- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Secondary bacterial infection in humans is one of the pathological conditions requiring clinical attention. In this study, we examined the effect of lipopolysaccharide (LPS) on
encephalomyocarditis virus (EMCV) infected mice. All mice inoculated with EMCV at 5 days before LPS challenge died within 24 h. LPS-induced TNF-α mRNA expression was significantly increased
in the brain and heart at 5 days after EMCV infection. CD11b+/TLR4+ cell population in the heart was remarkably elevated at 5 days after EMCV infection and sorted CD11b+ cells at 5 days
after EMCV infection produced a large amount of TNF-α on LPS stimulation _in vivo_ and _in vitro_. In conclusion, we found that the infiltration of CD11b+ cells into infected organs is
involved in the subsequent LPS-induced lethal shock in viral encephalomyocarditis. This new experimental model can help define the mechanism by which secondary bacterial infection causes a
lethal shock in viral encephalomyocarditis. SIMILAR CONTENT BEING VIEWED BY OTHERS MYOCARDITIS AND INFLAMMATORY CARDIOMYOPATHY: CURRENT EVIDENCE AND FUTURE DIRECTIONS Article 12 October 2020
HIV-1-ASSOCIATED LEFT VENTRICULAR CARDIAC DYSFUNCTION IN HUMANIZED MICE Article Open access 16 June 2020 STAT1 REGULATES NEUTROPHIL GELATINASE B-ASSOCIATED LIPOCALIN INDUCTION IN
INFLUENZA-INDUCED MYOCARDITIS Article Open access 15 May 2024 INTRODUCTION Polymicrobial infectious diseases show the involvement of 2 or more microbes, including viruses, bacteria, fungi or
parasites and these microbes act synergistically to mediate complex disease processes. In particular, a bacterial infection superimposed over an acute viral infection such as influenza is
well known as the aggravation factor in the infectious disease1,2. Lipopolysaccharide (LPS), the outer membrane of gram-negative bacteria, causes systemic inflammatory response syndrome,
endotoxic shock, disseminated intravascular coagulation and multi-organ failure3,4. LPS is recognized by Toll-like receptor 4 (TLR4)-expressing immunocompetent cells, mainly monocytes and
macrophages and induces the production of inflammatory cytokines through NF-κB activation3. Toxic effects of LPS are partially induced by the release and action of macrophage-derived
inflammatory cytokines. Especially, the mass production of tumor necrosis factor-α (TNF-α) causes septic shock with an abrupt reduction in blood pressure, leading to rapid aggravation of the
disease condition5,6. It is known that LPS-induced lethal shock is caused by a large quantity of TNF-α produced by immune cells activated by some kind of pre-stimulation7,8.
Encephalomyocarditis virus (EMCV), which is a single-stranded RNA virus and a member of _Cardiovirus_ in the Picornaviridae family, causes acute myocarditis and encephalitis in various
animal species9,10. In some reports, including our previous study, the death of mice by EMCV infection of high density (500 plaque-forming units (pfu)) occurred after 6 days and the mice
that survived for 12 days subsequently recovered. Additionally, remarkable inflammation in the brain and heart occurred at around 6 days after EMCV infection and thereafter the inflammation
reduced as the days progressed. Hence, these results suggested that inflammatory cells reach the activating stage around 6 days after EMCV infection11,12,13. It was also demonstrated that
various cells under the activating stage augment reactivity to LPS stimulation14,15,16. Previous studies showed that TNF-α is produced in large quantities by subsequent LPS stimulation
during adenovirus infection17, lymphocytic choriomeningitis virus and varicella-zoster virus infection18, but the original source of TNF-α is not elucidated. Moreover, it is unknown how LPS
affects in the viral encephalomyocarditis. The aim of our study is to examine the effect of LPS on EMCV-infected mice and to characterize the infiltrating cells, which possibly produce
TNF-α, into the heart in this model. RESULTS SURVIVAL OF MICE AFTER LPS STIMULATION DURING EMCV INFECTION The mice were intraperitoneally inoculated with 20 pfu of EMCV and were
intravenously injected with 10 μg LPS at 0, 2 and 5 days after the EMCV inoculation. At 5 days after EMCV infection, all mice died after LPS treatment within 24 h, but the mice subjected to
this treatment at 0 and 2 days after EMCV infection were alive (Table 1). Moreover, at 5 days after EMCV infection, LPS-induced lethal shock developed in the mice in an LPS dose-dependent
manner (Table 2). To compare the survival rate between WT and TNF-α KO mice after LPS treatment during EMCV infection, the mice were treated intravenously with 10 μg LPS at 5 days after EMCV
infection. All WT mice died after LPS treatment within 24 h at 5 days after EMCV infection, whereas TNF-α KO mice did not die. Moreover, LPS-induced lethality in WT mice was improved by
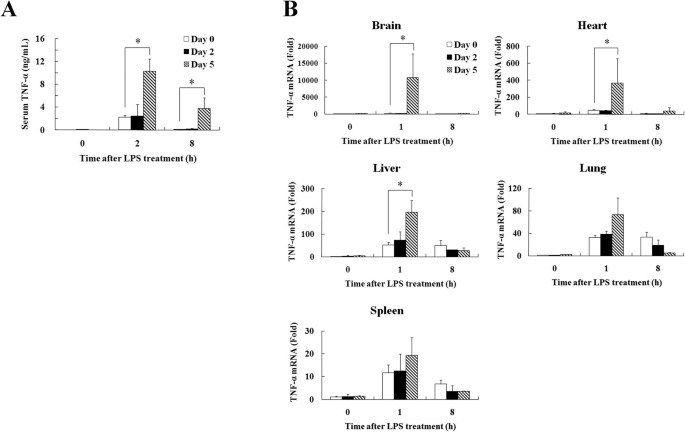
anti-TNF-α antibody (100 μg/mouse) treatment before LPS stimulation (Table 3). EFFECT OF EMCV INFECTION ON LPS-INDUCED TNF-Α PRODUCTION The concentration of serum TNF-α was measured at 0, 2
and 8 h after LPS treatment in each EMCV infection period. LPS-induced TNF-α concentration in the serum was markedly increased at 5 days after EMCV infection than at 0 and 2 days after EMCV
infection (Fig. 1A). For the localization of TNF-α production, TNF-α mRNA expression was determined in the brain, heart, liver, lung and spleen at 0, 1 and 8 h after LPS treatment. TNF-α
mRNA expression in the brain, heart and liver of mice was significantly increased at 5 days after EMCV infection as compared with that at 0 and 2 days after EMCV infection (Fig. 1B).
HISTOPATHOLOGICAL FINDING AND VIRAL LOAD OF THE TISSUES AFTER EMCV INFECTION Histological changes were observed in the brain and heart. The mice were inoculated intraperitoneally with 20 pfu
of EMCV. Neuronal cell death and the presence of some inflammatory cells in hippocampus or brain surface of the brain and marked infiltration of inflammatory cells in the heart were seen at
5 days after EMCV infection. The infiltrating cells mainly included macrophages and neutrophils (Fig. 2A). In the liver, a little infiltration of inflammatory cells was observed at 5 days
after EMCV infection and no remarkable change was seen in the lung during each infection period (Data not shown). All tissues at 8 h after LPS treatment in EMCV-infected mice were not
observed the significant increase of inflammatory cells in comparison with the tissues before LPS treatment (data not shown). We measured the viral load in the brain, heart, liver, lung and
spleen at 2 and 5 days after EMCV infection. Viral loads in all tissues at 5 days after EMCV infection were increased as compared with those at 2 days after EMCV infection. In particular,
viral loads in the brain and heart markedly increased at 5 days after the infection (Fig. 2B). EFFECT OF EMCV INFECTION ON MRNA EXPRESSION OF CHEMOKINES IN THE TISSUES Chemoattractant
protein-1 (MCP-1) is a major chemoattractant responsible for the recruitment of macrophages19,20. macrophage inflammatory protein-2 (MIP-2) and keratinocyte-derived chemokine (KC), which are
produced by macrophages, exhibit potent neutrophil chemotactic activity21,22. Because the infiltrated cells were mainly composed of macrophages and neutrophils as revealed by
histopathological findings (Fig. 2A), the mRNA expressions of MCP-1, MIP-2 and KC were determined in the brain and heart at 0, 2 and 5 days after EMCV infection. The mRNA expression of these
chemokines in the brain and heart on 5 days after EMCV infection was significantly up-regulated as compared with that at 0 and 2 days after EMCV infection (Fig. 3). MCP-1 mRNA expression at
5 days after EMCV infection was 808-fold in the brain and 125-fold in the heart based on 0 days. Similarly, MIP-2 mRNA expression was enhanced 254-fold in the brain and 44-fold in the heart
and KC mRNA expression was increased 23-fold in the brain and also in the heart. Although the expression of chemokines in the liver, lung and spleen had a tendency to up-regulated at 5 days
after EMCV infection, the up-regulation was not more remarkable than that in the brain or heart. EFFECT OF EMCV INFECTION ON TLR4 MRNA EXPRESSION IN TISSUES AND INFILTRATING CELLS IN THE
HEART The expression of TLR4 mRNA was examined in the brain, heart, liver, lung and spleen at 0, 2 and 5 days after EMCV infection. TLR4 mRNA expression in the brain, heart and liver at 5
days after EMCV infection was significantly increased. On the other hand, TLR4 mRNA expression in the lung and spleen showed no significant change after viral infection (Fig. 4A). Next, we
determined the phenotype of mononuclear cells (MNCs) from the heart at 5 days after EMCV infection. Although a lot of CD11b+TLR4+ cells were contained in MNCs from the heart at 5 days after
EMCV infection, few CD11c+, CD3+, CD19+ and CD49b+ cells were contained (Fig. 4B). LPS-INDUCED TNF-Α PRODUCTION IN CD11B+ CELLS FROM THE HEART _IN VIVO_ AND _IN VITRO_ Because MNCs from the
heart at 5 days after EMCV infection were mainly occupied by CD11b+ cells, we evaluated the ability of LPS-induced TNF-α production in CD11b+ cells _in vivo_. The mice were intravenously
inoculated with brefeldin A (250 μg/mouse) and LPS (10 μg/mouse) at 5 days after EMCV infection and MNCs from the heart were harvested at 1 h after LPS treatment. TNF-α producing cell in
gated CD11b+ cells on the basis of isotype control were increased by LPS treatment (Fig. 5A). CD11b+ and CD11b- cells from the heart at 5 days after EMCV infection were isolated by magnetic
cell sorting. Isolated cells and total cells before isolation were cultured at 1×105 cells/200 μL with LPS (1 μg/mL) for 24 h. LPS-induced TNF-α production in CD11b+ cells was significantly
increased as compared with that in total cells and CD11b− cells. CD11b− cells could not produce TNF-α after LPS stimulation (Fig. 5B). DISCUSSION In this study, we demonstrated that LPS
treatment at 5 days after EMCV infection induces an excess of TNF-α production in the brain and heart and lethal shock (Table 1, Fig. 1). Because this lethal effect of LPS was cancelled out
in TNF-α KO mice infected with EMCV (Table 3), it is likely that TNF-α from the brain and heart plays a critical role in the lethal effect of LPS on EMCV infection. Septic shock is
characterized by hypotension, decreased systemic vascular resistance and impaired vascular reactivity and TNF-α has been implicated as a principal mediator in the pathogenesis of septic
shock23. TNF-α is mainly produced by macrophages in the peripheral tissue after pathogen infection and is involved in the elimination of pathogens from the host24,25. A protective role of
TNF-α in EMCV-infected mouse model is previously reported11. In contrast, an excessive production of TNF-α induces hypothermia, hypotension, multiple organ failure, septic shock and
death5,6. In particular, previous reports demonstrated that the macrophages treated with some pre-stimulants produce a large amount of TNF-α on subsequent LPS stimulation. _Propionibacterium
acnes,_ an anaerobic gram-positive bacterium, exerts strong immunomodulatory activities and participates in the formation of intrahepatic granulomas and induction of hypersensitivity for
LPS in mice. Additionally, these activities depended on the recognition of bacteria via TLR9 and subsequent IL-12-mediated IFN-γ production26,27. EMCV infection also markedly increased the
LPS-induced TNF-α mRNA level and viral road in the brain and heart (Fig. 1B, 2B). Namely, the susceptibility to LPS was enhanced in the EMCV-infected site. TLR4 recognizes LPS from
gram-negative bacteria and its recognition is essential for the activation of the innate immune system. In humans with myocarditis, TLR4 mRNA is also increased in the heart28. In the present
study, the mRNA expression of TLR4 in the brain, heart and liver was increased after EMCV infection (Fig. 4A) and a significant increase in TNF-α mRNA expression was also confirmed after
subsequent LPS stimulation (Fig. 1B). Histological findings revealed neuronal cell death and the presence of some inflammatory cells in hippocampus or brain surface of the brain, remarkable
infiltration of inflammatory cells in the heart and a little infiltration of inflammatory cells in the liver at 5 days after EMCV infection; further, the infiltrating cells were mainly
composed of macrophages and neutrophils. These results indicate that the accumulation of inflammatory cells in the heart after EMCV infection is associated with this lethal septic shock
model. In fact, there is a correlation between TLR4 mRNA expression and the number of infiltrated TLR4 positive cells in the heart. A previous report also demonstrated the augmentation of
TLR4 mRNA expression by infiltration of TLR4-positive leukocytes into the liver in an IL-17-induced multiple tissue inflammation model29. Furthermore, our previous studies proved the
infiltration of TLR4-positive leukocytes into the liver on α-galactosylceramide administration and the excessive response to subsequent LPS stimulation8,15. In this study, CD11b+/TLR4+ cells
in the heart increased at 5 days after EMCV infection, but few CD11c+, CD3+, CD19+ and CD49b+ cells were contained (Fig. 4B). LPS-induced TNF-α was mainly produced by CD11b+ cells in the
heart at 5 days after EMCV infection _in vivo_ and _in vitro_ (Fig. 5). Therefore, it is suggested that the infiltration of CD11b+ cells into the heart involves in the up-reguration of TLR4
mRNA and subsequent LPS induced TNF-α production. mRNA expression of MCP-1, MIP-2 and KC in the brain and heart at 5 days after EMCV infection was markedly increased than that in uninfected
mice (Fig. 3). The enhancement of the mRNA expression of these chemokines, especially MCP-1, may be involved in the infiltration of inflammatory cells into the heart. MCP-1 acts as a potent
chemoattractant and activator of monocytes/macrophages19,20. Shiratsuchi et al30 showed that macrophages phagocytose influenza virus-infected HeLa cells in a manner mediated by
phosphatidylserine that appears on the surfaces of infected cells during the process of apoptosis. In addition, the inhibition of macrophage recruitment by MCP-1 augmented alveolar
epithelial damage and apoptosis during influenza pneumonitis31. Thus, MCP-1 has an effective role in viral clearance, but it also may aggravate lethal shock by subsequent LPS stimulation
under the EMCV-infected state. Although the mice treated with 500 μg LPS died within 48 to 72 h in the general LPS-induced shock model32,33, all mice treated with 10 μg LPS died within 24
hours in our model; this suggests that LPS susceptibility in mice markedly rises under the EMCV-infected condition. It is known that infection by the influenza virus, coxsackie virus and
adenovirus are involved in the onset of viral encephalitis or myocarditis34,35, but there are few reports showing the effect of the secondary bacterial superimposed infection on viral
encephalomyocarditis and its mechanism. In the present study, even a low-dose of LPS could cause the lethal septic shock in EMCV-infected mice. Thus, it is necessary to monitor serum
endotoxin levels and prevent bacterial infection in patients with encephalitis and myocarditis. In conclusion, we established a septic shock model using mice with viral encephalomyocarditis
and demonstrated that the infiltration of CD11b+ cells into tissues is involved in this lethal shock. From an immunological point of view, our animal model is useful for investigating the
pathological state of bacterial superimposed infection on viral encephalomyocarditis. METHODS REAGENTS LPS from _Escherichia coli_ O111:B5 and brefeldin A were purchased from Sigma-Aldrich
(St Louis, MO, USA). Mouse TNF-α antibody (monoclonal rat IgG1) for neutralize was purchased from R&D Systems (Minneapolis, MN, USA). MICE Mouse experiments were performed according to
the guidelines of the Animal Ethics Committee of Gifu University Graduate School of Medicine. C57BL/6J mice aged approximately 8–10 weeks were obtained from Japan SLC (Hamamatsu, Japan) and
used as wild-type (WT) mice. TNF-α gene knockout (TNF-α KO) mice of a C57BL/6J background were produced by gene targeting as described previously11,36,37. VIRAL INOCULATION AND LPS TREATMENT
A myocarditic variant of EMCV was generously provided by Dr. Seto (Keio University, Tokyo, Japan). The virus stock was stored at −80°C in Hanks' balanced salt solution (HBSS) with 0.1%
BSA required for use. The mice were inoculated intraperitoneally with 20 pfu of EMCV in 0.2 mL saline and were treated intravenously with 10 μg LPS in 0.2 mL saline at 0 (non-infected), 2
and 5 days after the viral inoculation unless otherwise noted. The lethality was determined at 24 h after LPS treatment. The experiments were performed according to the institutional
guidelines of Gifu University for microbiologic study. MEASUREMENT OF SERUM TNF-Α CONCENTRATION The concentration of TNF-α in the serum and culture supernatant was determined using a mouse
TNF-α Quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's recommendations. REAL-TIME RT-PCR ANALYSIS Real-time RT-PCR was used to quantify the
levels of TNF-α, TLR4, MCP-1, MIP-2, KC mRNA and EMCV RNA. Total RNA in the brain, heart, liver, lung and spleen was isolated using Isogen (Nippon Gene, Tokyo, Japan) and transcribed to cDNA
by using the High capacity cDNA transcription kit (Applied Biosystems, Foster City, CA, USA). Purified cDNA was used as the template for real-time PCR conducted using pre-designed
primer/probe sets for TNF-α, TLR4, MCP-1, MIP-2, KC and 18S rRNA (Applied Biosystems), according to the manufacturer's recommendations. The determination of EMCV RNA was performed by
the use of the LightCycler DNA Master SYBR Green I (Roche Diagnostic Systems, Indianapolis, IN, USA) and the following oligonucleotide primer pairs: EMCV sense, 5′-GTCGTGAAGGAAGCAGTTCC-3′,
antisense, 5′-CACGTGGCTTTTGGCCGCAGAGGC-3′11. 18S rRNA was used as an internal control. Real-time PCR was carried out using a Light-Cycler rapid thermal cycler system (Roche Diagnostic
Systems). HISTOLOGICAL EXAMINATION Histopathological examination of the brain and heart was performed at 0, 2 and 5 days after EMCV infection. The tissues were fixed in 10% formalin in PBS
for 48 h and embedded in paraffin. Tissue sections were deparaffinized, stained with H&E and examined under light microscopy. CELL ISOLATION IN HEART Single cells from the heart,
particularly non-parenchymal cells, were isolated in reference to a previous report38. The heart was minced with scissors and was shaken with 5 mL HBSS containing collagenase type 2 (600
U/mL) and deoxyribonuclease I (60 U/mL) (Worthington, Lakewood, NJ, USA) for 30 min at 37°C. The specimen was filtered through a stainless steel mesh and red blood cells were lysed. After
washing and filtration through a 70-μm cell strainer, the single cells were suspended in RPMI 1640 medium (Wako Pure Chemical Industries; Osaka, Japan) containing 10% heat-inactivated fetal
bovine serum (Thermo Fisher Scientific Inc, Waltham, MA, USA) and cultured at 37°C in a 5% CO2 atmosphere. FLOW CYTOMETRY ANALYSIS MNCs from the heart at 5 days after EMCV infection were
obtained by centrifugation of the single cells with Ficoll-Conray (IBL, Gunma, Japan). Flow cytometry was used to evaluate the expression level of CD3, CD11b, CD11c, CD19, CD49b and TLR4 in
MNCs from the heart at 5 days after EMCV infection. Following reaction with anti-CD16/CD32 antibody to suppress nonspecific binding, MNCs were stained with FITC-conjugated hamster anti-mouse
CD3ε antibody (clone 145-2C11; BD Biosciences, Franklin Lakes, NJ, USA), FITC-conjugated rat anti-mouse CD11b antibody (clone M1/70; eBioscience, San Diego, CA, USA), FITC-conjugated
hamster anti-mouse CD11c antibody (clone HL-3; BD Biosciences), FITC-conjugated mouse anti-mouse CD19 antibody (clone MB19-1; eBioscience), FITC-conjugated rat anti-mouse CD49b antibody
(clone DX5; eBioscience) and PE-conjugated rat anti-mouse TLR4/MD2 complex antibody (clone MTS510; eBioscience). The positive rate of the cells was made on the basis of isotype control. The
phenotypic characterization of the cells was carried out using FACSCanto II (BD Biosciences). INTRACELLULAR CYTOKINE STAINING _IN VIVO_ The mice were intravenously inoculated with brefeldin
A (250 μg/mouse) and LPS (10 μg/mouse) at 5 days after EMCV infection. MNCs from the heart at 1 h after LPS treatment were fixed and permeabilized with the Cytofix/Cytoperm buffer (BD
Biosciences) and were stained with PE-conjugated rat anti-TNF-α antibody (clone MP6-XT22; eBioscience). The cells were analyzed using FACSCanto II (BD Biosciences). ISOLATION OF CD11B+ CELLS
MNCs from the heart obtained at 5 days after EMCV infection were separated into CD11b+ and CD11b− cells using anti-CD11b-conjugated magnetic beads (Miltenyi Biotec GmbH, Bergisch Gladbach,
Germany). The magnetically labeled cells were purified using the QuadroMACS separation unit attached to a MACS multistand and LS columns (Miltenyi Biotec GmbH). STATISTICAL ANALYSIS In each
experiment, the results were expressed as the mean ± SD. The statistical significance of the difference in mean values was determined by Student's _t_ test or one-way analysis of
variance followed by Scheffe's test. _P_ values of less than 0.05 were considered significant. REFERENCES * Bakaletz, L. O. Developing animal models for polymicrobial diseases. Nat.
Rev. Microbiol. 2, 552–568 (2004). Article CAS Google Scholar * Brundage, J. F. Interactions between influenza and bacterial respiratory pathogens: implications for pandemic preparedness.
Lancet Infect. Dis. 6, 303–312 (2006). Article Google Scholar * Cohen, J. The immunopathogenesis of sepsis. Nature 420, 885–891 (2002). Article ADS CAS Google Scholar * Bannerman, D.
D. & Goldblum, S. E. Direct effects of endotoxin on the endothelium: barrier function and injury. Lab Invest 79, 1181–1199 (1999). CAS PubMed Google Scholar * Tracey, K. J. &
Cerami, A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med 45, 491–503 (1994). Article CAS Google Scholar * Brouckaert, P. & Fiers, W. Tumor
necrosis factor and the systemic inflammatory response syndrome. Curr Top Microbiol Immunol 216, 167–187 (1996). CAS Google Scholar * Gumenscheimer, M., Mitov, I., Galanos, C. &
Freudenberg, M. A. Beneficial or deleterious effects of a preexisting hypersensitivity to bacterial components on the course and outcome of infection. Infect Immun 70, 5596–5603 (2002).
Article CAS Google Scholar * Ito, H. et al. Lethal endotoxic shock using alpha-galactosylceramide sensitization as a new experimental model of septic shock. Lab Invest 86, 254–261 (2006).
Article CAS Google Scholar * Kishimoto, C., Kuribayashi, K., Masuda, T., Tomioka, N. & Kawai, C. Immunologic behavior of lymphocytes in experimental viral myocarditis: significance
of T lymphocytes in the severity of myocarditis and silent myocarditis in BALB/c-nu/nu mice. Circulation 71, 1247–1254 (1985). Article CAS Google Scholar * Topham, D. J., Adesina, A.,
Shenoy, M., Craighead, J. E. & Sriram, S. Indirect role of T cells in development of polioencephalitis and encephalomyelitis induced by encephalomyocarditis virus. J Virol 65, 3238–3245
(1991). CAS PubMed PubMed Central Google Scholar * Wada, H. et al. Tumor necrosis factor-alpha (TNF-alpha) plays a protective role in acute viralmyocarditis in mice: A study using mice
lacking TNF-alpha. Circulation 103, 743–749 (2001). Article CAS Google Scholar * Yamamoto, K. et al. Attenuation of virus-induced myocardial injury by inhibition of the angiotensin II
type 1 receptor signal and decreased nuclear factor-kappa B activation in knockout mice. J Am Coll Cardiol 42, 2000–2006 (2003). Article CAS Google Scholar * Nasu-Nishimura, Y. et al.
Cellular prion protein prevents brain damage after encephalomyocarditis virus infection in mice. Arch Virol 153, 1007–1012 (2008). Article CAS Google Scholar * Ito, H. et al. Augmentation
of lipopolysaccharide-induced nitric oxide production by alpha-galactosylceramide in mouse peritoneal cells. J Endotoxin Res 11,213–219 (2005). Article CAS Google Scholar * Ohtaki, H. et
al. Valpha14 NKT cells activated by alpha-galactosylceramide augment lipopolysaccharide-induced nitric oxide production in mouse intra-hepatic lymphocytes. Biochem Biophys Res Commun 378,
579–583 (2009). Article CAS Google Scholar * Ohtaki, H. et al. Interaction between LPS-induced NO production and IDO activity in mouse peritoneal cells in the presence of activated
Valpha14 NKT cells. Biochem Biophys Res Commun 389, 229–234 (2009). Article CAS Google Scholar * Fejer, G. et al. Adenovirus infection dramatically augments lipopolysaccharide-induced TNF
production and sensitizes to lethal shock. J Immunol 175, 1498–1506 (2005). Article CAS Google Scholar * Nansen, A. & Randrup Thomsen, A. Viral infection causes rapid sensitization
to lipopolysaccharide: central role of IFN-alpha beta. J Immunol 166, 982–988 (2001). Article CAS Google Scholar * Matsushima, K., Larsen, C. G., DuBois, G. C. & Oppenheim, J. J.
Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med 169, 1485–1490 (1989). Article CAS Google
Scholar * Jiang, Y., Beller, D. I., Frendl, G. & Graves, D. T. Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. J
Immunol 148, 2423–2428 (1992). CAS PubMed Google Scholar * Driscoll, K. E. Macrophage inflammatory proteins: biology and role in pulmonary inflammation. Exp Lung Res 20, 473–490 (1994).
Article CAS Google Scholar * Lira, S. A. et al. Expression of the chemokine N51/KC in the thymus and epidermis of transgenic mice results in marked infiltration of a single class of
inflammatory cells. J Exp Med 180, 2039–2048 (1994). Article CAS Google Scholar * Tracey, K. J. et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal
bacteraemia. Nature 330, 662–664 (1987). Article ADS CAS Google Scholar * Sergerie, Y., Rivest, S. & Boivin, G. Tumor necrosis factor-alpha and interleukin-1 beta play a critical
role in the resistance against lethal herpes simplex virus encephalitis. J Infect Dis 196, 853–860 (2007). Article CAS Google Scholar * Bekker, L. G. et al. Immunopathologic effects of
tumor necrosis factor alpha in murine mycobacterial infection are dose dependent. Infect Immun 68, 6954–6961 (2000). Article CAS Google Scholar * Tchaptchet, S. et al. Innate,
antigen-independent role for T cells in the activation of the immune system by Propionibacterium acnes. Eur J Immunol 40, 2506–2516 (2010). Article CAS Google Scholar * Inatsu, A. et al.
Novel mechanism of C-reactive protein for enhancing mouse liver innate immunity. Hepatology 49, 2044–2054 (2009). Article CAS Google Scholar * Satoh, M. et al. Expression of Toll-like
receptor 4 is associated with enteroviral replication in human myocarditis. Clin Sci (Lond) 104, 577–584 (2003). Article CAS Google Scholar * Tang, H. et al. TLR4 activation is required
for IL-17-induced multiple tissue inflammation and wasting in mice. J Immunol 185, 2563–2569 (2010). Article CAS Google Scholar * Shiratsuchi, A., Kaido, M., Takizawa, T. & Nakanishi,
Y. Phosphatidylserine-mediated phagocytosis of influenza A virus-infected cells by mouse peritoneal macrophages. J Virol 74, 9240–9244 (2000). Article CAS Google Scholar * Narasaraju,
T., Ng, H. H., Phoon, M. C. & Chow, V. T. MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. Am J Respir Cell Mol Biol 42,
732–743 (2010). Article CAS Google Scholar * Jung, I. D. et al. Blockade of indoleamine 2,3-dioxygenase protects mice against lipopolysaccharide-induced endotoxin shock. J Immunol 182,
3146–3154 (2009). Article CAS Google Scholar * Liu, J. et al. The circadian clock Period 2 gene regulates gamma interferon production of NK cells in host response to
lipopolysaccharide-induced endotoxic shock. Infect Immun 74, 4750–4756 (2006). Article CAS Google Scholar * Morishima, T. et al. Encephalitis and encephalopathy associated with an
influenza epidemic in Japan. Clin Infect Dis 35, 512–517 (2002). Article Google Scholar * Yajima, T. & Knowlton, K. U. Viral myocarditis: from the perspective of the virus. Circulation
119, 2615–2624 (2009). Article Google Scholar * Taniguchi, T., Takata, M., Ikeda, A., Momotani, E. & Sekikawa, K. Failure of germinal center formation and impairment of response to
endotoxin in tumor necrosis factor alpha-deficient mice. Lab Invest 77, 647–658 (1997). CAS PubMed Google Scholar * Ito, H. et al. Role of TNF-alpha produced by nonantigen-specific cells
in a fulminant hepatitis mouse model. J Immunol 182, 391–397 (2009). Article CAS Google Scholar * Pfister, O. et al. CD31- but Not CD31+ cardiac side population cells exhibit functional
cardiomyogenic differentiation. Circ Res 97, 52–61 (2005). Article CAS Google Scholar Download references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Informative Clinical
Medicine, Gifu University Graduate School of Medicine, 1-1 Yanagido, Gifu, 501-1194, Japan Hirofumi Ohtaki, Hiroyasu Ito, Masato Hoshi, Yosuke Osawa & Mitsuru Seishima * Department of
Pathology, Gifu University Graduate School of Medicine, 1-1 Yanagido, Gifu, 501-1194, Japan Manabu Takamatsu & Akira Hara * First Department of Internal Medicine, Gifu University
Graduate School of Medicine, 1-1 Yanagido, Gifu, 501-1194, Japan Hisataka Moriwaki * Department of Medical Technology, Nagoya University School of Health Sciences, 1-20 Daikominami-1-chome,
Higashi-ku, Nagoya, Aichi, 461-8673, Japan Tetsuya Ishikawa * Human Health Sciences, Graduate School of Medicine and Faculty of Medicine, Kyoto University, 53 Kawahara-cho, Shogoin, Sakyo,
Kyoto, 606-8507, Japan Kuniaki Saito Authors * Hirofumi Ohtaki View author publications You can also search for this author inPubMed Google Scholar * Hiroyasu Ito View author publications
You can also search for this author inPubMed Google Scholar * Masato Hoshi View author publications You can also search for this author inPubMed Google Scholar * Yosuke Osawa View author
publications You can also search for this author inPubMed Google Scholar * Manabu Takamatsu View author publications You can also search for this author inPubMed Google Scholar * Akira Hara
View author publications You can also search for this author inPubMed Google Scholar * Tetsuya Ishikawa View author publications You can also search for this author inPubMed Google Scholar *
Hisataka Moriwaki View author publications You can also search for this author inPubMed Google Scholar * Kuniaki Saito View author publications You can also search for this author inPubMed
Google Scholar * Mitsuru Seishima View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS HO, HI and MS designed the project. HO, HI, YO, AH, TI,
HM, KS and MS designed experiments. HO, MH and MT performed the experimental work. HO, HI and MS prepared the manuscript and figures. All authors reviewed the manuscript. ETHICS DECLARATIONS
COMPETING INTERESTS The authors declare no competing financial interests. RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0
Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ohtaki, H., Ito, H.,
Hoshi, M. _et al._ High susceptibility to lipopolysaccharide-induced lethal shock in encephalomyocarditis virus-infected mice. _Sci Rep_ 2, 367 (2012). https://doi.org/10.1038/srep00367
Download citation * Received: 16 March 2012 * Accepted: 29 March 2012 * Published: 16 April 2012 * DOI: https://doi.org/10.1038/srep00367 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative