- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Molecular docking is the most commonly used technique in the modern drug discovery process where computational approaches involving docking algorithms are used to dock small molecules into
macromolecular target structures. Over the recent years several evaluation studies have been reported by independent scientists comparing the performance of the docking programs by using
default ‘black box’ protocols supplied by the software companies. Such studies have to be considered carefully as the docking programs can be tweaked towards optimum performance by selecting
the parameters suitable for the target of interest. In this study we address the problem of selecting an appropriate docking and scoring function combination (88 docking algorithm-scoring
functions) for substrate specificity predictions for feruloyl esterases, an industrially relevant enzyme family. We also propose the ‘Key Interaction Score System’ (KISS), a more
biochemically meaningful measure for evaluation of docking programs based on pose prediction accuracy.
A key objective of the commonly used molecular docking programs is to predict the correct placement of small molecules or ligands within the binding pocket of an enzyme or protein and the
biological implications of this process. This knowledge is subsequently applied to identify novel ligands through virtual screening of compound libraries1,2. Several commercial and academic
softwares are available for molecular modeling and docking studies. A bundle of studies on the evaluation of molecular docking programs and scoring functions have been published focusing on
pose prediction (re-docking a compound with a known conformation and orientation into the target's active site followed by selection of the docking program that return poses below a
preselected Root Mean Square Deviation value from the known conformation) and virtual screening (docking a decoy set of inactive compounds that has been mixed with compounds with known
activity against the target in question followed by selection of the docking program based on enrichment studies)3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20. A very surprising and
interesting recent study by Cross et al (2009)21 on comparison of molecular docking programs for pose prediction and virtual screening accuracy showed that there is significant variability
on the performance of docking programs based on the target enzyme or protein family. The findings of Cross et al change the paradigm of traditional or previous evaluation studies that used
an array of diverse protein structures and standard datasets like DUD (Directory of Useful Decoys)22,23,24. Every molecular docking program or scoring function has a bias for particular
physical properties of the target protein or enzyme of interest. It has been proposed that the differences in performance of the molecular docking programs could be attributed to the
composition of the training sets used while developing particular docking programs that have different intended goals21. So, selection of a molecular docking program for a particular target
needs careful consideration, as each program gives results of varying quality depending on the target. A recent trend is to select docking programs that suit your protein of interest25,26
while conclusions from previous evaluation studies should be exploited as a rough guide for selecting a docking program rather than sticking to the statements of expected performance based
on diverse set of proteins or ligands.
In this study we start anew in the evaluation and selection of molecular docking programs suitable for a specific target of interest. We address the problem of selecting an appropriate
docking and scoring combination for substrate specificity predictions, specifically for the feruloyl esterase families, where each family possess both overlapping as well as unique
specificity to the individual substrates (Fig. 1). The framework presented here is applicable to select software packages for docking studies for every enzyme or protein family. We recently
proposed a novel classification system for feruloyl esterases (FAEs) that resulted in 12 families, which have the capability of acting on a large range of substrates for cleaving ester bonds
and synthesizing high-added value molecules through esterification and transesterification reactions27. As mentioned above, there is some overlapping in the substrate-activity maps of the
members of the various FAE families (FEFs) due to the flexibility of their residues in the FAE binding pocket. We therefore consider as the ultimate challenge for a docking program to
correctly predict the ‘sensitive’ substrate specificity profile of the FAE families, which will position it superior among the others and more suitable for enzymes with high flexibility. We
also propose an assessment measure, the Key Interaction Score System (KISS), to evaluate pose prediction accuracy. KISS carries both biological and chemical interaction information and it is
presented and discussed in detail below.
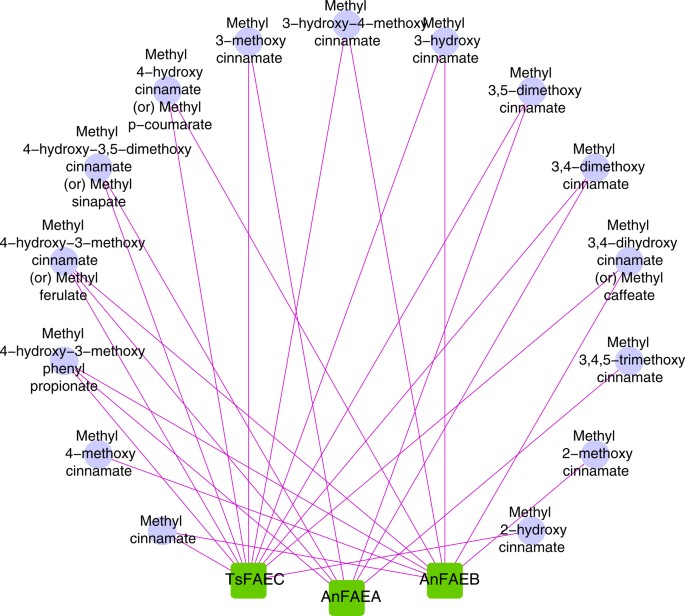
Overlapping substrate specificities among the members (TsFAEC, AnFAEA and AnFAEB) of different FAE families; the diagram was created using Cytoscape version 2.841,42.
The enzymes TsFAEC, AnFAEA and AnFAEB were capable of hydrolyzing 12, 7 and 9 substrates respectively.
Detailed substrate specificity spectra is available only for three enzymes viz., feruloyl esterase A (AnFAEA) and feruloyl esterase B (AnFAEB) from Aspergillus niger and feruloyl esterase C
(TsFAEC) from Talaromyces stipitatus (their experimental kinetic data are given in Supplementary Table S1, see Section A in Supplementary Information). In our earlier study on the
development of a FAE classification system27, pharmacophore models, based on key pharmacophore features of their substrate spectra, were proposed for those three FAEs and the respective
sub-families that they belong to. While the three-dimensional crystal structure of AnFAEA has been resolved28, the crystal structures of the other two enzymes are not available yet. In the
absence of any resolved X-ray or NMR structures, the three-dimensional atomic models for AnFAEB and TsFAEC were modeled from multiple threading alignments29 and iterative structural assembly
simulations using the I-TASSER algorithm, an extension of the previous TASSER method30,31,32,33,34. Structure refinement of the modeled structures was carried out using the Discovery Studio
software suite version 3.0 (Accelrys Inc, USA). Structural information and validation data (Supplementary Table S2) of the modeled FAEs are given in Section B of Supplementary Information.
The coordinates of the model structures (see Supplementary Fig. S1) were submitted to the Protein Model DataBase (PMBD)35.
Many evaluation studies have been performed using the default settings of the docking programs, which only provides a baseline performance of each program and lacks the insights of different
options provided in the respective software. This is a point that should be considered carefully when claiming performance differences between the
programs3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20. In the present study, docking programs were evaluated using the recommended optimized options in the respective software for a
particular task, which eliminated the user bias to particular software or results. Additional support was received from the lead application scientific specialists (see Acknowledgments) of
the respective software companies. This contribution also facilitated the elaboration of the observed variability in the results obtained by algorithms of the same program (e.g., Glide XP
and Glide SP for docking functions in Schrödinger suite). Since new versions of docking programs are frequently released, these must be evaluated by the community almost in an annual base.
To the best of our knowledge, this is not only the first evaluation study with the most recent versions (released in 2011) of popular state-of-the-art commercial docking suites, but probably
also the most complete with 88 docking algorithm-scoring function sets (involving 24 docking algorithms and 24 scoring functions). As briefly discussed above, the evaluation or selection of
the best docking program involves two major steps; first, to predict the pose of the ligand correctly when compared with the conformation in a co-crystallized protein or enzyme and second,
to predict binding affinities close to experimental observations.
The proposed Key Interaction Score System (KISS) is suggested as an improvement to the first step, namely pose selection, since the ability of a docking program to produce the correct
binding mode is a prerequisite to later predict a set of reliable binding affinities. Even though the traditional approach of evaluating the docking programs using the RMSD (Root Mean Square
Deviation) is commonly used, the main drawback is not taking into account the interactions between the ligand and the receptor. Hence, as an extension of the RMSD evaluation, we analyzed
here whether the docked ligand pose reproduced the same interactions with the receptor as those observed in the cognate-ligand crystal structure. The cognate ligand crystal structure of the
AnFAEA (PDB ID: 1UWC)28 was analyzed for key interactions (hydrogen bonds, polar and non-polar contacts, pi-interactions) of the ligand with the receptor. The most important point that
should be remembered when comparing the interactions of the docked and crystal structure pose is that the crystal structures do not contain the coordinates for hydrogen, so hydrogens must be
included before any comparison or simulation/docking process. The preprocessing of the protein structures is described in Section D of the Supplementary Information, while the observed
differences in the interactions of unprocessed and processed crystal structures of 1UWC (as illustrated in Supplementary Fig. S2) only reinforce our assessment for the utility of this step
before docking or simulation studies. For ranking the docking programs based on the KISS score, the hydrogen bond interactions in the ‘processed’ crystal structures were used as control
systems. The function for calculating the KISS score is given below:
where, Ir = Number of reproduced hydrogen bond interactions by the docked pose. Ic = Total hydrogen bond interactions present in the binding pose of processed cognate ligand crystal
structure. The hydrogen bond interactions between ligand and protein were explicitly taken into account when comparing the docked poses with the preprocessed cognate ligand crystal
structures for calculating the KISS score. No cut-offs were used in analyzing the docked poses for calculating the KISS score. Imposing cut-offs would result in overweighting or
underweighting of interactions or side chains or groups. Since no cut-offs were imposed, KISS score is extensible and could be included in various docking algorithms and scoring functions. A
high KISS score can be achieved if the docked pose of the ligand reproduces the ‘same’ hydrogen bond interactions with the receptor seen in the crystal structures irrespective of low or
high RMSD. Having a large RMSD between the experimental ligand pose and the computationally calculated pose by a docking program does not indicate a low quality of its force field
implementation or scoring algorithm implemented, if the overall binding modes and interactions are reproduced the same way as seen in the crystal structure. Despite the general speculation
that the lower the RMSD, the more likely the docked ligand will reproduce the interactions of the ligand in the crystal structure, this does not hold true for all cases. In this study we
consider and compare both RMSD and KISS, even though more focus is given to the latter due to its biological significance. RMSD and KISS score are inversely correlated for the docking
algorithms listed in Table 1. On the other hand, for approximately half of the docking algorithms in this study the lowest RMSD score does not correspond to the highest KISS score (Fig. 2a)
and vice versa (Fig. 2b). For example in the case of pose selection studies with AnFAEA, even though a high RMSD of 2.5 Å was observed from the binding mode seen in the crystal structure,
the docked pose 3 generated by the Alpha Triangle docking algorithm reached a KISS score of 0.66. Whereas, the best pose (pose rank 1) according to the low RMSD consideration (1.39 Å)
generated by the same Alpha Triangle docking algorithm was considered to be less accurate as it showed a KISS score of 0.5 (see Supplementary Table S3 and Supplementary Fig. S3). Similar
trend was observed for FA-1UWC docking with the Optimizer docking algorithm and the variants of the Surflex-Dock docking algorithm (Fig. 2).
Docking algorithms, where there is no correlation between RMSD and KISS score during cognate ligand docking accuracy studies on the crystal structure of the AnFAEA (PDB ID: IUSW).
(a) Lowest RMSD poses and their respective KISS scores. (b) Docked poses with highest KISS score and their respective RMSD.
In many of the docked poses generated from all the docking algorithms it was observed that the ligand establishes additional interactions with the amino acid residues of the binding pocket.
Even though those poses increase the number of ligand-receptor interactions, they were considered as incorrect due to lack of the original key interactions seen in the crystal structures.
From the examples discussed above, it is evident that having a low RMSD between the docked and the crystallographic pose does not necessarily mean that the ligand can actually form similar
interactions or similar binding modes and that a high RMSD value does not indicate a vice versa situation. Hence, when evaluating docking programs it is also essential to look into all of
scoring poses carefully. The high flexibility of the ligand/substrate and the flexibility of the binding pocket residues of FAEs27 increase the chances of high variability between the
experimental and docked poses; although the same interactions were reproduced by docking programs that showed a KISS score of 1. It should also be noted that the degree of implementation of
ligand and receptor flexibility varies widely between the docking algorithms. When we evaluated the docking algorithms for pose prediction accuracy just based on RMSD between the
computationally docked pose and the pose in the crystal structure, FlexX TM, FlexX SIS, Triangle Matcher and Proxy Triangle were ranked superior in generating low RMSD (









