than 50%
of the novel miRNAs. Many of these targets are of high confidence and thus reinforce the biological functionality of these novel miRNA genes. A global overview of all the targets of these
novel miRNAs indicates a wide range of biological activities ranging from gene regulation to metabolic pathways. Many of these targets are components of biochemical pathways (RiceCyc,
http://pathway.gramene.org/gramene/ricecyc.shtml). For example, _GLUTATHIONE S-TRANSFERASE_ targeted by n-118 is involved in ROS metabolism whereas _ARGINYL-TRNA SYNTHETASE_ targeted by
n-042 (class-II) is involved in ‘tRNA charging’ (Metabolic cluster pathway) wherein it catalyses the formation of L-arginyl tRNA from L-arginine with the use of an ATP molecule. Similarly,
_POLYGALACTURONASE_ targeted by n-173 (class-I) is involved in ‘homogalacturonan degradation pathway’_. CYCLOPROPANE-FATTY-ACYL-PHOSPHOLIPID SYNTHASE_ targeted by n-108 (class-II) is
involved in ‘Fatty acids and lipids biosynthesis pathway,’ specifically in cyclopropane and cyclopropene fatty acid biosynthesis. Other important targets include _PROTEIN PHOSPHATASE 2C_,
_SKIP/SNW_ protein, plant viral response protein, _F-BOX_ domain containing protein, _TYROSINE PROTEIN KINASE_ targeted by n-139, n-156, n-140, n-102, and n-098, respectively. The study
further explores the expression dynamic of these miRNAs during the reproductive phase (‘heading’ and ‘anthesis’) of the rice plant development and includes tissue such as flag leaf,
spikelets and roots, which have not been explored extensively in terms of miRNA expression dynamics. Flag leaf, which bears the panicle, is the major source of photosynthates to the
developing panicle and thus a very important tissue52,53. Novel miRNA n-024 and n-063, which putatively target a ‘_GLUTATHIONE S-TRANSFERASE’_ and an _‘EARLY FLOWERING PROTEIN’_, were found
to be significantly enriched in the flag leaf as compared to other tissues. Similarly, roots are important as they are the first to experience change in the soil moisture content. Studies on
comparative proteome studies in the roots of wild type and _DREB1A_ over-expression lines under drought stress conditions indicated that many stress and defense related proteins were
up-regulated in both plants but a novel protein R40C1 was up-regulated only in transgenic roots54. Root-specific over-expression of _OsNAC10_ increases grain yield significantly under field
drought conditions by enlarging roots and enhancing drought tolerance55. More than 70% of all the novel miRNA genes expressed appreciably in the mature roots. Under drought conditions almost
all novel miRNAs were found to be down-regulated in the mature roots, except n-170 that gets up-regulated and putatively targets an ‘_AUXIN RESPONSE FACTOR (ARFs)’._ Other known miRNAs such
as miR160 and miR167, which also target ARFs are known to be involved in the regulation of root development56,57. Another interesting developmentally regulated novel miRNA is n-001 that
targets “Hd16”. Hd16 is known to phosphorylate rice DELLA protein SLR1 and Ghd7, a CO-like protein which suppresses flowering by inhibiting the expression of Ehd1 (a floral activator)41,42.
Hd16/EL1 is a flowering repressor identified from a cross between Nipponbare and Koshihikari. It is postulated that Hd16 might act as a mediator between floral transition and other
developmental processes such as gibberellin signaling and tillering. Similarly, the drought responsive novel miRNAs target several important genes such as _GLUTATHIONE S-TRANSFERASE_
(n-024), _GLUTAREDOXIN 2_ (n-137), _EARLY FLOWERING PROTEIN_ (n-063)_, SKP1-LIKE PROTEIN 1B_ (n-192), _GPI-ANCHORED PROTEIN_ (n-002), _OsFBX213_ (n-006) and _CYCLIN-DEPENDENT KINASE
INHIBITOR_ (n-006) among others. Thus, expression profiling in the adult tissues of rice plant provided useful insight into the probable functionality of these novel miRNA genes. In summary,
rapid advances in deep sequencing technology has greatly assisted discovery of miRNA genes in rice14,15,16,58,59,60. Identification of 71 novel miRNAs and unreported orthologous miRNAs in
rice is significant as it expands the number of biological activities that are under the regulation of miRNAs. Moreover, it also implies that there could be several other miRNAs that remain
undiscovered. Thus, further studies should still be carried out on diverse tissue and growth conditions to identify novel miRNA genes in rice. Previous findings have also indicated that
there must be a large number of miRNAs that may be unique to the rice genome, and possibly many more miRNAs remain unidentified60. Moreover, while further investigations would need to be
done, nevertheless, it appears that there could be functional miRNA genes which do not fall within the confines of a classical miRNA gene definition. It would be very interesting to analyze
the activity (biogenesis and targeting) of such loci as it may help better understand the diversity of miRNA genes. METHODS PLANT GROWTH CONDITIONS AND STRESS TREATMENT Seeds of _Oryza
sativa_ L. ssp. _aus_ cultivar N22 were surface sterilized with 0.1% HgCl2 and Teepol, rinsed and then soaked in R.O. water overnight. Seeds were grown on muslin cloth tied over a tray
containing Yoshida rice growth medium {40 mg/L NH4NO3, 10 mg/L NaH2PO4.2H2O, 40 mg/L K2SO4, 40 mg/L CaCl2, 40 mg/L MgSO4.7H2O, 0.5 mg/L MnCl2.4H2O, 0.05 mg/L 0.2 mg/L H3BO3, 0.01 mg/L
ZnSO4.7H2O, 0.01 mg/L CuSO4.5H2O and 2 mg/L FeCl3.6H2O (in monohydrate citric acid) with pH adjusted to 5} for two weeks in culture room with 28 ± 2 °C for 16/8 h photoperiod and then
transplanted to field. Drought treatment was given to field grown mature plants by withholding water supply 10–15 days before the expected date for ‘heading’. Soil moisture was monitored
using Hydra Probe Soil Moisture Sensor. Tissue was collected when soil moisture content reached below 15% and plants showed distinct leaf rolling phenotype. Tissues (flag leaves, spikelets
and roots) were collected only from plants that were at an appropriate stage of panicle development (heading or anthesis). Drought induction was checked by estimating the transcript levels
of drought stress marker genes _Rubisco small subunit_ (_RBCS_)61 and _OsbZIP23_62 in the collected tissue. _IN-SILICO_ ANALYSIS TO IDENTIFY NOVEL MIRNAS The sRNA tags (19,555,985) from 5
sRNA libraries were mapped to N22 genome sequence (Indica Rice Database; www.genomeindia.org.in/irdb) with the help of ‘maq’ software. Simultaneously, the tags were also mapped to miRBase
and Rfam (excluding miRNAs) databases and reads that matched with ≥80% similarity with miRBase (http://www.mirbase.org/) and ≥90% with Rfam database (http://rfam.sanger.ac.uk/) were excluded
from downstream analysis. The mapping of 15,933,373 remaining tags was further analyzed to identify loci where significant numbers of tags were mapped within a window size of 5 bases with
respect to the 5′ end-mapping coordinate. 2,610,147 such clusters were identified, out of which 145,028 clusters had ≥10 reads per cluster. Further analysis was done on these clusters to
identify both intergenic and intronic putative novel miRNA genes. For intergenic novel miRNAs, clusters falling within the CDS, intron and UTR regions of rice genes (RGAP,
http://rice.plantbiology.msu.edu/) were removed to represent only the intergenic regions of genome. Flanking 100 bp genomic sequence on either side of such clusters (120,456) was extracted
from the N22 genome (Indica Rice Database or IRDB; www.genomeindia.org/irdb). These sequences (~200 bp) were later trimmed down to adequate length capable of forming the hairpin. The
secondary structures were generated using UNAFold and mfold softwares63. The resultant structure files (.ct) were initially screened with the help of in-house developed perl scripts for a
suitable hairpin structure. For finding intronic miRNAs, clusters falling within the introns were retained (57,688) and subjected to similar analysis as described above. It was followed by
checking the structures and mapping pattern of the sRNA reads on the precursor sequences to identify the region of high reads accumulation in a non-phasic manner so as to represent the
mature sequence. Screening criteria64 was followed as (i) read abundance of ≥10 from multiple independent experiments (ii) ability of flanking sequences to form a miRNA precursor-like
hairpin with miRNA:miRNA* duplex (iii) mapped reads should originate from non-coding regions (iv) reads annotated as mature miRNA should have a consistent 5′ end while the 3′-end may be more
variable (v) preferably presence of tags corresponding to both miRNA and miRNA*. The sample details of all the libraries used in this study are given in Supplementary Table S14.
IDENTIFICATION OF PUTATIVE ORTHOLOGOUS MIRNA GENES In order to identify orthologous miRNAs in N22, ‘miFam.dat’ file was downloaded from miRBase 21. Out of 7,057 plant specific precursors,
those specific to rice are 592 while the rest 6,465 belong to plants other than rice. 5,099 precursors are distributed among 525 different families while the rest 1,958 are not assigned to
any family. The distribution of 525 families is such that 35 are rice specific, 38 are common among rice and other plants while 452 families are not found in rice. With these remaining 452
families (comprising of 3,204 precursors), downstream analysis for ortholog identification was carried out. 3,204 precursor sequences of miRBase were mapped on N22 genome by performing
blastn. Twenty-two had ≥50% precursor sequence similarity, which were checked for the conservation of mature sequence region. 16 out of 22 had ≤3 mismatches in the mature region. Secondary
structures were generated using RNAfold65 (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). PHYLOGENETIC CONSERVATION AND DIVERGENCE OF NOVEL MIRNAS Small RNA libraries were downloaded from
NCBI SRA database (http://www.ncbi.nlm.nih.gov/sra/) for two dicots, _viz. Arabidopsis thaliana_ (SRX058635, SRX058636 and SRX058638) and _Glycine max_ (SRX031157 and SRX131086); and four
monocots: _Oryza sativa_ (ERX014997, SRX017641 and SRX024857), _Triticum aestivum_ (SRX131958 and SRX131964), _Zea mays_ (SRX015774) and _Hordeum vulgare_ (SRR513546, SRR513547 and
SRR513548). Trimmed sRNA reads were aligned to the mature novel miRNA sequences. EXPRESSION PROFILING ON TAG DENSITY The expression profile of the novel miRNAs was determined by mapping the
sRNA libraries from flag leaf, spikelets (heading and anthesis) and mature roots from control and drought treated N22 plants (Supplementary Tables S11 and S12). The number of tags mapping to
each novel miRNA was normalized and the expression values were represented as reads per million (RPM) of the total genome-mapped sRNA tags in the library. The statistical significance of
the fold change was calculated as reported earlier66,67. QUANTITATIVE RT-PCR Total RNA was extracted from harvested tissues using TRI Reagent (Sigma) followed by DNaseI treatment
(Fermentas). Small RNA samples enriched using 4M LiCl were polyadenylated using Poly(A) Tailing kit (Ambion) and 2 μg of each sample was reversed transcribed with miR_oligodT_RTQ primer for
first strand cDNA synthesis using SuperScript II Reverse Transcriptase (Invitrogen). To analyze the expression of the mature miRNAs, qRT-PCR was done using TaqMan Fast Universal PCR Master
Mix (ABI) with RTQ universal reverse primer, miRNA specific forward primer and fluorogenic probe68 in StepOnePlus Real-Time PCR System (ABI) according to the manufacturer’s protocol. The
expression level of miRNA was normalized using 5S rRNA’s expression as an endogenous control. ∆∆Ct method was employed to calculate relative fold change (2−∆∆Ct) in expression and standard
error was calculated. The detail of primers used for qRT-PCR expression analysis is given in Supplementary Table S15. DEGRADOME LIBRARY PREPARATION Degradome libraries were prepared38 from
three different tissues of N22, _viz_. heading spikelet, anthesis spikelet and flag leaf at heading stage. Briefly, polyA enriched mRNAs obtained by using Oligotex kit, (QIAGEN) were ligated
with 5′-RNA adapter having an _Mme_I site using T4 RNA Ligase (Ambion) and reversed transcribed with an extended oligo(dT) having a 3′ adapter sequence using Superscript II RT (Invitrogen).
A short PCR of 15 cycles was performed to amplify the cDNA and the PCR product was digested with _Mme_I (NEB) to generate equal-sized fragments that was recovered by 12% PAGE. Then a
double-stranded 3′-DNA adapter was ligated to the _Mme_I digested products. The resulting product was PCR-amplified (21 cycles), gel-purified, cloned in PUC19 to check the quality of library
prepared and then given for high-throughput sequencing by Illumina HiSeq 2000 platform. Details of all the primer and adapters used in degradome library preparation are given in
Supplementary Table S15. The degradome data was then analyzed with the help of CleaveLand (ver. 3.1.1) pipeline to identify target genes and generate t-plots, utilizing the mature sequences
of sRNAs and cDNA transcripts of RGAP (TIGR 7). As per the analysis, the targets are identified in 5 categories (category ‘0’ to category ‘4’)39. Category ‘0’ is the best while category ‘4’
is the least significant. Category ‘0’ indicates the best possible condition in which the cleaved site has the maximum number of degradome reads (>1) and there is only one position in the
transcript for this maximum value. Category ‘1’ means >1 read which is equal to the maximum number of reads on the transcript when there is >1 position at maximum value. Category ‘2’
means >1 read which is >average depth but not the maximum on the transcript. Category ‘3’ means >1 read but ≤average depth on transcript while category ‘4’ means a single read at
that position. P-value is the indication of statistical significance and is based on the number and distribution of the degradome reads on the target transcript with reference to the
miRNA-binding site. Alignment score is based on miRNA-mRNA alignments and is calculated on position-weighted scoring matrix39. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Mutum, R. D.
_et al._ Identification of novel miRNAs from drought tolerant rice variety Nagina 22. _Sci. Rep._ 6, 30786; doi: 10.1038/srep30786 (2016). REFERENCES * Chen, X. MicroRNA biogenesis and
function in plants. _FEBS Letters_ 579, 5923–5931 (2005). Article CAS PubMed PubMed Central Google Scholar * Jeong, D.-H. & Green, P. J. The Role of Rice microRNAs in Abiotic Stress
Responses. _J. Plant Biol._ 56, 187–197 (2013). Article CAS Google Scholar * Robert-Seilaniantz, A. et al. The microRNA miR393 re-directs secondary metabolite biosynthesis away from
camalexin and towards glucosinolates. _Plant J._ 67, 218–231 (2011). Article CAS PubMed Google Scholar * Liu, J. X. et al. Genetic variation in the sensitivity of anther dehiscence to
drought stress in rice. _Field Crop Res._ 97, 87–100 (2006). Article Google Scholar * Gorantla, M. et al. Identification of stress-responsive genes in an _indica_ rice (_Oryza sativa_ L.)
using ESTs generated from drought-stressed seedlings. _J. Exp. Bot._ 58, 253–265 (2007). Article CAS PubMed Google Scholar * Jagadish, S. V. K. et al. Physiological and proteomic
approaches to address heat tolerance during anthesis in rice (_Oryza sativa_ L.). _J. Exp. Bot._ 61, 143–156 (2009). Article PubMed Central Google Scholar * Mitsui, T., Shiraya, T.,
Kaneko, K. & Wada, K. Proteomics of rice grain under high temperature stress. _Front. Plant Sci._ 4, 1–5 (2013). Article Google Scholar * Mutum, R. D. et al. Evolution of
variety-specific regulatory schema for expression of osa-miR408 in _indica_ rice varieties under drought stress. _FEBS J._ 280, 1717–1730 (2013). Article CAS PubMed Google Scholar *
Kansal, S. et al. Unique miRNome during anthesis in drought-tolerant _indica_ rice var. Nagina 22. _Planta_ 241, 1543–1559 (2015). Article CAS PubMed Google Scholar * Lenka, S. K.,
Katiyar, A., Chinnusamy, V. & Bansal, K. C. Comparative analysis of drought-responsive transcriptome in _Indica_ rice genotypes with contrasting drought tolerance. _Plant Biotechnol J._
9, 315–327 (2010). Article PubMed Google Scholar * Joshi, T. et al. Prediction of novel miRNAs and associated target genes in _Glycine max_ . _BMC Bioinformatics_ 9, 1–9 (2010). Google
Scholar * Schreiber, A. W., Shi, B.-J., Huang, C.-Y., Langridge, P. & Baumann, U. Discovery of barley miRNAs through deep sequencing of short reads. _BMC Genomics_ 12, 129 (2011).
Article CAS PubMed PubMed Central Google Scholar * Zhao, C. Z. et al. Deep sequencing identifies novel and conserved microRNAs in peanuts (_Arachis hypogaea_ L.). _BMC Plant Biol._ 10,
1–12 (2010). Article Google Scholar * Barrera-Figueroa, B. E. et al. High throughput sequencing reveals novel and abiotic stress-regulated microRNAs in the inflorescences of rice. _BMC
Plant Biol._ 12, 1–11 (2012). Article Google Scholar * Peng, T. et al. Characterization and expression patterns of microRNAs involved in rice grain filling. _PLoS ONE_ 8, e54148 (2013).
Article ADS CAS PubMed PubMed Central Google Scholar * Yi, R. et al. Identification and expression analysis of microRNAs at the grain filling stage in rice (_Oryza sativa_ L.) via Deep
Sequencing. _PLoS ONE_ 8, e57863 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Axtell, M. J. Classification and comparison of small RNAs from plants. _Annu. Rev.
Plant Biol._ 64, 137–159 (2013). Article CAS PubMed Google Scholar * Kurihara, Y. & Watanabe, Y. _Arabidopsis_ micro-RNA biogenesis through Dicer-like 1 protein functions. _Proc.
Natl. Acad. Sci. USA_ 101, 12753–12758 (2004). Article ADS CAS PubMed Google Scholar * Rajagopalan, R., Vaucheret, H., Trejo, J. & Bartel, D. P. A diverse and evolutionarily fluid
set of microRNAs in _Arabidopsis thaliana_ . _Genes Dev._ 20, 3407–3425 (2006). Article CAS PubMed PubMed Central Google Scholar * Park, M. Y., Wu, G., Gonzalez-Sulser, A., Vaucheret,
H. & Poethig, R. S. Nuclear processing and export of microRNAs in _Arabidopsis_ . _Proc. Natl. Acad. Sci. USA_ 102, 3691–3696 (2005). Article ADS CAS PubMed Google Scholar * Yu, B.
et al. Methylation as a crucial step in plant microRNA biogenesis. _Science_ 307, 932–935 (2005). Article ADS CAS PubMed PubMed Central Google Scholar * Yang, Z., Ebright, Y. W., Yu,
B. & Chen, X. HEN1 recognizes 21–24 nt small RNA duplexes and deposits a methyl group onto the 2′ OH of the 39 terminal nucleotide. _Nucleic Acids Res._ 34, 667–675 (2006). Article CAS
PubMed PubMed Central Google Scholar * Vaucheret, H., Vazquez, F., Cre´te´, P. & Bartel, D. P. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway
are crucial for plant development. _Genes Dev._ 18, 1187–1197 (2004). Article CAS PubMed PubMed Central Google Scholar * Devers, E. A., Branscheid, A., May, P. & Krajinski, F. Stars
and symbiosis: microRNA- and microRNA*-mediated transcript cleavage involved in arbuscular mycorrhizal symbiosis. _Plant Physiol._ 156, 1990–2010 (2011). Article CAS PubMed PubMed
Central Google Scholar * Shao, C., Ma, X., Xu, X. & Meng, Y. Identification of the highly accumulated microRNA*s in _Arabidopsis_ (_Arabidopsis thaliana_) and rice (_Oryza sativa_).
_Gene_ 515, 123–127 (2013). Article CAS PubMed Google Scholar * Ambros, V. et al. A uniform system for microRNA annotation. _RNA_ 9, 277–279 (2003). Article CAS PubMed PubMed Central
Google Scholar * Jeong, D. H. et al. Massive analysis of rice small RNAs: mechanistic implications of regulated MicroRNAs and variants for differential target RNA cleavage. _Plant Cell_
23, 4185–4207 (2011). Article CAS PubMed PubMed Central Google Scholar * Meyers, B. C. et al. Criteria for annotation of plant MicroRNAs. _Plant Cell_ 20, 3186–3190 (2008). Article CAS
PubMed PubMed Central Google Scholar * Tong, Y. A. et al. Genome-wide analysis reveals diversity of rice intronic miRNAs in sequence structure, biogenesis and function. _PLoS ONE_ 8,
e63938 (2013). Article ADS PubMed PubMed Central Google Scholar * Peng, H. et al. MicroRNA profiles and their control of male gametophyte development in rice. _Plant Mol. Biol._ 80,
85–102 (2012). Article CAS PubMed Google Scholar * Creighton, C. J., Reid, J. G. & Gunaratne, P. H. Expression profiling of microRNAs by deep sequencing. _Brief Bioinform._ 10,
490–497 (2009). Article CAS PubMed PubMed Central Google Scholar * Wu, L. et al. Rice microRNA effector complexes and targets. _Plant Cell_ 21, 3421–3435 (2009). Article CAS PubMed
PubMed Central Google Scholar * Wu, L. et al. DNA Methylation mediated by a microRNA pathway. _Mol. Cell_ 38, 465–475 (2010). Article CAS PubMed Google Scholar * Mi, S. et al. Sorting
of small RNAs into _Arabidopsis_ Argonaute complexes is directed by the 5′ terminal nucleotide. _Cell_ 33, 116–127 (2008). Article Google Scholar * Takeda, A., Iwasaki, S., Watanabe, T.,
Utsumi, M. & Watanabe, Y. The mechanism selecting the guide strand from small RNA duplexes is different among Argonaute proteins. _Plant Cell Physiol._ 49, 493–500 (2008). Article CAS
PubMed Google Scholar * Roberts, J. T., Cardin, S. E. & Borchert, G. M. Burgeoning evidence indicates that microRNAs were initially formed from transposable element sequences. _Mob.
Genet. Elements_ 4, e29255 (2014). Article PubMed PubMed Central Google Scholar * Addo-Quaye, C., Eshoo, T. W., Bartel, D. P. & Axtell, M. J. Endogenous siRNA and miRNA targets
identified by sequencing of the _Arabidopsis_ degradome. _Curr. Biol._ 18, 758–762 (2008). Article CAS PubMed PubMed Central Google Scholar * German, M. A., Luo, S., Schroth, G.,
Meyers, B. C. & Green, P. J. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. _Nat. Protoc._ 4, 356–362
(2009). Article CAS PubMed Google Scholar * Addo-Quaye, C., Miller, W. & Axtell, M. J. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets.
_Bioinformatics_ 25, 130–131 (2009). Article CAS PubMed Google Scholar * Zheng, Y., Li, Y. F., Sunkar, R. & Zhang, W. SeqTar: an effective method for identifying microRNA guided
cleavage sites from degradome of polyadenylated transcripts in plants. _Nucleic Acids Res._ 40, e28 (2012). Article CAS PubMed Google Scholar * Hori, K. et al. Hd16, a gene for casein
kinase I, is involved in the control of rice flowering time by modulating the day-length response. _Plant J._ 76, 36–46 (2013). CAS PubMed PubMed Central Google Scholar * Lee, Y. S.
& An, G. Complex regulatory networks of flowering time in rice. _Rice Res_. 3, 1–9 (2015). CAS Google Scholar * Wu, G. & Poethig, R. S. Temporal regulation of shoot development in
_Arabidopsis thaliana_ by miR156 and its target SPL3. _Development_ 133, 3539–3547 (2006). Article CAS PubMed PubMed Central Google Scholar * Wu, G. Plant MicroRNAs and Development. _J.
Genet. Genomics_ 40, 217–230 (2013). Article CAS PubMed Google Scholar * Piriyapongsa, J. & Jordan, I. K. Dual coding of siRNAs and miRNAs by plant transposable elements. _RNA_ 14,
814–821 (2008). Article CAS PubMed PubMed Central Google Scholar * Maher, C., Stein, L. & Ware, D. Evolution of _Arabidopsis_ microRNA families through duplication events. _Genome
Res._ 16, 510–519 (2006). Article CAS PubMed PubMed Central Google Scholar * Arikit, S. et al. An Atlas of Soybean Small RNAs Identifies Phased siRNAs from Hundreds of Coding Genes.
_Plant Cell_ 26, 4584–4601 (2014). Article CAS PubMed PubMed Central Google Scholar * Axtell, M. J. & Bowman, J. L. Evolution of plant microRNAs and their targets. _Trends Plant
Sci._ 13, 343–349 (2008). Article CAS PubMed Google Scholar * Wang, Y. et al. Genomic dissection of small RNAs in wild rice (_Oryza rufipogon_): lessons for rice domestication. _New
Phytol._ 196, 914–925 (2012). Article CAS PubMed Google Scholar * Li, Y. F., Zheng, Y., Addo-quaye, C. & Liu, J.-Y. Transcriptome-wide identification of microRNA targets in rice.
_Plant J._ 168, 742–759 (2010). Article Google Scholar * Zhou, M. et al. Degradome sequencing reveals endogenous small RNA targets in rice (_Oryza sativa L_. ssp. _indica_). _Front Biol_.
5, 67–90 (2010). Article CAS Google Scholar * Yoshida, S. _Fundamentals of Rice Crop Science._ IRRI, Los Banos, Philippines, Ch. 1, 1–61 (1981). * Nakano, H., Makino, A. & Mae, T.
Effects of panicle removal on the photosynthetic characteristics of the flag leaf of rice plants during the ripening stage. _Plant Cell Physiol._ 36, 653–659 (1995). CAS Google Scholar *
Paul, S., Gayen, D., Datta, S. K. & Datta, K. Dissecting root proteome of transgenic rice cultivars unravels metabolic alterations and accumulation of novel stress responsive proteins
under drought stress. _Plant Sci._ 234, 133–143 (2014). Article Google Scholar * Jeong, J. S. et al. Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice
under field drought conditions. _Plant Physiol._ 153, 185–197 (2010). Article CAS PubMed PubMed Central Google Scholar * Turner, M. et al. Ectopic expression of miR160 results in auxin
hypersensitivity, cytokinin hyposensitivity, and inhibition of symbiotic nodule development in Soybean. _Plant Physiology_ 162, 2042–2055 (2013). Article CAS PubMed PubMed Central Google
Scholar * Kinoshita, N. et al. IAA-Ala Resistant3, an evolutionarily conserved target of miR167, mediates _Arabidopsis_ root architecture changes during high osmotic stress. _Plant Cell_
24, 3590–3602 (2012). Article CAS PubMed PubMed Central Google Scholar * Peng, T. et al. Differential expression of the microRNAs in superior and inferior spikelets in rice (_Oryza
sativa_). _J. Exp. Bot._ 62, 4943–4954 (2011). Article CAS PubMed Google Scholar * Wei, L. Q., Yan, L. F. & Wang, T. Deep sequencing on genome-wide scale reveals the unique
composition and expression patterns of microRNAs in developing pollen of _Oryza sativa_ . _Genome Biol._ 12, R53 (2011). Article CAS PubMed PubMed Central Google Scholar * Sunkar, R.,
Zhou, X., Zheng, Y., Zhang, W. & Zhu, J. K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. _BMC Plant Biol._ 8, 1–17 (2008). Article Google Scholar
* Vu, J. C. V., Gesch, R. W., Allen, L. H. Jr., Boote, K. J. & Bowes, G. CO2 Enrichment Delays a Rapid, Drought-Induced Decrease in Rubisco Small Subunit Transcript Abundance. _J.
Plant Physiol._ 155, 139–142 (1999). Article CAS Google Scholar * Nijhawan, A., Jain, M., Tyagi, A. K. & Khurana, J. P. Genomic Survey and Gene Expression Analysis of the Basic
Leucine Zipper Transcription Factor Family in Rice. _Plant Physiol._ 146, 333–350 (2008). Article CAS PubMed PubMed Central Google Scholar * Markham, N. R. & Zuker, M. UNAFold:
software for nucleic acid folding and hybridization. _Methods Mol. Biol._ 453, 3–31 (2008). Article CAS PubMed Google Scholar * Kozomara, A. & Griffiths-Jones, S. miRBase:
integrating microRNA annotation and deep-sequencing data. _Nucleic Acids Res._ 39, D152–D157 (2011). Article CAS PubMed Google Scholar * Hofacker, I. L. Vienna RNA secondary structure
server. _Nucleic Acids Res._ 31, 3429–3431 (2003). Article CAS PubMed PubMed Central Google Scholar * Niu, S. et al. Transcriptome/Degradome-Wide Discovery of MicroRNAs and Transcript
Targets in Two _Paulownia australis_ Genotypes. _PLoS ONE_ 9, e106736 (2014). Article ADS PubMed PubMed Central Google Scholar * Audic, S. & Claverie, J.-M. The Significance of
Digital Gene Expression Profiles. _Genome Res._ 7, 986–995 (1997). Article CAS Google Scholar * Ro, S., Park, C., Jin, J., Sanders, K. M. & Yan, W. A PCR-based method for detection
and quantification of small RNAs. _Biochem. Biophys. Res. Commun._ 351, 756–763 (2006). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work
was supported by the Department of Biotechnology (DBT), Government of India (Grant no. BT/PR10071/AGR/36/31/2007 and BT/PR628/AGR/36/674/2011). R.D.M. acknowledges fellowships from UGC
(University Grants Commission) and University of Delhi, S. Kumar from DBT, S.B. from University of Delhi, S. Kansal from CSIR (Council of Scientific and Industrial Research), India. AUTHOR
INFORMATION AUTHORS AND AFFILIATIONS * Department of Plant Molecular Biology, University of Delhi South Campus, Benito Juarez Road, New Delhi, 110021, India Roseeta Devi Mutum, Santosh
Kumar, Sonia Balyan, Shivani Kansal & Saurabh Raghuvanshi * National Institute of Plant Genome Research, Aruna Asaf Ali Marg, New Delhi, 110067, India Saloni Mathur Authors * Roseeta
Devi Mutum View author publications You can also search for this author inPubMed Google Scholar * Santosh Kumar View author publications You can also search for this author inPubMed Google
Scholar * Sonia Balyan View author publications You can also search for this author inPubMed Google Scholar * Shivani Kansal View author publications You can also search for this author
inPubMed Google Scholar * Saloni Mathur View author publications You can also search for this author inPubMed Google Scholar * Saurabh Raghuvanshi View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS R.D.M did the analysis, identification and expression profiling of novel miRNAs and orthologous miRNAs in N22, prepared and
analyzed degradome libraries and compiled the manuscript. S. Kumar assisted in screening of molecules and performing of real time analysis. S.B. provided small RNA libraries, S. Kansal
helped in initial screening. S.M. generated the genome sequence of N22 while S.R. conceptualized and supervised the study. CORRESPONDING AUTHOR Correspondence to Saurabh Raghuvanshi. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION (PDF 11478 KB) SUPPLEMENTARY TABLE S1 (XLSX 32 KB)
SUPPLEMENTARY TABLE S2 (XLSX 13 KB) SUPPLEMENTARY TABLE S3 (XLSX 10 KB) SUPPLEMENTARY TABLE S4 (XLSX 35 KB) SUPPLEMENTARY TABLE S5 (XLSX 53 KB) SUPPLEMENTARY TABLE S6 (XLSX 8 KB)
SUPPLEMENTARY TABLE S7 (XLSX 9 KB) SUPPLEMENTARY TABLE S8 (XLSX 8 KB) SUPPLEMENTARY TABLE S9 (XLSX 64 KB) SUPPLEMENTARY TABLE S10 (XLSX 12 KB) SUPPLEMENTARY TABLE S11 (XLSX 84 KB)
SUPPLEMENTARY TABLE S12 (XLSX 64 KB) SUPPLEMENTARY TABLE S13 (XLSX 12 KB) SUPPLEMENTARY TABLE S14 (XLSX 11 KB) SUPPLEMENTARY TABLE S15 (XLSX 9 KB) RIGHTS AND PERMISSIONS This work is
licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license,
unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce
the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Mutum, R., Kumar, S., Balyan, S.
_et al._ Identification of novel miRNAs from drought tolerant rice variety Nagina 22. _Sci Rep_ 6, 30786 (2016). https://doi.org/10.1038/srep30786 Download citation * Received: 11 February
2016 * Accepted: 11 July 2016 * Published: 08 August 2016 * DOI: https://doi.org/10.1038/srep30786 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative







