- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Indian tasar silkmoth, _Antheraea mylitta_ is an economically important wild silkmoth species distributed across India. A number of morphologically and ethologically well-defined
ecotypes are known for this species that differ in their primary food plant specificity. Most of these ecotypes do not interbreed in nature, but are able to produce offspring under captive
conditions. Microsatellite markers were developed for _A. mylitta_ and out of these, ten well-behaved microsatellite loci were used to analyze the population structure of different ecoraces.
A total of 154 individual moths belonging to eight different ecoraces, were screened at each locus. Hierarchical analysis of population structure using Analysis of MOlecular VAriance
(AMOVA) revealed significant structuring (FST = 0.154) and considerable inbreeding (FIS = 0.505). A significant isolation by distance was also observed. The number of possible population
clusters was investigated using distance method, Bayesian algorithm and self organization maps (SOM). The first two methods revealed two distinct clusters, whereas the SOM showed the
different ecoraces not to be clearly differentiated. These results suggest that although there is a large degree of phenotypic variation among the different ecoraces of _A. mylitta_,
genetically they are not very different and the phenotypic differences may largely be a result of their respective ecology. SIMILAR CONTENT BEING VIEWED BY OTHERS GENETIC DIVERSITY,
POPULATION STRUCTURE, AND DNA FINGERPRINTING OF _AILANTHUS ALTISSIMA_ VAR. _ERYTHROCARPA_ BASED ON EST-SSR MARKERS Article Open access 07 November 2023 GENETIC DIVERSITY AND CONSERVATION OF
SIBERIAN APRICOT (_PRUNUS SIBIRICA_ L.) BASED ON MICROSATELLITE MARKERS Article Open access 11 July 2023 DNA FINGERPRINTING, FIXATION-INDEX (FST), AND ADMIXTURE MAPPING OF SELECTED BAMBARA
GROUNDNUT (_VIGNA SUBTERRANEA_ [L.] VERDC.) ACCESSIONS USING ISSR MARKERS SYSTEM Article Open access 15 July 2021 INTRODUCTION Populations of several species are further classified by
taxonomists into subspecies, races, demes, clines and so on, of which only cline and deme have non-arbitrary definitions1. Ecotypes consist of genetically distinct subsets of populations in
a species, that are specialized to particular environments2,3. More generally, ecotypes can be defined as subsets of populations within a species that have different fundamental niches4. The
Indian tasar silkworm, _Antheraea mylitta_ is a natural fauna of tropical India, distributed in different geographical locations and habitats in this country. Possibly, because of the
distinct ecological conditions prevailing in these different localities, several morphological variants, traditionally called ecoraces, have been identified in _A. mylitta_5. As high as 44
ecoraces are reported in this species, which feed primarily on _Terminalia_ (Family: Combretaceae) species and _Shorea robusta_ (Family: Dipterocarpaceae) and also on a number of secondary
food plants5. The genus _Terminalia_ and _Shorea_ are quite far phylogenetically. Although both are Rosids, _Terminalia_ belong to order Myrtales and _Shorea_ to Malvales6. The ecoraces are
uni, bi or trivoltine depending upon the geo-ecological conditions and differ from each other in several qualitative and quantitative traits7, such as cocoon weight and colour, larval colour
and so on. Although most of these ecotypes do not interbreed in nature, some of them produce offspring when mated in captivity8. Tasar cocoons are reported to be the largest among all the
silk-producing insects in the world9. The silk fibre produced by _A. mylitta_ has its own distinctive colour and is coarser than _Bombyx mori_ silk. However, the tasar silk has higher
tensile strength, elongation and stress-relaxation values than the silk secreted by the domesticated silkworm _B. mori._ These properties have made tasar silk as competent and desirable as
_B. mori_ silk10,11. Since, _A. mylitta_ primarily inhabits forested habitats, it is expected that with the gradual depletion of forest cover due to surge in the human activities the habitat
lost its continuity and resulted in geographic isolation. This isolation might have allowed the populations to continue separately for generations, resulting in different ecoraces. There is
quite a bit of ambiguity in naming of these ecoraces. As the boundaries between the ecoraces are often fuzzy and the races do not obey the concept of static boundaries, the racial identity
seems to be arbitrary at times. Quite surprisingly, two crops of the same population are sometimes named as two different ecoraces12. There is a lack of well-characterized molecular markers
to study _A. mylitta_ ecoraces. Although few studies have been carried out with RFLP13, RAPD14, SCAR15, ISSR12,16 and other DNA markers17, except for RFLP, the rest are dominant markers and
hence the estimation of allele frequencies are based on the assumption that the loci are in Hardy-Weinberg equilibrium. Also, barring a couple of reports on _Antheraea assama_18,19,20, there
are no reported studies that describe the detailed population genetics of saturniid silkmoths, using SSR markers. The pronounced phenotypic and behavioral variation of _A. mylitta_ ecoraces
have made it difficult for researchers to identify ecorace specific phenotypic markers8. Therefore, there is a need to identify genetic markers for a specific phenotype to differentiate
ecoraces. Are these ecoraces genetically distinct from each other? Do the ecoraces form structured population? Are any of these ecoraces in decline? These questions are of considerable
importance to the biology of _A. mylitta_. With these points in mind, we developed 32 microsatellite markers and screened eight _A. mylitta_ ecoraces collected from different geographical
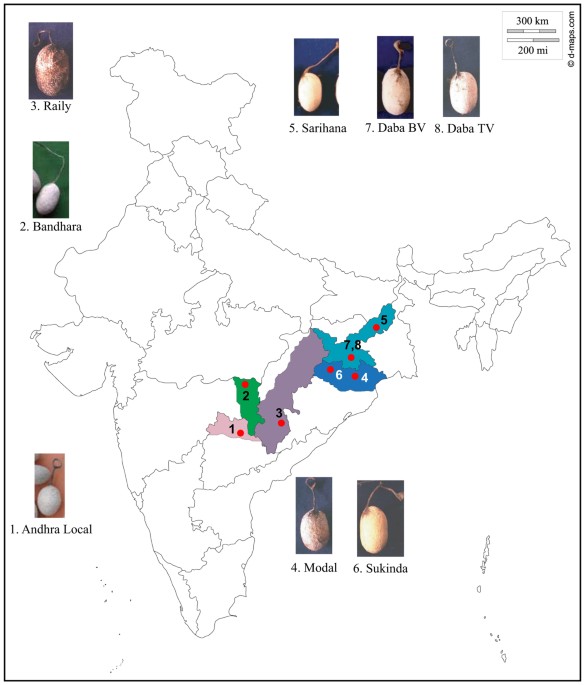
locations across India to obtain insights into the population genetics of these ecoraces (Fig. 1 and Table 1). RESULTS GENETIC VARIATION DESCRIPTIVE STATISTICS Considerable variation was
observed at all microsatellite loci. Except Bhandara, which was monomorphic at the locus Amysat013, all other populations showed polymorphism at all the 10 loci (Supplementary data 1).
Number of alleles ranged from 1 (Locus: Amysat013, Population: Bhandara) to 15 (Locus: Amysat023, Population: Modal) across the ten microsatellite loci studied among the eight ecoraces. The
average number of alleles per locus ranged between 2.5 ± 0.76SD (Amysat013) and 11.75 ± 2.05SD (Amysat023). The minimum average number of alleles for a population across all 10 loci was 3.7
± 1.77SD for Sukinda and the maximum was 6.6 ± 4.24SD for Modal. Average observed heterozygosity per locus (Ho) taking all the populations together ranged from 0 (Amysat013) to 0.81 ± 0.12
(Amysat023). The minimum average Ho across all the loci was observed in the population Daba Trivoltine (0.25 ± 0.32) and maximum in Sukinda (0.37 ± 0.34). HWE Hardy-Weinberg exact tests were
done for all the ten loci and inferences were drawn based on the sequential Bonferroni corrected α (Supplementary data 2, Table 2). No test could be performed for locus Amysat013 for the
Bhandara population, as the locus was monomorphic in this population. Also GENEPOP did not return any result for the locus Amysat037 in case of the populations Modal and Daba Bivoltine.
Closer inspection of the data revealed that in these two populations, the expected (He) and observed heterozygosities (Ho) for this locus are exactly the same. Generally, the more similar He
and Ho are, the higher is the probability that the locus is in HWE. This is supported by the observations in all other loci. A similar exact test21,22 was also performed in ARLEQUIN 3.023,
where the p-value of the HWE test in both these cases was 1.0. Hence, these two scenarios were inferred with a p-value of 1. Six of the ten loci were in HWE in at least two populations.
Amysat026 was in equilibrium in all the eight populations and Amysat001 was in equilibrium in seven populations. Both Amysat023 and Amysat037 were in equilibrium in six populations. Only
Amysat013 was not in equilibrium in any of the populations. Seven out of ten loci were in equilibrium for the Sukinda population and for Sarihana, only two of the loci were in equilibrium.
Among other reasons, departures from HWE expectations may occur in a case where a locus contains null alleles. MICROCHECKER was used to evaluate the effect of null alleles on the results.
Except four loci (Amysat001, Amysat023, Amysat026, Amysat037), all other loci were observed to have significant homozygote excess indicating the presence of possible null alleles. LD Exact
test for genotypic disequilibrium was performed using Markov chain simulations to identify significant association between pairs of loci. Tests for each population as well as global tests
were carried out. As there are 10 loci in all, there can be 45 possible pairs. However, due to lack of polymorphism for Amysat013 in the Bhandara population, only 36 tests could be performed
for this population. Out of these 351 total comparisons, significant LD, following sequential Bonferroni correction, could be observed in only three cases, i.e. Loci Amysat023 &
Amysat032, Amysat026 & Amysat032 and Amysat023 & Amysat026, all in Sukinda population. In case of the global test, following sequential Bonferroni correction, only three pairs of
loci (Amysat023 & Amysat032, Amysat023 & Amysat026 and Amysat026 & Amysat032), out of 45 comparisons were found to be under LD. All other loci segregated independently of each
other (Supplementary data 3). F-STATISTICS AND AMOVA The locus specific fixation indices are shown in Table 2. Except Amysat026 and Amysat032, all other loci had a significant FST value. FIS
values were significant for all loci, except Amysat026. Result of the AMOVA, along with the fixation indices and their bootstrap confidence intervals are shown in Table 3. As the proportion
of missing data was very less, the results of the haplotype as well as the locus by locus analysis were very similar. Here, results of only the locus by locus analysis are reported.
Weighted average of the locus by locus FIS value was considerably high (0.505), suggesting significant inbreeding within these populations. FST was also observed to be significant (0.154).
In accordance with these values, 15.4% of the covariance was assigned to the across populations and 43% to the within population component. About 42% of the covariance could be assigned to
the within individuals component. MANTEL TEST The samples for Daba Bivoltine and Daba Trivoltine were collected from the same locality and hence geographic distance between them was zero.
The maximum pairwise physical distance, i.e., 1034.4 km was between Sarihana and Andhra Local populations. Pairwise FST values ranged from 0.015 (between Sarihana and Daba Bivoltine
populations) to 0.339 (between Bhandara and Sukinda populations). All pairwise genetic distance values were significant except the one between Sarihana and Daba Bivoltine populations (FST =
0.015, p = 0.44) (see Supplementary data 4). The Mantel test revealed significant positive correlation between genetic and physical distance r = 0.59, p = 0.026 (Fig. 2). The Mantel test
between the residuals of a km-FST regression, however, was found not to be significant (r = 0.35, p = 0.055) suggesting a lack of migration-drift equilibrium (Fig. 3). BOTTLENECK ANALYSIS In
the BOTTLENECK analysis, none of the populations showed significant excess of simulated gene diversity compared to the observed gene diversity either under IAM or TPM, suggesting lack of
recent bottleneck in any of these eight populations. POPULATION ANALYSIS USING DISTANCE METHODS The NJ tree constructed based on Nei _et al._’s DA distance, showed two major clusters (Figure
S1), one with Andhra Local and Bhandara populations and another included remaining six ecoraces (Raily, Modal, Sukinda, Sarihana, Daba Bivoltine and Daba Trivoltine). NJ tree did not show
any specific relation among these six ecoraces. Although the two Daba ecoraces clustered together, the bootstrap value was found to be too less to make any strong conclusion. BAYESIAN
ANALYSIS OF PROBABLE POPULATION CLUSTERS The results of STRUCTURE analysis were very similar to that of the phylogenetic analysis. The modal value of ΔK was found to be at K = 2, suggesting
that the data is best explained consisting of only two populations. One group containing two ecoraces (Andhra Local and Bhandara) of southern and central India and the second group consisted
of remaining six ecoraces (Raily, Modal, Sarihana, Sukinda, Daba Bivoltine and Daba Trivoltine) mainly from eastern India (Fig. 4). ARTIFICIAL NEURAL NETWORK The SOM output layer consisted
of a 11 × 6 grid (Fig. 5). The map had a topographical error of 0. The quantization error was also reasonably small (1.792) suggesting the map quality to be good. The distances between
neighbouring units on this map are shown in Fig. 6a. One can also visually inspect the level of clustering by comparing Fig. 6a and Figure S3. A better visualization can be obtained by
looking at the hit histograms (Figure S2 in Supplementary file). From Fig. 5b, it appears that ecoraces Andhra Local (red) and Bhandara (green) are clustered together. However, this is a
crude way of clustering the data as boundaries of each cluster are difficult to visualize in this way. Therefore, this SOM was further subjected to cluster analysis using Ward’s linkage
(minimum variance criterion). The resulting dendrogram is shown in Fig. 6 (A higher resolution version can be found in Supplementary file, Figure S4). The Davies-Boulding clustering index24
indicated the best clustering to be 7 (Minimum value of the index 0.954 at level 7). The cluster boundaries are also indicated in Figure S3. If a cut-off is taken at squared Euclidean
distance of 5, Group 1 and the rest forms two distinct clusters, Group 1 consist of the highest number of neurons among these 7 groups and is dominated by Andhra Local and Bhandara
populations. However, these two ecoraces are not exclusive to this group and can be found in other groups as well. DISCUSSION In this paper, we have characterized the microsatellites of _A.
mylitta_ and used them to study the genetic structure of its different ecoraces. Like most other species studied, we observed dinucleotide microsatellites to be the most abundant, followed
by tri- and tetra nucleotide. Among the dinucleotide microsatellites, those with (CA) motifs were the most abundant. CA/GT repeats are generally the most common dinucleotide repeat in a wide
variety of vertebrates and arthropods25,26. Insects27,28, including lepidopterans29,30,31,32,33,34,35,36,37 are no exception. There have been studies investigating the nature and
distribution of genetic variation in wild lepidopterans. However, these have mainly focused on threatened or declining species38,39,40 or pests41,42,43,44. Among other silkworms the genetic
structure of _A. assama_, a species with a very restricted distribution, has been studied and its populations were found to be reasonably differentiated18,20. Different strains and lines of
the domesticated silkworm _B. mori_ have also been subjected to this kind of analysis32,45,46,47, however these studies are arguably not comparable to wild lepidopteran species. A couple of
studies using ISSR markers to analyze the intra-race diversity in Raily16 and Daba12 ecoraces of _A. mylitta_ revealed considerable genetic differentiation across and within the populations
of both the ecoraces. In another work, using RAPD markers, genetic variation among the different ecoraces of this moth were assessed14. However, none of the previous works did a detailed
study of the genetic structure of the different _A. mylitta_ ecoraces. Here, we have studied the genetic structure of _A. mylitta_ for a large part of its distribution. Eight ecoraces
distributed as far as 1000 km apart were collected and their population genetics was investigated using ten polymorphic microsatellite loci. There was significant deviation from HWE in some
of the loci in a few populations. This can be because of several reasons- 1) There was an overall excess of homozygotes than one would expect just by chance alone as reflected by the high
locus specific FIS values as well as its weighted average (0.505) across loci in the AMOVA (Table 3). As has been reported in the ‘Methods’ section, the sample cocoons were collected within
about 500 m radius of forest area and may consist of offspring of the same mother. This may be a possible reason of observing such a high FIS value. 2) The sample sizes for each population
was consistently low (Maximum N = 22, Minimum N = 15) and may not represent every genotype adequately and 3) Presence of null alleles, as was found using MICROCHECKER, which in turn may
result in the excess of homozygote in all the loci. Significant association was absent in almost all locus pairs in all the populations. Even globally, i.e., when all the populations were
included together, there was significant association between only three of the possible 45 pairs of loci. AMOVA revealed significant structuring of _A. mylitta_ populations in the region
under study (FST = 0.154). Although result of the Mantel test was significant (r = 0.59, p = 0.026), there was an absence of migration-drift equilibrium as reflected by the lack of
significant correlation between the residuals of a km-FST regression and km (r = 0.35, p = 0.055). Excluding the localities with non-significant pairwise FST values, i.e., Daba Bivoltine and
Sarihana, either sequentially or together, did not change this pattern, i.e., the km-FST correlation was consistently significant, but the km-residual Mantel was not significant. This
suggests a lack of migration-drift equilibrium in the region studied. One may posit from the extent of scatter of individual points in Figs 2 and 3 that random genetic drift has been
relatively more important than migration in shaping the current genetic structure of _A. mylitta_. However, as there are not too many points in the short distance range, this lack of
equilibrium can also be an artifact of the sampling regime48. Both distance method and Bayesian analysis divided _A. mylitta_ populations into two groups. One group consisted of one
population each from southern and central India (Andhra Local and Bhandara). Another consisted of populations from eastern India. To investigate this pattern further, Andhra Local and
Bhandara was taken as one group and the rest of the six populations were taken as another and an AMOVA was performed, giving 17% of the covariance assigned to the “Among groups” component
and 5% to the “Among populations within groups” component, as compared to the overall “among populations” component of 15.41%, suggesting the possibility of substructuring. This is in full
agreement with the STRUCTURE analysis. Remarkably, the two Daba ecoraces, though occurring sympatrically was found to be genetically distinct (FST = 0.06452). This is in line with the
previous report14, wherein they have used RAPD markers to differentiate 10 ecoraces of _A. mylitta_. In the NJ tree (Figure S1), except for the cluster consisting of Andhra Local and
Bhandara ecoraces, all other bootstrap values were quite low (33 to 53). We further investigated this pattern using Kohonen maps. Although the Bayesian clustering algorithm and SOM have been
shown to behave similarly for multilocus genotype data49, in the present study, the results of the two methods were not exactly the same. A hit map of the eight ecoraces revealed samples
from Andhra Local and Bhandara sharing the same or nearby neurons (Fig. 5b and Supplementary Figure S3). However in the hierarchical cluster analysis the pattern observed was not as clear as
that of the phylogenetic and Bayesian analysis. Although many samples from Andhra Local and Bhandara ecoraces did cluster together [Cluster 1 (Group I) in Fig. 6], there were other clusters
in which other samples from these ecoraces were distributed (Fig. 5b and Supplementary Figure S3). Clustering of Andhra Local and Bhandara has also been shown previously using RAPD
markers14. Andhra Local and Bhandara have adjacent distributions with no obvious geographic barrier between these two and the other six ecoraces (Fig. 1). This relationship shown in the
phylogenetic tree is in line with its distribution geographically. The _A. mylitta_ ecoraces have considerably different niches. They have different host plants, are behaviourally different,
have different cocoon colours and commercial characteristics17. The differences are so marked that they are popular with their unique local names in particular regions. Though there is a
notion that each ecotype is specific to a particular region, overlapping distribution across the contact zones cannot be ruled out50. Experiments carried out to evaluate the effect of
different food plants on cocoon characteristics revealed that food plants also have a considerable effect on these characteristics51. Though different morphologically identifiable ecoraces
are found in different geographical regions of India, we speculate that these may be the result of a particular ecological condition prevailing in that region and not because of their
distinct genotype. Considering the lack of ecorace specific structure observed in this study, we opine that there may be less number of genetically distinct ecoraces than reported earlier.
There seems to be a greater influence of environment such as host plants, climatic conditions, geographical locations etc., in shaping the phenotype of these ecoraces. Therefore, it is
likely that most of these ecoraces are genetically similar but differ in certain morphology. _A. mylitta_ is a highly polyphagous species and displays a wide range of adaptation.
Lepidopteran populations are known to be affected by habitat quality52 and food plants53. Either or both of these factors could be responsible for the variation in phenotype observed among
these ecoraces. In this current work, we present a detailed genetic study of eight different ecoraces of a wild silkworm _A. mylitta_. However, one should keep in mind that we do not have
replicates of the ecoraces in our dataset, i.e., each ecorace has been sampled only from a single locality and hence the effect of ecorace and locality might be confounded in the pattern we
have observed. Future work should concentrate on doing a more detailed behavioural and ecological study to understand the natural history of _A. mylitta_ in greater detail. Cross feeding
different ecoraces with the primary food plants of each other will help in confirming or ruling out the nature of host plant as a cause of phenotypic variation. Other ecological factors,
similarly, can be tested, either sequentially or together, to get into the root cause of the origin of these ecoraces. Recent developments in statistical modeling such as the generalized
linear model and the suit of different mixed models, allows one to carry out such investigations with great efficiency and cogency. Moreover, performing rigorous mating experiments may help
us to understand the cause and nature of reproductive barrier among the different ecoraces. From the perspective of its genetics, identifying the hybrid zones of different ecoraces and
studying the population structure including those regions may give a finer understanding of their population genetics. Development of additional microsatellite markers and employing a more
extensive sampling regime including samples at a finer distance scale would improve the resolution of the understood population genetic structure and, intra- and inter-race diversity of this
species. METHODS COLLECTION OF SAMPLES OF DIFFERENT ECORACES _A. mylitta_ is an exceptionally versatile species as its ecoraces vary, in terms of phenotype, voltinism, distribution and also
in their choice of food plants and some of the ecoraces do not freely interbreed54. For example, the ecoraces Andhra Local, Bhandara and Sarihana are notable for their smaller cocoons than
the others. On the other hand, the ecoraces Raily and Modal produce silk with significantly longer filament length than the other ecoraces. Wild populations of Daba can be both bi and
trivoltine in nature, while semi-domestic counterparts are exclusively either bivoltine or trivoltine. Ecoraces Modal and Raily are wild in nature and are found exclusively in natural
habitats12. The cocoon characteristics, sampling locations and information on primary food plants of all the eight ecoraces (Fig. 1) reported in the presented study are given in Table 1.
Live pupae were collected from their natural habitats in eight geographic locations representing eight ecoraces (Table 1 and Fig. 1). At each sampling site, wild cocoons were collected from
the respective host plants. All sampling at each site was done within ~500 m radius of forest area. CONSTRUCTION OF REPEAT ENRICHED GENOMIC LIBRARY AND SEQUENCING For the construction of
genomic library, DNA was isolated from _A. mylitta_ pupae using the method described earlier55. Genomic DNA was enriched using an oligonucleotide mix of (ATT)8, (GAGT)2, (CA)10, (GA)10,
(GATA)10, (CAC)7 and (AGC)7, following previously reported protocol56. In brief, DNA was digested with _Rsa_I and _Xmn_I restriction enzymes, ligated to double stranded superSNX linkers,
hybridized with biotinylated microsatellite oligonucleotides and captured on streptavidin coated magnetic beads. Unhybridized DNA was washed away and captured DNA was recovered by polymerase
chain reaction (PCR) using the single stranded superSNX-F as primer. The PCR products were ligated into the pCR®4-TOPO® TA vector (Invitrogen) and transformed into the XL1-Blue competent
cells. Colony PCR was performed to select the amplicons of size 500–1000 bp. In total, 155 of the 240 colonies screened contained inserts of size more than 500 bp, which were sequenced using
M13 forward and reverse primers, on ABI Prism 3100 Genetic Analyzer (Applied Biosystems). Among these, 46 had non-redundant microsatellite repeats, out of which a majority were found to
harbor dinucleotide repeat motifs (65.2%), followed by tri- (17.4%), tetra- (6.5%) and complex (10.9%) repeat motifs. Among dinucleotide repeat motifs CA was found to be more abundant (60%)
followed by GA (30%) and AT (10%). No CG motif was observed. A total of 16 clones were eliminated because the microsatellite sequences did not have sufficient flanking regions and so primer
sequences could not be designed for amplification. Therefore, primers were designed for the rest of the 30 genomic microsatellite loci. Out of which, eight loci either did not yield expected
product size or produced multiple non-specific bands. After the initial PCR assays, 22 microsatellite loci could be successfully amplified. EST SSRS _A. mylitta_ unigene EST sequences57
were downloaded from WildSilkBase. Out of 720 non-redundant EST sequences 50 were found to contain microsatellite repeats and 11 loci had insufficient flanking sequences to design the primer
pairs. A total of 14 loci were selected based on the length and type of repeat motifs (di, tri, tetra and penta) for primer synthesis. PCR AMPLIFICATION AND GENOTYPING OF SSRS We used
Primer3 program58 to design primers flanking SSRs in the EST clusters and microsatellite enriched sequences. PCR was carried out in a Mastercycler Gradient (Eppendorf, Germany) in a 10 μl
reaction containing 1X PCR buffer (MBI Fermentas), 100 μM dNTPs, 1.0 to 3.0 mM MgCl2, 5 pmole of each primers, 10 ng of genomic DNA as template and 0.5 U _Taq_ polymerase (MBI Fermentas).
The PCR conditions were initially at 94 °C for 3 minutes as initial denaturation and 35 cycles of: 94 °C denaturation for 30 seconds, appropriate annealing temperature (established
empirically) for 30 seconds and 72 °C extension for 45 seconds and 72 °C for 10 minutes as final extension. Of the 14 EST-SSRs and 30 genomic SSRs, 10 and 22 respectively, gave amplification
of exact size (Remaining were with multiple bands) as assessed by 1.5% agarose gel electrophoresis (Supplementary Table S1). To identify microsatellite loci that are polymorphic, we
randomly selected two individuals from each of the eight ecoraces (Fig. 1 and Table 1) and screened all the 32 microsatellite markers (Table 1 in Supplementary file). Out of these, we
finally selected 10 microsatellite marker loci based on polymorphism and absence of non-specific PCR amplifications. In all, 154 individuals from the eight ecoraces were genotyped using 10
microsatellite markers. For all the amplifications we used FAM-labeled fluorescent forward primer and unlabeled reverse primer. The PCR products were run on an ABI PRISM 3730 DNA analyzer,
with Pop-7 as sieving matrix, HiDi-Formamide as single-stranded DNA stabilizer and GeneScan 500 ROX as a size standard. Subsequently, ABI PRISM GeneMapper software version 3.0 was used to
size the alleles. Data on allele sizes in each individual was tabulated for genetic analysis. STATISTICAL ANALYSIS DESCRIPTIVE STATISTICS AND HARDY-WEINBERG EQUILIBRIUM For each locus,
number of alleles (_A_), number of alleles in each polymorphic locus (_Ap_) (polymorphism cut-off = 99%), expected heterozygosity as Nei’s unbiased estimates of genetic diversity (H_E_)59,
observed heterozygosity (H_O_) and Weir and Cockerham’s60 estimate of inbreeding coefficient (f) were assessed using Genetic Data Analysis 1.061. Exact test for Hardy-Weinberg equilibrium
(HWE) with the explicit alternative hypothesis of heterozygote deficiency62,63 was performed for each locus in each population using GENEPOP 4.0.5.364,65. Locus specific global tests taking
all the populations together were also performed with the same software. All tests were performed with the default parameters of 1000 dememorization steps and 20 batches per locus, with 5000
iterations per batch. All inferences were drawn following sequential Bonferroni correction66. The probability of null allele occurrence was estimated using MICROCHECKER67 in which null
alleles were considered to occur at a locus if an overall significant excess of homozygotes is seen, distributed evenly across the homozygote classes. LINKAGE DISEQUILIBRIUM Exact test for
genotypic composite linkage disequilibrium (LD)68 for each locus pair in each population was performed in GENEPOP with the default settings of 1000 dememorization steps, 20 batches per test
and 5000 iterations per batch64,65. The null hypothesis of an equilibrium was accepted or rejected following sequential Bonferroni correction. POPULATION DIFFERENTIATION AND MANTEL TEST
Pairwise FST values and permutation test to estimate their significance was performed in ARLEQUIN 3.5 using 10,000 permutations. Pairwise physical distance in kilometers using the GPS
coordinates of the sampling sites was estimated using GEOGRAPHIC DISTANCE MATRIX GENERATOR69. Correlation between Slatkin’s linearized estimate of FST70 and genetic distance, i.e. isolation
by distance (IBD), was tested with Mantel test71 in Mantel Tester, which is the program zt, with a user friendly GUI72. Mantel test was inferred following 10,000 permutations. In addition, a
Mantel test was done between the absolute residuals of a regression of FST/(1- FST) upon physical distance (km) and physical distance, to check for the existence of migration-drift
equilibrium, one of the basic assumptions of IBD48,73. BOTTLENECK ANALYSIS The BOTTLENECK program74 was used to determine if any signal of past bottleneck could be detected. The program was
run under infinite alleles model (IAM) and two-phase mutation model (TPM)75 with probabilities of single step and multi step mutations to be 0.9 and 0.1 respectively76. When a population
undergoes reduction in population size it consequently loses its genetic diversity. This is reflected in both loss in number of alleles as well as observed gene diversity. In a population
that has experienced such a reduction recently, the former parameter is lost faster than the latter. Therefore in such a population one would expect observed gene diversity to be higher than
expected gene diversity estimated from the number of alleles (k)77,78. BOTTLENECK is equipped with three different statistical methods to test for this, i.e., i) sign test, ii) standardized
difference test and iii) Wilcoxon sign-rank test. The first suffers from very low statistical power, the second can only be performed if the number of loci is more than 20, the third is
recommended for mot situations where there are reasonable numbers of markers and samples. Therefore Wilcoxon sign-rank test was used to compare the simulated gene diversity with the observed
gene diversity. ANALYSIS OF MOLECULAR VARIANCE Hierarchical population structure was evaluated with the AMOVA60,68,79 using the software ARLEQUIN 3.0. The analysis was performed choosing
two main hierarchical structures; 1) Taking all the eight populations together and including the individual level and 2) Taking the Andhra Local and Bhandara populations together in one
group and the rest six populations in another group (Rationale for doing so is in the ‘Discussion’ section). As the frequency of missing data was very low (maximum frequency of missing data
was 3% for Amysat025), both haplotype and locus by locus analyses were performed and their results compared. “Pairwise differences” was chosen as the distance method and 10,000 permutations
were used to test for the significance of covariance components and fixation indices. ARLEQUIN also returned bootstrap percentile values for the fixation indices. More than 20,000 bootstraps
were carried out for each fixation index. POPULATION ANALYSIS USING DISTANCE METHODS There are several methods of estimating distances among populations from microsatellite allele
frequencies and construct a tree from it. There are traditional distance estimates such as Nei’s standard genetic distance DST80, Cavalli-Sforza and Edward’s chord distance DC81 etc., which
are designed for loci mutating following IAM. Along with these, there are measures of genetic distance that have been explicitly defined to estimate distances based on microsatellite data,
such as Goldstein _et al._’s δμ81,82 and Shriver _et al._’s DSW83. All these distance measures have their own advantages and disadvantages. However, it has been shown using both computer
simulation84 as well as with real data85 that for constructing phylogenetic trees, the efficiency of (δμ)2 or DSW, is not very good. Rather, it was shown that the probability of the
traditional distance methods in returning the correct tree topology is more. In these studies, the measure DA86 was found to perform better than the traditional measures as well as the
microsatellite specific measures. And it was found to be efficient with both classical markers as well as microsatellites. Therefore, DA was selected as the distance measure to construct a
dendrogram for the microsatellite data of these eight populations. Neighbour joining (NJ) tree with 10000 bootstrap resampling of the loci87 was constructed using POPTREE288. BAYESIAN
ANALYSIS OF PROBABLE POPULATION CLUSTERS Phylogenetic clustering method using individual genotype data although widely used suffers from several limitations. The actual tree topology is
dependent on the genetic distance used and obtaining a reliable confidence interval with a few number of loci is not easy89. Therefore, we examined the number of distinct groups into which
individuals from these eight populations can be clustered using the Bayesian clustering program STRUCTURE 2.3.189,90,91. STRUCTURE uses a model based clustering technique to estimate the
number of potential genetic clusters into which the individuals in a sample can be grouped. Given a series of potential clusters (K), STRUCTURE estimates the likelihood of each K to the
data, which is achieved by estimating the number of groups that are in HWE and linkage equilibrium (LE). The K at which the likelihood reaches a plateau is taken as the number of optimal
cluster number for the dataset. STRUCTURE was run assuming admixture and correlated allele frequency models. The populations were assumed to be in LE. The sampling locations were taken as
putative population origin for the individuals. For the MCMC runs 100000 burn in period, followed by 100000 replications were used as recommended in the STRUCTURE manual. Value of K was
taken from 1 to 15 and for each K, 20 iterations were performed. The most probable K was inferred from the modal value of rate of change of the LnP(D) value between successive runs (ΔK) as
suggested by Evanno _et al._92. This “Evanno method” has been implemented in the web based program STRUCTURE HARVESTER and it was used to estimate ΔK for each value of K and identify the
mode93. ARTIFICIAL NEURAL NETWORKS We also analysed the data using unsupervised neural networks in the form self-organizing maps (SOM) or Kohonen maps94,95. This method allows one to explore
and visualize a multidimensional dataset into a two dimensional grid. It also allows one to look for possible clusters in the data96. It has been shown in a recent study that under moderate
differentiation, like in case of our data, classical clustering techniques like NJ, are not perfect in finding the correct number of clusters in the data. In such cases, Bayesian clustering
techniques like STRUCTURE and SOM were more efficient49. SOM has been used extensively in ecology97,98,99,100,101,102,103, but there have been very few instances of its application to
genetic data44,53,104. The network consists of two layers, -i) the input layer containing as many nodes as there are markers in a dataset, each of which is connected to all the individual
genotypes and ii) the output layer, which contains the neurons arranged in a two dimensional grid of hexagonal lattice. Each of these neurons is represented by weight vectors (also known as
the codebook vector), whose dimension is same as the number of nodes in the input. During the training of the map, distance between each input vector and all the weight vectors are
calculated and the hexagon closest to the input vector is called its best matching unit (BMU). Following this, all the weight vectors are update so that the BMU moves closer to the input
vector in the input layer. The topological neighbours of this neuron are also dragged along. This updating is continued in iterations. At the end of this process, individuals with similar
multilocus genotypes will be placed closer on the SOM. This then can be visualized using different techniques. Detailed account of the theoretical background as well as the algorithm to make
these maps can be found elsewhere95,104,105. Our data consisted of 131 different alleles across the 10 loci, which formed the number of nodes in our input layer. For each of these 131
markers an individual was coded as 1, 0.5, or 0, if it was either homozygote, heterozygote or lacks this marker respectively103. Individuals who had missing data for a particular locus were
coded as 0 for all alleles in that locus. We used a linear initialization of the SOM and trained it using a batch algorithm. The training was done in two phases–i) rough training phase with
large neighbourhood radii and ii) fine-tuning phase with smaller radii. We further subjected this SOM to hierarchical cluster analyses using an agglomerative clustering method106. These
analyses were performed using the SOM toolbox for MATLAB107 and SOMVIS108. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Chakraborty, S. _et al._ Genetic analysis of Indian tasar silkmoth
(_Antheraea mylitta_) populations. _Sci. Rep._ 5, 15728; doi: 10.1038/srep15728 (2015). REFERENCES * Mayr, E. & Ashlock, P. D. Principles of systematic zoology. (McGraw-Hill, 1991). *
Turesson, G. The genotypical response of the plant species to the habitat. Hereditas 3, 211–350 (1922). Article Google Scholar * Turesson, G. The species and variety as ecological units.
Hereditas 3, 100–113 (1922). Article Google Scholar * Begon, M., Townsend, C. R. & Harper, J. L. Ecology: from individuals to ecosystems. (Blackwell, 2006). * Jolly, M. S., Sen, S. K.
& Ahsan, M. M. Tasar Culture. (Ambika Publishers, 1974). * Wang, H. et al. Rosid radiation and the rapid rise of angiosperm-dominated forests. Proc. Natl. Acad. Sci. USA 106, 3853–3858
(2009). Article CAS ADS PubMed PubMed Central Google Scholar * Srivastava, A. K. et al. In Advances in Indian Sericulture Research (eds Dandin, S. B. Gupta, V. P. ). pp. 387–394.
(Central Sericultural Research and Training Institute, Mysore, India, 2002). * Singh, B. M. K. & Srivastava, A. K. In Current Technology Seminar on Non-mulberry Sericulture Base paper.
6, pp. 1–39. (CTR&TI, Ranchi, India, 1997). * Akai, H. Cocoon filament characters and post-cocoon technology. Int. J. Wild Silkmoth Silk 5, 255–259 (2000). Google Scholar * Iizuka, E.
Physical properties of silk thread from cocoons of various wild silkmoths including domestic silk moth. Int. J. Wild Silkmoth Silk 5, 266–269 (2000). Google Scholar * Rajkhowa, R. Structure
property correlation of non-mulberry and mulberry silk fibres. Int. J. Wild Silkmoth Silk 5, 287–298 (2000). Google Scholar * Kar, P. K. et al. Genetic Variability and genetic strucuture
of wild and semi-domestic populations of tasar silkworm (_Antheraea mylitta_) ecorace Daba as revealed through ISSR markers. Genetica 125, 173–183 (2005). Article CAS PubMed Google
Scholar * Mahendran, B., Padhi, B., Ghosh, S. K. & Kundu, S. Genetic variation in ecoraces of tropical tasar silkworm, _Antheraea mylitta_ D. using RFLF technique. Curr. Sci. 90, 100
(2006). CAS Google Scholar * Saha, M., Mahendran, B. & Kundu, S. C. Development of Random Amplified Polymorphic DNA markers for tropical tasar silkworm _Antheraea mylitta_. J Econ.
Entomol. 101, 1176–1182 (2008). Article CAS PubMed Google Scholar * Saha, M. & Kundu, S. C. Molecular Identification of tropical tasar silkworm (_Antheraea mylitta_) ecoraces with
RAPD and SCAR markers. Biochem. Genet. 44, 75–88 (2006). Article CAS PubMed Google Scholar * Srivastava, A. K., Kar, P. K., Sinha, R., Sinha, M. K. & Vijayaprakash, N. B. Assessment
of genetic diversity in different populations of Raily ecorace of Indian tasar silkworm, _Antheraea mylitta_ using ISSR marker. Int. J. Indust. Entomol. 19, 249–253 (2009). Google Scholar *
Mahendran, B., Acharya, C., Dash, R., Ghosh, S. K. & Kundu, S. C. Repetitive DNA in tropical tasar silkworm _Antheraea mylitta_. Genetica 370, 51–57 (2006). CAS Google Scholar *
Arunkumar, K. P. et al. Genetic diversity and population structure of Indian golden silkmoth (_Antheraea assama_). PLoS One 7, e43716, D:10.1371/journal.pone.0043716 (2012). Article ADS
CAS PubMed PubMed Central Google Scholar * Arunkumar, K. P., Kifayathullah, L. & Nagaraju, J. Microsatellite markers for the Indian golden silkmoth, _Antheraea assama_ (Saturniidae:
Lepidoptera). Mol. Ecol. Resour. 9, 268–270, doi: 10.1111/j.1755-0998.2008.02414.x (2009). Article CAS PubMed Google Scholar * Singh, Y. T. et al. Genetic variation within native
populations of endemic silkmoth _Antheraea assamensis_ (Helfer) from Northeast India indicates need for _in situ_ conservation. PLoS One 7, e49972, doi: 10.1371/journal.pone.0049972 (2012).
Article CAS ADS PubMed PubMed Central Google Scholar * Guo, S. W. & Thompson, E. A. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics, 48,
361–372 (1992). Article CAS PubMed MATH Google Scholar * Levene, H. On a matching problem arising in genetics. Ann. Math. Stat. 20, 91–94 (1949). Article MathSciNet MATH Google
Scholar * Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour., 10,
564–567 (2010). Article PubMed Google Scholar * Davies, D. L. & Bouldin, D. W. A Cluster Separation Measure. IEEE Trans. Pattern. Anal. Mach. Intell. 1(2), (1979). * Beckmann, J. S.
& Weber, J. L. Survey of human and rat microsatellites. Genomics 12, 627–631 (1992). Article CAS Google Scholar * Tóth, G., Gáspári, Z. & Jurka, J. Microsatellites in different
eukaryotic genomes: survey and analysis. Genome Res. 10, 967–981 (2000). Article PubMed PubMed Central Google Scholar * Schug, M. D. et al. The mutation rates of di-, tri- and
tetranucleotide repeats in _Drosophila melanogaster_. Mol. Biol. Evo.l 15, 1751–1760 (1998). Article CAS Google Scholar * Butcher, R., Hubbard, S. & Whitfield, W. Microsatellite
frequency and size variation in the parthenogenetic parasitic wasp _Venturia canescens_ (Gravenhorst) (Hymenoptera: Ichneumonidae). Insect Mol. Biol. 9, 375–384 (2000). Article CAS PubMed
Google Scholar * Reddy, K. D., Abraham, E. & Nagaraju, J. Microsatellites in the silkworm, _Bombyx mori_: abundance, polymorphism and strain characterization. Genome 42, 1057–1065
(1999). Article CAS PubMed Google Scholar * Luque, C., Legal, L., Staudter, H., Gers, C. & Wink, M. ISSR (inter simple sequence repeats) as genetic markers in Noctuids (Lepidoptera).
Hereditas 136, 251–253 (2002). Article CAS PubMed Google Scholar * Kumar, L. S., Sawant, A. S., Gupta, V. S. & Ranjekar, P. K. Comparative analysis of genetic diversity among Indian
populations of _Scirpophaga incertulas_ by ISSR-PCR and RAPD-PCR. Biochem. Genet. 39, 297–309 (2001). Article CAS PubMed Google Scholar * Reddy, K. D., Nagaraju, J. & Abraham, E. G.
Genetic characterization of the silkworm _Bombyx mori_ by simple sequence repeat (SSR)-anchored PCR. Heredity 83, 681–687 (1999). Article CAS PubMed Google Scholar * Bogdanowicz, S.,
Mastro, V., Prasher, D. & Harrison, R. Microsatellite DNA variation among Asian and North American gypsy moths (Lepidoptera: Lymantriidae). Ann. Entomol.Soc. Am. 90, 768–775 (1997).
Article CAS Google Scholar * Meglécz, E. et al. Microsatellite flanking region similarities among different loci within insect species. Insect Mol. Biol. 16, 175–185 (2007). Article CAS
PubMed Google Scholar * Hundsdoerfer, A. K. & Wink, M. New source of genetic polymorphisms in Lepidoptera? Z. Naturforsch. C 60, 618–624 (2005). Article CAS PubMed Google Scholar
* Meglécz, E. & Solignac, M. Microsatellite loci for _Parnassius mnemosyne_ (Lepidoptera). Hereditas 128, 179–180 (1998). Article Google Scholar * Palo, J., Varvio, S. L., Hansk, I.
& VäinÖlä, R. Deveploping Microsatellite Markers for Insect Population Structure: Complex Variation in a Checkerspot Butterfly. Hereditas 123, 295–300 (1995). Article CAS PubMed
Google Scholar * Keyghobadi, N., Roland, J. & Strobeck, C. Influence of landscape on the population genetic structure of the alpine butterfly _Parnassius smintheus_ (Papilionidae). Mol.
Ecol. 8, 1481–1495 (1999). Article CAS PubMed Google Scholar * Meglécz, E., Nève, G., Pecsenye, K. & Varga, Z. Genetic variations in space and time in _Parnassius mnemosyne_ (L.)
(Lepidoptera) populations in north-east Hungary: implications for conservation. Biol. Conserv. 89, 251–259 (1999). Article Google Scholar * Packer, L. et al. Population biology of an
endangered butterfly, _Lycaeides melissa samuelis_ (Lepidoptera; Lycaenidae): genetic variation, gene flow and taxonomic status. Can. J. Zool. 76, 320–329 (1998). Article Google Scholar *
Bourguet, D., Bethenod, M. T., Pasteur, N. & Viard, F. Gene flow in the European corn borer _Ostrinia nubilalis_: implications for the sustainability of transgenic insecticidal maize.
Proc. R. Soc. B 267, 117–122 (2000). Article CAS PubMed PubMed Central Google Scholar * Zhou, X., Faktor, O., Applebaum, S. W. & Coll, M. Population structure of the pestiferous
moth _Helicoverpa armigera_ in the Eastern Mediterranean using RAPD analysis. Heredity 85, 251–256 (2000). Article PubMed Google Scholar * Martel, C., Réjasse, A., Rousset, F., Bethenod,
M. & Bourguet, D. Host-plant-associated genetic differentiation in Northern French populations of the European corn borer. Heredity 90, 141–149 (2003). Article CAS PubMed Google
Scholar * Roux, O. et al. ISSR-PCR: Tool for discrimination and genetic structure analysis of _Plutella xylostella_ populations native to different geographical areas. Mol. Phylogenet.
Evol. 43, 240–250 (2007). Article CAS PubMed Google Scholar * Nagaraju, J., Reddy, K. D., Nagaraja, G. M. & Sethuraman, B. N. Comparison of multilocus RFLPs and PCR-based marker
systems for genetic analysis of the silkworm, Bombyx mori. Heredity 86, 588–597 (2001). CAS PubMed Google Scholar * Furdui, E. M. et al. Genetic Characterization of _Bombyx mori_
(Lepidoptera: Bombycidae) Breeding and Hybrid Lines With Different Geographic Origins. J. Insect Sci. 14(1) doi: 10.1093/jisesa/ieu073 (2014). * Pereira, N. et al. Biological and molecular
characterization of silkworm strains from the Brazilian germplasm bank of _Bombyx mori_. Genet. Mol. Res. 12, 2138–2147 (2013). Article CAS PubMed Google Scholar * Hutchison, D. W. &
Templeton, A. R. Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability.
Evolution 53, 1898–1914 (1999). Article PubMed Google Scholar * Peña-Malavera, A., Bruno, C., Fernandez, E. & Balzarini, M. Comparison of algorithms to infer genetic population
structure from unlinked molecular markers. Stat. Appl. Genet. Molec. Biol. 13, 391–402 (2014). Article MathSciNet Google Scholar * Pandey, S. K. Silk Culture: A Biochemical Approach. 254
(APH Publishing, 2005). * Kar, P. K., Srivastava, A. K. & Naqvu, A. H. Changes in voltinism in populations of _Antheraea mylitta_: Response to photoperiod. Int. J. Wild Silkmoths 5,
176–178 (2000). Google Scholar * Clarke, R., Thomas, J., Elmes, G. & Hochberg, M. E. The effects of spatial patterns in habitat quality on community dynamics within a site. Proc. R.
Soc. B 264, 347–354 (1997). Article ADS PubMed Central Google Scholar * Luque, C. et al. Apparent influences of host-plant distribution on the structure and the genetic variability of
local populations of the Purple Clay (_Diarsia brunnea_). Biochem. Syst. Ecol. 37, 6–15 (2009). Article CAS Google Scholar * Arora, G. S. & Gupta, I. J. Taxonomic studies of some of
the Indian non-mulberry silkmoths (Lepidoptera: Saturniidae: Saturniinae). Memoirs Zool. Surv. India 16, 1–63 (1979). Google Scholar * Prasad, M. D. & Nagaraju, J. A comparative
phylogenetic analysis of full-length mariner elements isolated from the Indian tasar silkmoth, _Antheraea mylitta_ (Lepidoptera: saturniidae). _J_. Bioscience 28, 443–453 (2003). Article
CAS Google Scholar * Glenn, T. C. & Schable, N. A. Isolating microsatellite DNA loci. Method. Enzymol. 395, 202–222 (2005). Article CAS Google Scholar * Arunkumar, K. P., Tomar, A.,
Daimon, T., Shimada, T. & Nagaraju, J. WildSilkbase: an EST database of wild silkmoths. BMC Genomics 9, 338, doi: 10.1186/1471-2164-9-338 (2008). Article CAS PubMed PubMed Central
Google Scholar * Rozen, S. & Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Method. Mol. Biol. 132, 365–386 (2000). CAS Google Scholar * Nei, M.
Molecular Evolutionary Genetics (Columbia University Press, New York, 1987). * Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution
38, 1358–1370 (1984). CAS PubMed Google Scholar * Lewis, P. O. & Zaykin, D. Genetic Data Analysis: Computer program for the analysis of allelic data. _Version 1.0 (d16c)._ (2001). *
Rousset, F. & Raymond, M. Testing heterozygote excess and deficiency. Genetics 140, 1413–1419 (1995). CAS PubMed PubMed Central Google Scholar * Raymond, M. & Rousset, F. An
exact test for population differentiation. Evolution 49, 1280–1283 (1995). Article PubMed Google Scholar * Rousset, F. genepop’007: a complete re-implementation of the genepop software
for Windows and Linux. Mol. Ecol. Resour. 8, 103–106, (2008) Article PubMed Google Scholar * Raymond, M. & Rousset, F. Genepop (version 1.2), population genetics software for exact
tests and ecumenicism. J. Hered. 86, 248–249 (1995). Article Google Scholar * Rice, W. R. Analyzing tables of statistical tests. Evolution 43, 223–225 (1989). Article PubMed Google
Scholar * van Oosterhout, C., Hutchison, W. F., Wills, D. P. M. & Shipley, P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol.
Notes 4, 535–538 (2004). Article CAS Google Scholar * Weir, B. S. Genetic Data Analysis II: Methods for Discrete Population Genetic Data. (Sinauer Associates, Inc., 1996). * Ersts, P.
Geographic distance matrix generator (version 1.2. 3). American Museum of Natural History, Center for Biodiversity and Conservation (2012). * Slatkin, M. A measure of population subdivision
based on microsatellite allele frequencies. Genetics 139, 457–462 (1995). CAS PubMed PubMed Central Google Scholar * Mantel, N. The detection of disease clustering and a generalized
regression approach. Cancer Res. 27, 209–220 (1967). CAS PubMed Google Scholar * Bonnet, E. & Van De Peer, Y. zt: a software tool for simple and partial Mantel tests. J. Stat.
Software 7, 1–12 (2002). Article Google Scholar * Wright, S. Isolation by distance. Genetics 28, 114 (1943). CAS PubMed PubMed Central Google Scholar * Cornuet, J. M. & Luikart, G.
Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014 (1996). CAS PubMed PubMed Central Google
Scholar * Di Rienzo, A. et al. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 91, 3166–3170 (1994). Article CAS ADS PubMed PubMed
Central Google Scholar * Luikart, G., Allendorf, F., Piry, S. & Cornuet, J. Molecular genetic test identifies endangered populations. Conserv. Biol. 12, 228–237 (1998). Article Google
Scholar * Cornuet, J. M. & Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014
(1996). CAS PubMed PubMed Central Google Scholar * Maruyama, T. & Fuerst, P. A. Population bottlenecks and nonequilibrium models in population genetics. II. Number of alleles in a
small population that was formed by a recent bottleneck. Genetics 111, 675–689 (1985). CAS PubMed PubMed Central Google Scholar * Excoffier, L., Smouse, P. E. & Quattro, J. M.
Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491 (1992). CAS PubMed PubMed
Central Google Scholar * Nei, M. Genetic disatance between populations. Am. Nat. 106, 283–292 (1972). Article Google Scholar * Cavalli-Sforza, L. L. & Edwards, A. W. Phylogenetic
analysis. Models and estimation procedures. Am. J. Hum. Genet. 19, 233–257 (1967). CAS PubMed PubMed Central Google Scholar * Goldstein, D. B., Ruiz Linares, A., Cavalli-Sforza, L. L.
& Feldman, M. W. Genetic absolute dating based on microsatellites and the origin of modern humans. Proc. Natl. Acad. Sci. USA 92, 6723–6727 (1995). Article CAS ADS PubMed PubMed
Central Google Scholar * Shriver, M. D. et al. A novel measure of genetic distance for highly polymorphic tandem repeat loci. Mol. Biol. Evol. 12, 914–920 (1995). CAS PubMed Google
Scholar * Takezaki, N. & Nei, M. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144, 389–399 (1996). CAS PubMed PubMed Central Google
Scholar * Takezaki, N. & Nei, M. Empirical tests of the reliability of phylogenetic trees constructed with microsatellite DNA. Genetics 178, 385–392 (2008). Article PubMed PubMed
Central Google Scholar * Nei, M., Tajima, F. & Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J. Mol. Evol. 19, 153–170 (1983).
Article CAS ADS PubMed Google Scholar * Felsenstein, J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791 (1985). Article PubMed Google Scholar
* Takezaki, N., Nei, M. & Tamura, K. POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface.
Mol. Biol. Evol. 27, 747–752, (2010). Article CAS PubMed Google Scholar * Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype
data. Genetics 155, 945–959 (2000). CAS PubMed PubMed Central Google Scholar * Hubisz, M., Falush, D., Stephens, M. & Pritchard, J. Inferring weak population structure with the
assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332 (2009). Article PubMed PubMed Central Google Scholar * Falush, D., Stephens, M. & Pritchard, J. K. Inference
of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003). CAS PubMed PubMed Central Google Scholar * Evanno,
G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–20 (2005). Article CAS PubMed Google
Scholar * Earl, D. & vonHoldt, B. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361
(2012). Article Google Scholar * Kohonen, T. Self-organized formation of topologically correct feature maps. Biol. Cybernet. 43, 59–69 (1982). Article MathSciNet MATH Google Scholar *
Kohonen, T. Self-Organizing Maps. (Springer Berlin Heidelberg, 2001). * Kohonen, T. Essentials of the self-organizing map. Neural Net. 37, 52–65 (2013). Article Google Scholar * Lek, S.
& Guégan, J.-F. Artificial neuronal networks: application to ecology and evolution. (Springer, 2000). * Lek, S. et al. Application of neural networks to modelling nonlinear relationships
in ecology. Ecol. Model. 90, 39–52 (1996). Article Google Scholar * Giraudel, J. & Lek, S. A comparison of self-organizing map algorithm and some conventional statistical methods for
ecological community ordination. Ecol. Model. 146, 329–339 (2001). Article Google Scholar * Park, Y. S., Chang, J., Lek, S., Cao, W. & Brosse, S. Conservation strategies for endemic
fish species threatened by the Three Gorges Dam. Conserv. Biol. 17, 1748–1758 (2003). Article Google Scholar * Park, Y.-S., Céréghino, R., Compin, A. & Lek, S. Applications of
artificial neural networks for patterning and predicting aquatic insect species richness in running waters. Ecol. Model. 160, 265–280 (2003). Article Google Scholar * Worner, S. &
Gevrey, M. Modelling global insect pest species assemblages to determine risk of invasion. J. Appl. Ecol. 43, 858–867 (2006). Article Google Scholar * Chon, T.-S., Park, Y. S., Moon, K. H.
& Cha, E. Y. Patternizing communities by using an artificial neural network. Ecol. Model. 90, 69–78 (1996). Article Google Scholar * Giraudel, J., Aurelle, D., Berrebi, P. & Lek,
S. In Artificial Neuronal Networks (eds Lek, S. & Guégan, J. F. ) Ch. 15, 187–202 (Springer, 2000). * Kohonen, T. The self-organizing map. Neurocomputing 21, 1–6, (1998). Article MATH
Google Scholar * Ward Jr, J. H. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58, 236–244 (1963). Article MathSciNet Google Scholar * Vesanto, J., Himberg,
J., Alhoniemi, E. & Parhankangas, J. Self-organizing map in Matlab: the SOM Toolbox. in _Proceedings of the Matlab DSP conference, Espoo, Finland, November 16–17, pp. 35–40_ (1999). *
Rauber, A. _Data mining with SOMVIS._ (2009) Available at: http://ifs.tuwien.ac.at/dm/somvis-matlab/index.html. (Accessed: 15th June 2015) * Dalet, D. d-maps.com (2007). Available at:
http://d-maps.com/carte.php?num_car=24853&lang=en (Accessed: 5th October 2015). Download references ACKNOWLEDGEMENTS This work is dedicated to the memory of Dr. J. Nagaraju, who
unexpectedly passed away during the preparation of the manuscript. This work was supported by the Centre of Excellence on Genetics and Genomics of Silkmoths (grant number BT/01/COE/05/12)
from the Department of Biotechnology, Government of India. AUTHOR INFORMATION Author notes * Chakraborty Saikat and Muthulakshmi M contributed equally to this work. * Deceased. AUTHORS AND
AFFILIATIONS * Centre of Excellence for Genetics and Genomics of Silkmoths, Laboratory of Molecular Genetics, Centre for DNA Fingerprinting and Diagnostics, Tuljaguda, Nampally, Hyderabad,
500001, India Saikat Chakraborty, M Muthulakshmi, Deena Vardhini, J Nagaraju & K. P. Arunkumar * Centre for Ecological Sciences, Indian Institute of Science, Bangalore, 560012, India
Saikat Chakraborty * Muga silkworm seed organization and Eri silkworm seed organization, Central Silk Board, Guwahati, 781006, India P Jayaprakash Authors * Saikat Chakraborty View author
publications You can also search for this author inPubMed Google Scholar * M Muthulakshmi View author publications You can also search for this author inPubMed Google Scholar * Deena
Vardhini View author publications You can also search for this author inPubMed Google Scholar * P Jayaprakash View author publications You can also search for this author inPubMed Google
Scholar * J Nagaraju View author publications You can also search for this author inPubMed Google Scholar * K. P. Arunkumar View author publications You can also search for this author
inPubMed Google Scholar CONTRIBUTIONS K.P.A. and J.N.: conceived and designed the project. S.C., M.M., D.V., P.J. and K.P.A.: performed the experiments and data analysis. K.P.A. and S.C.
wrote the manuscript. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY INFORMATION SUPPLEMENTARY
DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4 RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images
or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the
Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Chakraborty, S., Muthulakshmi, M., Vardhini, D. _et al._ Genetic analysis of Indian
tasar silkmoth (_Antheraea mylitta_) populations. _Sci Rep_ 5, 15728 (2015). https://doi.org/10.1038/srep15728 Download citation * Received: 17 March 2015 * Accepted: 30 September 2015 *
Published: 29 October 2015 * DOI: https://doi.org/10.1038/srep15728 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative




