- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The pleiotropic Src kinase Lyn has critical roles in host defense in alveolar macrophages against bacterial infection, but the underlying mechanism for Lyn-mediated inflammatory
response remains largely elusive. Using mouse _Pseudomonas aeruginosa_ infection models, we observed that Lyn−/− mice manifest severe lung injury and enhanced inflammatory responses,
compared with wild-type littermates. We demonstrate that Lyn exerts this immune function through interaction with IL-6 receptor and cytoskeletal protein Ezrin via its SH2 and SH3 domains.
Depletion of Lyn results in excessive STAT3 activation, and enhanced the Src homology 2-containing inositol-5-phopsphatase 1 (SHIP-1) expression. Deletion of SHIP-1 in Lyn−/− mice (double
knockout) promotes mouse survival and reduces inflammatory responses during _P. aeruginosa_ infection, revealing the rescue of the deadly infectious phenotype in Lyn deficiency.
Mechanistically, loss of SHIP-1 reduces NF-κB-dependent cytokine production and dampens MAP kinase activation through a TLR4-independent PI3K/Akt pathway. These findings reveal Lyn as a
regulator for host immune response against _P. aeruginosa_ infection through SHIP-1 and IL-6/STAT3 signaling pathway in alveolar macrophages. SIMILAR CONTENT BEING VIEWED BY OTHERS ZKSCAN3
IN SEVERE BACTERIAL LUNG INFECTION AND SEPSIS-INDUCED IMMUNOSUPPRESSION Article 09 September 2021 MAFB REGULATES NLRP3 INFLAMMASOME ACTIVATION BY SUSTAINING P62 EXPRESSION IN MACROPHAGES
Article Open access 16 October 2023 EXTRACELLULAR CIRP DYSREGULATES MACROPHAGE BACTERIAL PHAGOCYTOSIS IN SEPSIS Article Open access 05 December 2022 INTRODUCTION _Pseudomonas aeruginosa_ is
an opportunistic bacterium causing acute and chronic infection in immunocompromised people,1 such as patients with cystic fibrosis, chronic obstructive pulmonary disease, severe burns and
cancer. _P. aeruginosa_ infection is the major cause of the morbidity and mortality of these diseases.2 Innate immune responses, including inflammatory cytokine production, immune cell
recruitment and phagocytic clearance by neutrophils and macrophages, have critical roles in host defense during the early stages of infection and profoundly influence the generation of the
adaptive immune responses and disease outcomes.3–5 The pattern recognition receptors are important factors for recognizing pathogen-associated molecular patterns, such as lipopolysaccharide
(LPS), and initiating innate (frontline) immunity to battle against pathogens, and subsequently transmitting signals for antigen-specific adaptive immune responses to provide a second layer
of protection.6 Lyn, a member of the Src family of nonreceptor tyrosine kinases, is an important regulator of immune homeostasis and pattern recognition receptor-induced responses.7 Previous
studies have determined that Lyn is involved in B-cell responses,8 proliferation and degranulation of mast cells,9 integrin signaling in neutrophils,10 M2 macrophage polarization,11
dendritic cell function, and NK cell activation.12 Lyn is activated upon ligand binding to a wide variety of cell surface receptors that are essential for promoting or limiting immune
responses.13,14 A recent study showed that Lyn can activate Abl2/Arg to facilitate IgG-mediated phagocytosis and _Leishmania_ infection.15 Another work indicated that Lyn is also relevant to
microbiota-dependent intestinal inflammation and susceptibility to enteric pathogens.16 Lyn has roles during pulmonary infections. Previous reports from us and others showed that Lyn is
located on the inner leaflet of the plasma membrane and in the proximity of lipid rafts, and can thus be translocated into the activated membrane domains to transmit cellular signals for
either facilitating phagocytosis or regulating inflammatory responses.17–19 Upon _Klebsiella pneumoniae_ infection, Lyn negatively regulates inflammatory responses via the p38/NF-κB
signaling pathway to maintain a balanced inflammation.20 Lyn is also critical for _P. aeruginosa_ internalization into lung cells.21 Lyn is activated to be associated with lipid rafts and
TLR2, playing an important role in the initial stages of _P. aeruginosa_ infection in both alveolar macrophages (AMs) and lung epithelial cells. Our more recent study showed that Lyn
facilitates the delivery of both _K. pneumoniae_ and _P. aeruginosa_ to lysosomes for eradication through TLR2-initiated autophagy-related phagocytosis.22 However, the molecular mechanism in
Lyn-mediated signaling pathway during _P. aeruginosa_ infection is still largely unclear. Using murine gene knockout models, here we set up to elucidate the molecular mechanism by which Lyn
regulates host defense during _P. aeruginosa_-induced acute pneumonia. We demonstrate that Lyn directly binds to interleukin-6 receptor (IL-6R) to regulate IL-6/STAT3 signaling pathway in
AMs. Furthermore, we found that Lyn regulates nuclear factor-κB (NF-κB) and MAP kinase (MAPK) activation by controlling the activation of cytosolic phosphatase SH2-containing
inositol-5′-phosphatase 1 (SHIP-1). During _P. aeruginosa_ infection, SHIP-1 positively regulates NF-κB signaling pathway, but negatively regulates MAPK signaling pathway through Akt
activation. These results provide new insight into the function of Lyn in bacterial infection. MATERIALS AND METHODS MICE C57BL/6J mice (6–8 weeks) were obtained from the Jackson Laboratory
(Bar Harbor, ME, USA). Lyn−/− and Ship-1−/− mice that are constructed based on C57BL/6J mice were kindly provided by Dr S Li (University of Massachusetts School of Medicine)23 and Dr C Baran
(Ohio State University), respectively.24 Lyn−/−/Ship-1−/− (short as L/S−/−) mice were generated by cross-breeding Lyn−/− mice with Ship-1−/− mice in our facility using a standard protocol
(no abnormal phenotypes noticed). Mice were genotyped by PCR and/or western blotting to confirm the gene deletion before use. Animals were kept in a specific pathogen-free facility of
University of North Dakota.25 All animal studies were approved by the University of North Dakota Institutional Animal Care and Use Committee (IACUC) and performed in accordance with the
animal care and institutional guidelines (IACUC approval #1204-4). The animal experimental procedures including treatment, care and end point choice were followed the Animal Research:
Reporting _In Vivo_ Experiment guidelines.25 PRIMARY CELLS AND CELL LINES Mice were killed, and the thoracic cavity and trachea were dissected. A small incision was made in the trachea via
1-ml syringe with an angiocath (BD Biosciences, Franklin Lakes, NJ, USA), and recovered to a sterile tube. The lungs were lavaged three times with 1 ml of phosphate-buffered saline
containing 1% fetal bovine serum (Life Technologies, Grand Island, NY, USA). The retained bronchoalveolar lavage fluid (BALF) was centrifuged at 600 _g_ for 5 min at 4 °C. A small smear was
made from the BALF and stained with HEMA-3 (ThermoFisher Scientific, Waltham, MA, USA) for cell differential counting according to the manufacture’s instruction.26 The cell pellets were
resuspended in RPMI 1640 medium (Life Technologies) supplemented with 10% fetal bovine serum and incubated on culture plate for 1 h at 37 °C/5% CO2 incubator to allow attachment of
macrophages. Non-adherent cells were removed by washing with normal saline. Murine MH-S AM cells were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured
following the manufacturer’s instructions.25 BACTERIA PREPARATION AND INFECTION EXPERIMENTS The _P. aeruginosa_ wild-type (WT) strain, PAO1, was kindly provided by Dr S Lory (Harvard
University).27 PAK was obtained from Dr G Pier (Harvard University).28 Bacteria were grown for about 16 h in lysogeny broth at 37 °C with 220 r.p.m. shaking. The bacteria were pelleted by
centrifugation at 5000 _g_. Various mammalian cells were changed to antibiotic-free medium and infected by bacteria in an multiplicity of infection (MOI) of 20:1 bacteria–cell ratio. Mice
were anesthetized with 45 mg kg−1 ketamine and intranasally instilled 1×107 clonal-forming units (CFU) of PAO1 or 5×106 CFU of PAK in 50 μl phosphate-buffered saline (10 mice per group for
survival rates test and three mice per group for other assays), respectively.17 Mice were monitored for symptoms and killed when they were moribund. RNA ISOLATION AND QUANTITATIVE REVERSE
TRANSCRIPTION-PCR RNA was isolated from indicated MH-S cells. A 50-ng DNA-free RNA was used for the first strand of cDNA synthesis using a SuperScript III first-strand synthesis system (Life
Technologies). Quantitative reverse transcription-PCR was performed using the iTag Universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) and gene-specific primers (Supplementary Table
1, synthesized in Integrated DNA Technologies, Coralville, IA, USA), in a CFX Connect Real-time PCR Detection System (Bio-Rad). Relative transcript levels were first normalized CT values to
GAPDH, and then normalized to the indicated control (2−ΔΔCT).25 TRANSFECTION OF SIRNA, PLASMIDS, ACTIVATORS AND INHIBITORS Lyn (SC-35828), Ship-1 (SC-36491), Ezrin (SC-35350) and siNC
(SC-37007) short interfering RNAs (siRNAs) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). MH-S cells were transfected with siRNA (5 pM), p-NF-κB-luc (100 ng) plasmid29
using LipofectAmine 2000 (Life Technologies) for 24 h following the manufacture’s instruction. Two hours before PAO1 infection as indicated, MH-S cells were treated with 20 μm STAT3
inhibitor VI (Merck Millipore, Bellerica, MA, USA), NF-κB inhibitor Sulfasalazine (R&D Systems, Minneapolis, MN, USA) and Akt inhibitor VI (Santa Cruz Biotechnology), respectively.
BACTERIAL BURDEN ASSAY AMs from BALF and ground lung, spleen, liver and kidney tissues were homogenized with phosphate-buffered saline and spread on lysogeny broth dishes to enumerate
bacterial numbers. The dishes were cultured in a 37°C incubator overnight, and colonies were counted. Triplicates were done for each sample and control.25 DIHYDRODICHLOROFLUORESCEIN
DIACETATE ASSAY Dihydrodichlorofluorescein diacetate dye (Life Technologies) does not normally fluoresce but emits green fluorescence upon reaction with superoxide inside cells. AMs were
treated as above and an equal amount of dye was added. After 10-min incubation, fluorescence was measured using a fluorometer, using a 485-nm excitation and 528-nm emission filter.30
3-(4,5-DIMETHYLTHIAZOL-2-YL)-2,5-DIMETHYLTETRAZOLIUM BROMIDE ASSAY This assay measures the color change of 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (Sigma-Aldrich, St
Louis, MO, USA) upon reduction by enzymes to assess the viability of cells. Cells were treated as above, and equal amount of dye was added. After one hour incubation, reaction was terminated
by stop solution and left at room temperature overnight for complete dissolution of formazan and absorbance at 560 nm was recorded using a multiscan plate reader to quantify the
concentration of superoxide anion.29 HISTOLOGICAL ANALYSIS Lung tissues of three independent mice were fixed in 10% formalin (Sigma-Aldrich) for 24 h and then embedded in paraffin using a
routine histologic procedure. Five-micrometer sections were cut, stained by standard hematoxylin and eosin and examined for differences in morphology post infection.25 INFLAMMATORY CYTOKINE
PROFILING Cytokine concentrations of tumor necrosis factor-α (TNF-α), IL-6, IL-1β, MIP-2 and IL-4 were measured by enzyme-linked immunosorbent assay kits stained from eBioscience Co. (San
Diego, CA, USA), in samples of BALF collected at the indicated times after infection. BALFs were collected and 100-μl aliquots of samples were added to the coated microtiter wells. The
cytokine concentrations were determined with corresponding detection horseradish peroxidase-conjugated antibodies (Abs). The values were read at 450 nm.25 IMMUNOBLOTTING Mouse monoclonal Abs
against GAPDH (SC-47724), STAT3 (SC-8019), p-STAT3 (SC-8059), Ezrin (SC-58758), IL-6R (SC-374259), GST (SC-374171), SHIP-1 (SC-271426), NF-κB p50 (SC-166588), p-NF-κB p50 (SC-271908), p-p38
(SC-166182), p-ERK (SC-7383), TLR4 (SC-293072) and p-Akt (SC-293125), polyclonal Abs against rabbit polyclonal Abs against Lyn (SC-28790), and goat polyclonal Abs against IL-6 (SC-1265)
were obtained from Santa Cruz Biotechnology. The samples derived from cells and lung homogenates were lysed in RIPA buffer, separated by electrophoresis on 12% SDS-polyacrylamide gel
electrophoresis gels and transferred to nitrocellulose transfer membranes (GE Amersham Biosciences, Pittsburgh, PA, USA). Proteins were detected by western blotting using primary Abs at a
concentration of 1/200 (Santa Cruz Biotechnology) and were incubated overnight. Labeling of the first Abs was detected using relevant secondary Abs conjugated to horseradish peroxidase
(Santa Cruz Biotechnology),31 detected using ECL regents (Santa Cruz Biotechnology). Phosphorylated and total protein levels were determined and quantified by three independent successive
immunoblotting membranes. IMMUNOPRECIPITATION To obtain whole-cell lysates, indicated AMs and MH-S cells were homogenized in lysis buffer containing phosphatase inhibitor (1:10 000) and
protease inhibitors (1:50, ThermoFisher Scientific). Then, total cell lysate were mixed with indicated immunoprecipitation antibody, which were coupled to agarose beads (A/G, 50:50,
ThermoFisher Scientific). Immunoprecipitates were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose transfer membranes. Proteins were detected using
detective Abs and were incubated overnight. Labeling of the first Abs was detected using relevant secondary Abs conjugated to horseradish peroxidase and detected using ECL regents.25 GST-LYN
PULL-DOWN ASSAY GST-Lyn constructs with different functional domains were originally obtained from Dr O Miura (Tokyo Medical and Dental University, Tokyo, Japan) and transformed into
_Escherichia coli_ BL21 DE3 strain. The GST-Lyn fragments were extracted using immobilized glutathione columns following the manufacturer’s instructions.22 Equal whole-cell lysates were
incubated with GST-Lyn peptide for protein interactions. The pull-down products were analyzed by immunoblotting with indicated Abs. SUCROSE DENSITY GRADIENTS Triton-soluble and
Triton-insoluble cell fractions were prepared using the previously described methods.32 In brief, MH-S cells were treated with PAO1 (MOI=20, 30 min) and then lysed in TN1 buffer containing
0.5% Triton X-100. One mililiter the lysate was mixed with 1 ml of 85% sucrose and loaded at the bottom of a Beckman centrifuge tube (Beckman Coulter, Fullerton, CA, USA), and overlaid with
7 ml of 30% sucrose followed by 3.5 ml of 5% sucrose. The gradients were centrifuged for 17 h at 38 000 r.p.m. at 4 °C in a Beckman SW40Ti rotor. Nine fractions (~1.4 ml each) were collected
from the top of the gradient and were used for analysis by immunoblotting. IMMUNOSTAINING Lyn-silenced and control MH-S cells were infected with PAO1 (MPI=20) for 0, 30 and 60 min. Cells
were individually incubated with primary anti-SHIP-1 Ab, and then the second fluorescein isothiocyanate-conjugated Abs as described.25 Localization of SHIP-1 was observed under an LSM 510
Meta Confocal Microscope (Zeiss, Jena, Thuringia, Germany). STATISTICAL ANALYSIS Most experiments were conducted in triplicate. Differences between two groups were compared by one-way
analysis of variance (Tukey’s _post hoc_) using GraphPad Prism 5 software (GraphPad Software, San Diego, CA, USA), whereas mice survival rates were calculated using Kaplan–Meier curve.21
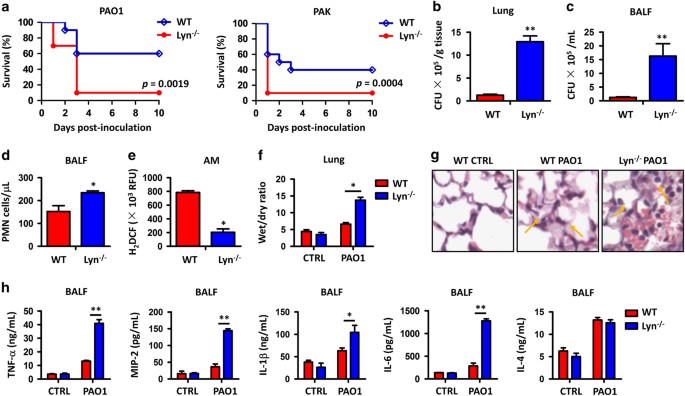
RESULTS LYN−/− MICE SHOW INCREASED INFLAMMATORY RESPONSES AND SEVERE LUNG INJURY FOLLOWING PAO1 INFECTION To investigate the physiological relevance of Lyn in _P. aeruginosa_ infection, we
intranasally instilled laboratory strain PAO1 at 1×107 CFU for each Lyn−/− mouse (Supplementary Figure 1A) as well as WT control mouse (with otherwise similar genetic backgrounds) to
establish an acute pneumonia model and compared the survival rates of these two groups of mice (10 mice per group). Figure 1a showed that Lyn−/− mice had a significantly increased mortality
compared with WT mice: after 3 days post PAO1 infection, only one Lyn−/− mouse survived, whereas >50% of WT mice remained alive. This result is represented by Kaplan–Meier survival curves
(_P_=0.0019, log-rank test). We also examined the survival rate of mice infected by another _P. aeruginosa_ strain PAK, which is reported to be more toxic to rodents than PAO1. Similar to
PAO1 strain, Lyn−/− mice succumbed to PAK (5×106 CFU per mouse, 10 mice per group) infection compared with WT mice (Figure 1a, _P_=0.0004, log-rank test). These data suggest that Lyn is
physiologically relevant to _P. aeruginosa_ infection in acute pneumonia models. To further analyze the cause of infection lethality in Lyn−/− mice, we examined bacterial burdens in the
lung, liver, spleen, kidney and BALF from 24 h post PAO1-infected mice. Bacterial CFU increased significantly in the organs of Lyn−/− mice compared with the same organs of WT mice (Figures
1b and c; Supplementary Figures 1B–D), demonstrating severe lung injury and pneumonia associated with Lyn deficiency. Increased recruitment of polymorphonuclear neutrophils (PMNs) into the
lung, which is assumed for bacterial clearance, also contributes to severe lung injury and systemic bacterial infection.33 To test this idea, we quantified PMN infiltration and observed
increased PMNs in the BALF of Lyn−/− mice compared with WT mice (Figure 1d). Phagocyte-released reactive oxygen species (ROS) are crucial for host defense against pathogenic infection by
either augmenting antibiotic activity or directly participating in bacterial killing in lysosomes. As determined by dihydrodichlorofluorescein diacetate assay, AMs of Lyn−/− mice showed a
significant decrease in oxidative stress at 24 h post infection compared with WT mice (Figure 1e). To assess the extent of acute lung injury, we measured wet/dry ratio of mouse lungs, and
found an approximately threefold increase in wet/dry ratio of Lyn−/− mouse lungs versus WT mouse lungs (Figure 1f). In addition, lung histology was examined 24 h post PAO1 infection as a
direct indicator of lung injury. Although both Lyn−/− and WT mice showed signs of pneumonia, histological alterations were more severe in the lungs of Lyn−/− mice (Figure 1g). The arrows
showed serious inflammatory response due to increased PMN penetration. Finally, we measured cytokine concentrations in BALF 24 h post PAO1 infection to gauge the inflammatory response. The
pro-inflammatory cytokines, including TNF-α, MIP-2, IL-1β and IL-6 levels, were significantly elevated in BALF from Lyn−/− mice compared to WT mice, but anti-inflammatory cytokine IL-4 was
not altered (Figure 1h), indicating that Lyn preferentially impacts pro-inflammatory cytokine production. Collectively, these data suggest that the severe lung injury in Lyn−/− mice is
caused by excessive ROS accumulation and overly active inflammatory response. LYN REGULATES IL-6/STAT3 SIGNALING PATHWAY BY BINDING WITH IL-6R AND EZRIN In Figure 1h, PAO1 infection
significantly altered inflammatory cytokine IL-6 expression in BALF of Lyn−/− mice, compared with that of WT mice. Considering that the mortality of PAO1-infected mice is highly
AM-dependent,34 and AM is one of the primary cell population in BALF,35 we assumed a critical role of IL-6 regulation by Lyn in these cells during bacterial infection. To confirm this, we
transfected murine lung macrophages (MH-S cells) with a siRNA against Lyn to inhibit Lyn expression, as well as with siNC (negative control siRNA) as a negative control (Supplementary Figure
S1E). Thirty minutes post PAO1 infection, compared with negative controls, IL-6 production was significantly increased in Lyn-silenced MH-S cells, which was detected by enzyme-linked
immunosorbent assay, quantitative PCR and immunoblotting, respectively (Figures 2a–c; Supplementary Figure S2A). However, IL-6R was not significant changed (Figure 2c; Supplementary Figure
S2A). As IL-6 activates transcription factor STAT3 to initiate the expression of other inflammatory factors,36 we studied whether Lyn regulates STAT3 in AMs during bacterial infection.
Immunoblotting showed that after PAO1 infection, the levels of both total and phosphorylated STAT3 were increased in Lyn-silenced MH-S cells, indicating that Lyn regulates the IL-6/STAT3
signaling pathway (Figure 2d; Supplementary Figure S2B). As Lyn is localized on the cell membrane,37 we proposed that Lyn may function via the IL-6/STAT3 signaling pathway by protein–protein
interactions with IL-6R (or other cell membrane proteins). To test this potential association mechanism, we lysed control and Lyn-silenced MH-S cells and performed immunoprecipitation
assays with anti-Lyn and anti-IL-6R Abs. Although it was not found in lysates from control MH-S cells (without PAO1 infection), a stable interaction between Lyn and IL-6R was detected in
siNC-treated MH-S cells after PAO1 infection, but was not found in Lyn-silenced MH-S cells (Figure 2e). Considering Lyn is activated upon ligand binding to a wide variety of cell surface
receptors that are essential for immune responses,13,14 such as cytoskeletal proteins CDC42 and actin, we presumed that these cell surface factors in AMs may also have an impact on IL-6R/Lyn
interaction during PAO1 infection. The co-immunoprecipitation data showed that most results were negative (data not shown), but unexpectedly, Ezrin, a cytosol migration regulator,38 was
detected in anti-Lyn Abs pulled down products in PAO1-infected WT MH-S cells, but was not found in anti-IL-6R Abs pulled down products in Lyn-silenced MH-S cells or control cells (Figure
2e), suggesting that an Ezrin–Lyn–IL-6R complex is formed in regulation of inflammatory responses against PAO1, and Lyn may bind to both IL-6R and Ezrin. We then tried to determine which
domain(s) of Lyn is/are critical for the interaction with IL-6R. Purified Lyn-GST peptides with distinct functional domains (see schematic, Figure 2f) were prepared as described
previously.20,22 The Lyn-GST fragments were coated on immobilized glutathione agarose beads to pull-down interacting partners from PAO1-infected MH-S cell lysates by anti-GST Abs, and probed
using IL-6R Abs. The presence of both Src homolog 2 (SH2) and SH3 domains of Lyn was responsible for interacting with IL-6R upon PAO1 infection in MH-S cells, whereas the kinase domain was
not (Figure 2g). We then asked whether Lyn regulates STAT3 activation through the IL-6R–Lyn–Ezrin complex. Considering IL-6R is critical for IL-6 recognition and binding, here we only
treated MH-S cells with siRNAs against Ezrin, to elucidate the inhibitory role of IL-6R–Lyn–Ezrin complex (Figure 2h). Immunoblotting showed that after PAO1 infection, the levels of total
and phosphorylated STAT3 were increased in the Ezrin-silenced MH-S cells (Figure 2i; Supplementary Figure S2C), indicating that Lyn indeed regulates the STAT3 activation by binding Ezrin. In
addition, 2 h post PAO1 infection, 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide assay showed a significant decrease in the survivals of Lyn- and Ezrin-silenced MH-S cells,
respectively (Figure 2j), indicating that the IL-6R–Lyn–Ezrin complex axis negatively regulates _P. aeruginosa_-induced host inflammatory responses. LYN DEFICIENCY RESULTS IN ENHANCED
PAO1-INDUCED SHIP-1 EXPRESSION AND MEMBRANE TRANSLOCATION The cytosolic phosphatase SHIP-1, SH2-containing inositol-5′-phosphatase, has critical roles in microphages, including generation of
alternatively activated macrophages,39 regulation of LPS-induced macrophage inflammatory responses40 and modulation of myeloproliferation.11 Diverse cellular functions have been previously
investigated in association of Lyn and SHIP-1,41,42 but whether Lyn is involved in SHIP-1 expression or membrane translation43 remains unclear. To investigate it, we measured SHIP-1
expression in Lyn-silenced and control MH-S cells at 0, 30 and 60 min post infection of PAO1. Immunoblotting results showed that SHIP-1 expression was induced in control MH-S cells, but
excessively induced Lyn-silenced MH-S cells (Figure 3a; Supplementary Figure S2D), indicating that PAO1-dependent SHIP-1 expression in AMs is regulated by Lyn. Further, to elucidate whether
Lyn has roles in membrane translocation of SHIP-1, Triton-soluble and Triton-insoluble fractions (lipid rafts) were isolated from Lyn-silenced and control MH-S cells using sucrose density
gradients. The fractions were separated by SDS-polyacrylamide gel electrophoresis and examined for the presence of SHIP-1 by immunoblotting. Thirty min post infection, only small portion of
SHIP-1 was translocated to the plasma membrane in control MH-S cells, but almost 100% translocated to the plasma membrane in Lyn-silenced MH-S cells (Figure 3b). Confocal laser scanning
microscopy also showed an enhanced expression and fast membrane translocation of SHIP-1 in Lyn-silenced MH-S cells, compared with that of control MH-S cells (Figure 3c), again demonstrating
that Lyn influences the expression and membrane translocation of SHIP-1. In addition, we successfully blocked its expression by treating Lyn-silenced and control MH-S cells with STAT3
inhibitor (Figure 3d; Supplementary Figure S2E). Immunoblotting showed that when STAT3 expression was inhibited, the expression of SHIP-1 was consistently repressed (Figure 3d; Supplementary
Figure S2E), indicating that excessive SHIP-1 expression in Lyn-silenced MH-S cells is caused by Lyn-dependent STAT3 activation. SHIP-1 DEFICIENCY RESULTS IN INCREASED SURVIVAL AND REDUCED
INFLAMMATORY RESPONSES IN LYN−/− MICE AGAINST PAO1 INFECTION To further characterize the critical role of SHIP-1 and Lyn during bacterial infection, we cross-bred Ship-1−/− mice with Lyn−/−
mice to generate a double gene knockout mouse (Lyn−/−/Ship-1−/−, short as L/S−/−, Supplementary Figure 1A), and then challenged with PAO1 (10 mice per group). Unexpectedly, although
containing a higher mortality compared with WT mice (_P_=0.021, log-rank test), L/S−/− mice had a significantly decreased mortality compared with Lyn−/− mice (_P_=0.046, log-rank test;
Figure 4a). We also found that 24 h post PAO1 infection, L/S−/− mice exhibited decreased bacterial burdens in the lungs (Figure 4b), BALF (Figure 4c) and other organs including liver, spleen
and kidney, compared with that of Lyn−/− mice (Supplementary Figures 1B–D). PMNs in the BALF of L/S−/− mice were also reduced (Figure 4d), whereas ROS in the AMs of L/S−/− mice were
increased (Figure 4e). In addition, the pro-inflammatory cytokines, including TNF-α, MIP-2, IL-1β and IL-6 levels, were significantly reduced in BALF from L/S−/− mice compared with Lyn−/−
mice, whereas anti-inflammatory cytokine IL-4 was increased (Figure 4f). These data collectively demonstrated that knockout of SHIP-1 in Lyn−/− mice reversed, at least in part, severe lung
damage and inflammatory responses that caused by Lyn deficiency, indicating that SHIP-1 may exert its function in host defense in a Lyn-dependent manner. SHIP-1 DEFICIENCY LEADS TO REDUCED
NF-ΚB-DEPENDENT TRANSCRIPTION IN LYN−/− AMS IN PAO1 INFECTION PAO1 infection of macrophages induces expression of transcription factor NF-κB, promotes its nucleic translocation and activates
its downstream cytokine gene transcription.44 It has previously been reported that SHIP-1 positively regulates NF-κB-dependent gene transcription in macrophage RAW264.7 in response to LPS
stimulation.45 Therefore, we asked whether SHIP-1 and Lyn influenced gene expression driven by NF-κB in AMs in response to PAO1 infection. Immunoblotting results showed that levels of total
and phosphorylated NF-κB were increased in the Lyn-silenced MH-S cells, but were inhibited in the Lyn and SHIP-1 double-silenced MH-S cells (Figure 5a; Supplementary Figure S2F).
Lyn-silenced, Lyn and SHIP-1 double-silenced, and control MH-S cells were transiently transfected with luciferase reporter plasmids that were dependent on NF-κB binding (NF-κB-luc).
NF-κB-dependent transcription of luciferase in these transfected cells were induced 30 min post PAO1 infection (Figure 5b). Compared with that in control MH-S cells, activity of luciferase
in Lyn-silenced MH-S cells was significantly higher, but was not changed in Lyn and SHIP-1 double-silenced cells (Figure 5b). Confocal laser scanning microscopy data showed that p-NF-κB was
slowly activated and translocated into the nuclei in Lyn and SHIP-1 double-silenced cells, compared with Lyn-silenced MH-S cells (Figure 5c). These data demonstrated that knockdown of SHIP-1
in Lyn-deficient AMs weakened PAO1-induced activation, phosphorylation and nuclear translocation of NF-κB. We then measured NF-κB-dependent gene transcription in indicated MH-S cells.
Quantitative reverse transcription-PCR data showed that 30 min post PAO1 infection, transcriptions of all tested NF-κB-dependent genes, including IL-1α, IL-1β, IL-6, IFN-γ, MCP-2, MIP1α,
TNF-α and TLR2, were significantly increased in Lyn-silenced MH-S cells (Figure 5d). Compared with Lyn-silenced MH-S cells, increase of these gene transcription in Lyn and SHIP-1
double-silenced MH-S cells was dampened (Figure 5d), indicating that knockdown of SHIP-1 in Lyn-deficient AMs abolished NF-κB-dependent gene excessive activation.
3-(4,5-Dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide assay showed that 30 min post PAO1 infection, cell viability of Lyn-silenced MH-S cells was significantly hampered compared with
control cells (Figure 5e). The reduced cell viability was partially restored to the level of control MH-S cells when SHIP-1 was also silenced in Lyn-silenced cells (Figure 5e). In addition,
when NF-κB was inhibited, the viability of Lyn-silenced MH-S cells increased to the similar level of Lyn and SHIP-1 double-silenced or control MH-S cells (Figure 5e), indicating a critical
role of Lyn, SHIP-1 and NF-κB in AM cell survival during PAO1 infection. PAO1-INDUCED MAPK PHOSPHORYLATION IS DOWNREGULATED IN SHIP-1 DEFICIENCY AMS MAPK phosphorylation in macrophages is
thought to be TLR4-dependent in _P. aeruginosa_ infection.46 To examine the molecular details of Lyn and SHIP-1 in regulating TLR4 signaling, we next assessed the activation of MAPK in
Lyn-silenced, Lyn and SHIP-1 double-silenced, and control MH-S cells. Immunoblotting results indicated that robust phosphorylation of ERK1/2 and p38 was significantly induced in Lyn-silenced
MH-S cells, compared with control cells (Figure 6a; Supplementary Figure S2G). In contrast, the enhanced phosphorylation of ERK1/2 and p38 was partially impeded in Lyn and SHIP-1
double-silenced MH-S cells (Figure 6a; Supplementary Figure S2G). To ensure that the signaling differences observed were not due to the differences in the expression of TLR4, we measured
TLR4 by immunoblotting and found a similar expression level in all indicated MH-S cells (Figure 6b; Supplementary Figure S2H). These results indicate that Lyn negatively while SHIP-1
positively regulates the activation of ERK and p38, and that this regulation is TLR4-independent. Previous studies demonstrate that Akt activation leads to a downregulation of LPS-induced
MAPK phosphorylation.40 To test whether the altered phosphorylation of MAPK is accompanied with different Akt activation, phosphorylation of Akt (Ser473) was compared in Lyn-silenced, Lyn
and SHIP-1 double-silenced, and control MH-S cells. The results shown in Figure 6c (and Supplementary Figure S2I) indicate that Akt phosphorylation is induced in control MH-S cells 30 min
post PAO1 infection. In contrast, Akt phosphorylation is significantly reduced in Lyn-silenced MH-S cells, but is substantially enhanced in Lyn and SHIP-1 double-silenced MH-S cells after
PAO1 infection (Figure 6c; Supplementary Figure S2I). These results suggest that Lyn and SHIP-1 may regulate PAO1-induced MAPK activation by changing Akt phosphorylation. In addition, to
test whether induction of Akt in Lyn and SHIP-1 double-silenced MH-S cells would restore PAO1-induced MAPK phosphorylation, MH-S cells were treated with the PI3K inhibitor wortmannin for 1 h
before infection of PAO1. The results shown in Figure 6d (and Supplementary Figure S2J) suggest that MAPK phosphorylation was restored in Lyn and SHIP-1 double-silenced MH-S cells of which
Akt phosphorylation was suppressed. DISCUSSION In this study, we report a critical regulatory role of Lyn in host defense against _P. aeruginosa_ infection, which is exerted in an IL-6/STAT3
signaling axis. We show that Lyn−/− mice have a high susceptibility to _P. aeruginosa_ infection manifested with decreased bacterial clearance, heightened pro-inflammatory cytokines and
severe lung injury. Built on the situation in _K. pneumoniae_ infection as seen in our previous report,20 Lyn regulates an IL-6/STAT3 signaling pathway in AMs during _P. aeruginosa_
infection by direct binding with IL-6R and cytoskeletal protein Ezrin through its SH2 and SH3 domains. Importantly, our data reveal a mechanism that SHIP-1 was in cooperation with Lyn in
regulation of host immune responses. Deficiency in Lyn results in excessive STAT3 activation and thus enhancing SHIP-1 expression and membrane translocation. Lack of SHIP-1 in Lyn−/− mice
promotes survival and thereby attenuating inflammatory responses during _P. aeruginosa_ infection. Mechanistically, loss of SHIP-1 reduces NF-κB-dependent cytokine production, dampened MAPK
activation through a PI3K/Akt pathway (summarized in Figure 6e). Thus, these finding strongly attest that Lyn negatively regulates AM inflammation against _P. aeruginosa_ infection through
the SHIP-1 and IL-6/STAT3 signaling pathway. This study again supports a concept that Lyn is critically involved in host inflammatory responses during _P. aeruginosa_ infection. Our data
showed that disease-related phenotypes in _P. aeruginosa_ infected Lyn−/− mice were similar to that in _K. pneumoniae_ infected Lyn−/− mice,20 including increased mortality, severe lung
injury, elevated inflammatory cytokines (IL-6 and TNF-α), increased phosphorylation of p38 and NF-κB. In addition, these indicated phenotypes are also in line with observations in house dust
mite-treated Lyn−/− mice,31 suggesting that Lyn may render a variety of immune responses to a wide range of microorganisms and other immunogens. On the other hand, bacterial
species-dependent roles of Lyn in regulation of immune responses have previously been demonstrated. However, ROS in AMs of Lyn−/− mice were significantly reduced during _P. aeruginosa_
infection (Figure 1e), but markedly increased during _K. pneumoniae_ infection,20 suggesting that Lyn’s influence on ROS production also varies with bacterial species. As Lyn−/− mice were
germline knocked out, all these indicated typical disease phenotypes, including decreased bacterial clearance, heightened pro-inflammatory cytokines and severe lung injury in Lyn−/− mice
(Figure 1), were on the impact of combined roles of Lyn in various cell types. For example, inhibition of Lyn’s function prevented _P. aeruginosa_ internalization in lung epithelial cells,21
which may accelerate bacterial dissemination to other organs and caused secondary injury (Supplementary Figure 1). Importantly, Lyn deficiency also significantly inhibited bacterial
phagocytosis and timely clearance by AMs,17,22 which resulted in the higher bacterial burden in BALF and lung tissue of Lyn−/− mice (Figure 1). In this study, we focused on the investigation
of Lyn’s role in AMs, because the mortality of PAO1-infected mice is highly AM-dependent.34 As one of the primary inflammatory cytokine source in the lung and BALF,35 excessive activation
of AMs increases inflammation and stimulates immune system,45 leading to programmed cell death and mice mortality.45,47 Previous studies showed that both Lyn and pro-inflammatory cytokine
IL-6 were involved in several autoimmune diseases. For example, Lyn-deficient leukocytes, notably B cells, overproduce IL-6, and this establishes an inflammatory environment leading to the
activation of B cells and cellular components, thus developing severe autoimmune pathology.8 In human multiple myeloma cells, the association of CD45 and Lyn requires the presence of IL-6.48
Nevertheless, how Lyn regulates IL-6- and IL-6-dependent signaling pathway is largely unknown. Using co-immunoprecipitation assays, we observed a stable association between IL-6R, Lyn and
Ezrin in mouse AMs after PAO1 infection (Figure 2e). Lyn is dominant in this protein complex because in Lyn-deficient AMs, the interactions between IL-6R and Ezrin are diminished (Figure
2e). Using purified peptides containing different Lyn functional domains, we clarified that both of Lyn SH2 and SH3 domains can bind to IL-6R, whereas Lyn kinase domain cannot (Figure 2g).
During _P. aeruginosa_ infection, an interaction of IL-6 and IL-6R in AMs activates transcript STAT3 to initiate the expression of other inflammatory factors.36 Our data showed that
interactions between IL-6R, Lyn and Ezrin negatively regulate the IL-6/STAT3 signaling pathway, because respective inhibition of each of these components results in enhanced expression and
phosphorylation of STAT3 after PAO1 infection (Figures 2d and i). This indirect regulation of STAT3 by Lyn in AMs is different from that in other cell types. For example, Lyn directly
phosphorylates STAT3 to impact proliferation, differentiation or growth arrest in B cells.49 In our study, inhibition of Lyn and Ezrin, respectively, resulted in a decreased cell viability
after PAO1 infection (Figure 2j), indicating that this complex has a critical role in host defense against _P. aeruginosa_. In addition, we noticed that the IL-6 production was significantly
elevated in Lyn-silenced MH-S cells (Figures 2a–c), suggesting that the activation of IL-6/STAT3 pathway may also result from IL-6 stimulation. However, our data showed that expression of
IL-6R in Lyn-silenced cells was not changed compared with that of control cells (Figure 2c). We believe that IL-6R expression limits the direct binding of IL-6 and IL-6R in Lyn-silenced MH-S
cells, even IL-6 was over produced, indicating that Lyn signaling to IL-6 contributes significantly to the STAT3 activation. In this study, we identified the mechanism of SHIP-1 along with
Lyn in immune responses to _P. aeruginosa_ infection. Diverse cellular functions have been previously investigated in association with both Lyn and SHIP-1.41,42 SHIP-1 and Lyn are involved
in regulation of integrin alpha IIb-β3 signaling in platelets,41 and in respect to Ag-triggered degranulation in mast cells.42 In macrophages, Lyn and SHIP-1 negatively regulate macrophage
colony-stimulating factor-induced Akt activity.24 A previous study demonstrated that activation of macrophages by the stimulation of LPS is negatively regulated by a Lyn/PI3K axis and
promoted by SHIP-1.50 However, the internal relationship between Lyn and SHIP-1 remains unclear. Direct phosphorylation and activation of SHIP-1 by Lyn has been reported with a loss of
SHIP-1 activity in Lyn-deficient mast cells.51 Our data for the first time showed that the expression and membrane translocation of SHIP-1 is dependent on Lyn in AMs (Figures 3a–c) during
_P. aeruginosa_ infection, which is an indirect role. Interaction of Lyn and IL-6R inhibits the activation of transcript factor STAT3 (Figure 2d), which further downregulates SHIP-1
expression (Figure 3d). Unlike the STAT3-independent pathway through a SHIP-1/IL-10/TNF-α axis,52 our findings indicate a STAT3-dependent SHIP-1 regulation during immune responses.
Importantly, we observed that an mitigated disease phenotype in Lyn and SHIP-1 double knockout mice after PAO1 infection, including increased survival, decreased PMN and reduced inflammatory
responses, compared with Lyn−/− mice (Figure 4), indicating that SHIP-1 positively regulates host immune responses, which is in line with other studies.50 Studies have investigated the
molecular mechanisms by which SHIP-1 regulates variable types of inflammation responses, including allergic airway inflammation,53 chronic inflammation in the myeloid compartment,54
triptolide ameliorates lieocolonic anastomosis inflammation53 and LPS-induced inflammatory responses.50 Compared with the previous studies, we demonstrate that Lyn collaborating with SHIP-1
acts through both the similar and unique mechanisms to regulate multiple signaling pathways. Our previous and current findings showed that Lyn negatively regulates the activation and nuclear
translocation of NF-κB in AMs after both _P. aeruginosa_ and _K. pneumoniae_ infection. Here we validated that Lyn regulates NF-κB activation and NF-κB-dependent gene transcription through
a Lyn/SHIP-1/NF-κB axis (Figure 5). We further demonstrate that Lyn positively regulates bacteria-induced Akt phosphorylation and thus negatively regulates Akt-dependent activation of MAPK
in AMs through SHIP-1 (Figure 6). Similar roles for Lyn and SHIP-1 in regulation of Akt were observed in B lymphocytes55 and bone marrow-derived macrophages.40,50 Studies showed a role for
Lyn in controlling MAPK activation is TLR4-dependent.50,56 However, our findings showed that expression of TLR4 was not significantly changed in AMs when Lyn was knocked out or Lyn and
SHIP-1 were both knocked out (Figure 6), suggesting that Lyn and SHIP-1 also controls MAPK activation in a TLR4-independent way. Despite being more dominant in this infection model for Lyn,
SHIP-1 also exhibited a critical role. In summary, we found that Lyn is required for full resistance to _P. aeruginosa_ infection and its deficiency contributes to elevated inflammatory
cytokine responses,57,58 which resulted in a severe susceptibility to this infection. We for the first time demonstrate that during bacterial infection, a Lyn–IL-6R–Ezrin complex negatively
regulates the IL-6/STAT3 signaling pathway in murine AMs. Further, we unraveled that SHIP-1 is under Lyn/STAT3 regulation and has critical roles in host inflammatory responses. Lyn regulates
NF-κB activation and NF-κB-dependent gene transcription through a Lyn/SHIP-1/NF-κB axis. In addition, the Lyn/SHIP-1 axis controls MAPK activation in a TLR4-independent manner. Overall, our
findings provide deeper insight into the role of Lyn in the regulation of host immune response against _P. aeruginosa_ infection and may help identify novel therapeutic approaches to
combatting against bacterial infection. REFERENCES * Lyczak J, Cannon C, Pier G . Establishment of _Pseudomonas aeruginosa_ infection: lessons from a versatile opportunist. _Microbes Infect_
2000; 2: 1051–1060. Article CAS Google Scholar * Sousa AM, Pereira MO . _Pseudomonas aeruginosa_ diversification during infection development in cystic fibrosis lungs—a review.
_Pathogens_ 2014; 3: 680–703. Article Google Scholar * Li R, Fang L, Tan S, Yu M, Li X, He S et al. Type I CRISPR-Cas targets endogenous genes and regulates virulence to evade mammalian
host immunity. _Cell Res_ 2016; 26: 1273–1287. Article CAS Google Scholar * Lovewell RR, Patankar YR, Berwin B . Mechanism of phagocytosis and host clearance of _Pseudomonas aeruginosa_.
_Am J Physiol Lung Cell Mol Physiol_ 2014; 306: L591–L603. Article CAS Google Scholar * Tosi MF . Innate immune responses to infection. _J Allergy Clin Immunol_ 2005; 116: 241–249, quiz
250. Article CAS Google Scholar * Suresh R, Mosser DM . Pattern recognition receptors in innate immunity, host defense, and immunopathology. _Adv Physiol Educ_ 2013; 37: 284–291. Article
Google Scholar * Lim YJ, Koo JE, Hong EH, Park ZY, Lim KM, Bae ON et al. A Src-family-tyrosine kinase, Lyn, is required for efficient IFN-beta expression in pattern recognition receptor,
RIG-1, signal pathway by interacting with IPS-1. _Cytokine_ 2015; 72: 63–70. Article CAS Google Scholar * Tsantikos E, Oracki SA, Quilici C, Anderson GP, Tarlinton DM, Hibbs ML .
Autoimmune diseases in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. _J Immunol_ 2010; 184: 1348–1360. Article CAS Google Scholar * Hernandez-Hansen
V, Mackay GA, Lowell CA, Wilson BS, Oliver JM . The Src kinase Lyn is a negative regulator of mast cell proliferation. _J Leukoc Biol_ 2004; 75: 143–151. Article CAS Google Scholar *
Pereira S, Lowell C . The Lyn tyrosine kinase negatively regulates neutrophil integrin signaling. _J Immunol_ 2003; 171: 1319–1327. Article CAS Google Scholar * Xiao W, Hong H, Kawakami
Y, Lowell CA, Kawakami T . Regulation of myeloproliferation and M2 macrophage programming in mice by Lyn/Hck, SHIP, and Stat5. _J Clin Invest_ 2008; 118: 924–934. CAS PubMed PubMed Central
Google Scholar * Krebs DL, Chehal MK, Sio A, Huntington ND, Da ML, Zilten P et al. Lyn-dependent signaling regulates the innate immune response by controlling dendritic cell activation of
NK cells. _J Immunol_ 2012; 188: 5094–5105. Article CAS Google Scholar * Ingley E . Functions of the Lyn tyrosine kinase in health disease. _Cell Commun Signal_ 2012; 10: 21. Article
CAS Google Scholar * Scapini P, Pereira S, Zhang H, Lowell CA . Multiple roles of Lyn kinase in myeloid cell signaling and function. _Immunol Rev_ 2009; 228: 23–40. Article CAS Google
Scholar * Wetzel DM, Rhodes EL, Li S, McMahon-Pratt D, Koleske AJ . The Src kinases Hck, Fgr, and Lyn activate Abl2/Arg to facilitate IgG-mediated phagocytosis and Leishmania infection. _J
Cell Sci_ 2016; 129: 3130–3143. Article CAS Google Scholar * Roberts ME, Bishop JL, Fan X, Beer JL, Kum WW, Krebs DL et al. Lyn deficiency leads to increased microbiota-dependent
intestinal inflammation and susceptibility to enteric pathogens. _J Immunol_ 2014; 193: 5249–5263. Article CAS Google Scholar * Kannan S, Audet A, Huang H, Chen LI, Wu M .
Cholesterol-rich membrane rafts and Lyn are involved in phagocytosis during _Pseudomonas aeruginosa_ infection. _J Immunol_ 2008; 180: 2396–2408. Article CAS Google Scholar * Kannan S,
Huang H, Seeger D, Audet A, Chen Y, Huang C et al. Alveolar epithelial type II cells activate alveolar macrophages and mitigate _P. aeruginosa_ infection. _PLoS one_ 2009; 4: e4891. Article
Google Scholar * Young RM, Holowka D, Baird B . A lipid raft environment enhances Lyn kinase activity by protecting the active site tyrosine from dephosphorylation. _J Biol Chem_ 2003;
278: 20746–20752. Article CAS Google Scholar * Li X, Zhou X, Ye Y, Li Y, Li J, Privrasky B et al. Lyn regulates inflammatory responses in _Klebsiella pneumoniae_ infection via the
p38/NF-kappaB pathway. _Eur J Immunol_ 2014; 44: 763–773. Article CAS Google Scholar * Kannan S, Audet A, Knittel J, Mullegama S, Gao GF, Min W . Src kinase Lyn is crucial for
_Pseudomonas aeruginosa_ internalization into lung cells. _Eur J Immunol_ 2006; 36: 1739–1752. Article CAS Google Scholar * Li X, He S, Zhou X, Ye Y, Tan S, Zhang S et al. Lyn delivers
bacterial to lysosomes for eradication through TLR2-initiated autophage related phagocytosis. _PLoS pathog_ 2016; 12: e1005363. Article Google Scholar * Hu Y, Liu Y, Pelletier S,
Buchdunger E, Warmuth M, Fabbro D et al. Requirement of Src kinase Lyn, Hck and Fgr for BCR-ABL1-induced B lymphoblastic leukemia but not chronic myeloid leukemia. _Nat Genet_ 2004; 36:
453–461. Article CAS Google Scholar * Baran CP, Tridandapani S, Helgason CD, Humphries RK, Krystal G, Marsh CB . The inositol 5’-phosphatase SHIP-1 and the Src kinase Lyn negatively
regulate macrophage colony-stimulating factor-induced Akt activity. _J Biol Chem_ 2003; 278: 38628–38636. Article CAS Google Scholar * Li R, Tan S, Yu M, Jundt MC, Zhang S, Min W .
Annexin A2 regulates antophagy in _Pseudomonas aeruginosa_ infection through the Akt1-mTOR-ULK1/2 signaling pathway. _J Immunol_ 2015; 195: 3901–3911. Article CAS Google Scholar * Guo Q,
Shen N, Yuan K, Li J, Wu H, Zeng Y et al. Caveolin-1 plays a critical role in host immunity against _Klebsiella pneumoniae_ by regulating STAT5 and Akt activity. _Eur J Immunol_ 2012; 42:
1500–1511. Article CAS Google Scholar * Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S et al. Analysis of _Pseudomonas aeruginosa_ diguanylate cyclases and
phosphodiesterases reveals a role for bis-(3’-5’)-cyclic-GMP in virulence. _Proc Natl Acad Sci USA_ 2006; 103: 2839–2844. Article Google Scholar * Pribe GP, Brinig MM, Hatano K, Grout M,
Coleman FT, Pier GB et al. Construction and characterization of a live, attenuated aroA deletion mutant of _Pseudomonas aeruginosa_ as a candidate intranasal vaccine. _Infect Immun_ 2002;
70: 1507–1517. Article Google Scholar * Zhou X, Li X, Ye Y, Zhao K, Zhuang Y, Li Y et al. MicroRNA-302b augments host defense to bacteria by regulating inflammatory responses via feedback
to TLR/IRAK4 circuits. _Nat Commun_ 2014; 5: 3619. Article Google Scholar * Wu M, Huang H, Zhang W, Kannan S, Weaver A, Mckibben M et al. Host DNA repair proteins in response to
_Pseudomonas aeruginosa_ in lung epithelial cells and in mice. _Infect Immun_ 2011; 79: 75–87. Article CAS Google Scholar * Li G, Fox J 3rd, Liu Z, Gao GF, Jin Y et al. Lyn mitigates
mouse airway remodeling by downregulating the TGF-beta3 isoform in house dust mite models. _J Immunol_ 2013; 191: 5359–5370. Article CAS Google Scholar * Triantafilou M, Miyake K,
Golenbock DT, Triantafilou K . Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. _J Cell Sci_ 2002;
115: 2603–2611. CAS PubMed Google Scholar * Langeneis JD . Neutrophil integrin affinity regulation in adhesion, migration, and bacterial clearance. _Cell Adh Migr_ 2013; 7: 476–481.
Google Scholar * Kooguchi K, Hashimoto S, Kobayashi A, Kitamura Y, Kudoh I, Wiener-Kronish J et al. Role of alveolar macrophages in initiation and regulation of inflammation in _Pseudomonas
aeruginosa_. _Infect Immun_ 1998; 66: 3164–3169. CAS PubMed PubMed Central Google Scholar * Guillemot L, Medina M, Pernet E, Leduc D, Chignard M, Touqui L et al. Cytosolic phospholipase
A2alpha enhances mouse mortality induced by _Pseudomonas aeruginosa_ pulmonary infection via interleukin 6. _Biochimie_ 2014; 107 PT A: 95–104. Article Google Scholar * Hodge DR, Hurt EM,
Farrar WL . The role of IL-6 and STAT3 in inflammation and cancer. _Eur J Cancer_ 2005; 41: 2502–2512. Article CAS Google Scholar * Kovarova M, Tolar P, Arudchandran R, Draberoval L,
Draber P . Structure-function analysis of Lyn kinase association with lipid rafts and initiation of early signaling events after Fcepsilon receptor I aggregation. _Mol Cell Biol_ 2001; 21:
8318–8328. Article CAS Google Scholar * Yi B, Chen L, Zeng J, Cui J, Wang G, Qian G et al. Ezrin regulating the cytoskeleton remodeling is required for hypoxia-induced Myofibroblast
proliferation and migration. _Front Cardiovasc Med_ 2015; 2: 10. Article Google Scholar * Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V et al. SHIP represses the generation of
alternatively activated macrophages. _Immunity_ 2005; 23: 361–374. Article CAS Google Scholar * Fang H, Pengal RA, Cao X, Ganesan LP, Wewers MD, Marsh CB et al. Lipopolysaccharide-induced
macrophages inflammatory response is regulated by SHIP. _J Immunol_ 2004; 173: 360–366. Article CAS Google Scholar * Maxwell MJ, Yuan Y, Anderson KE, Hibbs ML, Salemen HH, Jacson SP .
SHIP1 and Lyn kinase negatively regulate integrin alpha IIb beta 3 signaling in platelets. _J Biol Chem_ 2004; 279: 32196–32204. Article CAS Google Scholar * Nunes de Miranda SM, Wilhelm
T, Huber M, Zorn CN . Differential Lyn-dependence of SHIP1-deficient mast cell phenotype. _Cell Commun Signal_ 2016; 14: 12. Article Google Scholar * Krystal G . Lipid phosphatases in the
immune system. _Semin Immunol_ 2000; 12: 397–403. Article CAS Google Scholar * Zhang J, Wu XY, Yu FS . Inflammatory responses of corneal epithelial cells to _Pseudomonas aeruginosa_
infection. _Curr Eye Res_ 2005; 30: 527–534. Article CAS Google Scholar * Arango DG, Descoteaus A . Macrophage cytokines: involvement in immunity and infectious diseases. _Front Immunol_
2014; 5: 491. Google Scholar * Mclsaac SM, Stadnyk AW, Lin TJ . Toll-like receptors in the host defense against _Pseudomonas aeruginosa_ respiratory infection and cystic fibrosis. _J Leukoc
Biol_ 2012; 92: 977–985. Article Google Scholar * Sadikot RT, Blackwell TS, Christman JW, Prince AS . Pathogen-host interactions in _Pseudomonas aeruginosa_ pneumonia. _Am J Respir Crit
Care Med_ 2005; 171: 1209–1223. Article Google Scholar * Zhou Q, Yao Y, Ericson SG . The protein tyrosine phosphatase CD45 is required for interleukin 6 signaling in U226 meyloma cells.
_Int J Hematol_ 2004; 79: 63–73. Article CAS Google Scholar * Wang L, Kurosaki T, Corey SJ . Engagement of the B-cell antigen receptor activates STAT through Lyn in a Jak-dependent
pathway. _Oncogene_ 2007; 26: 2851–2859. Article CAS Google Scholar * Keck S, Freudenberg M, Huber M . Activation of murine macrophages via TLR2 and TLR4 is negatively regulated by a
Lyn/PI3K module and promoted by SHIP1. _J Immunol_ 2010; 184: 5809–5818. Article CAS Google Scholar * Hernandenz-Hansen V, Smith AJ, Surviladze Z, Chigaev A, Mazel T, Kalesnikoff J et al.
Dysregulation FcepsilonRI signaling and altered Fyn and SHIP activities in Lyn-deficient mast cell. _J Immunol_ 2004; 173: 100–112. Article Google Scholar * Chan CS, Ming-Lum A, Golds GB,
Lee SJ, Anderson RJ, Mui AL . Interleukin-10 inhibits lipopolysaccharide-induced tumor necrosis factor-alpha translation through a SHIP1-dependent pathway. _J Biol Chem_ 2012; 287:
38020–38027. Article CAS Google Scholar * Gold MJ, Hughes MR, Antignano F, Hirota JA, Zaph C, McNagny KM . Lineage-specific regulation of allergic airway inflammation by the lipid
phosphatase Src homology 2 domain-containing inositol 5-phosphatase (SHIP-1). _J Allergy Clin Immunol_ 2015; 136: 725–726. Article CAS Google Scholar * Maxwell MJ, Srivastava N, Park MY,
Tsantikos E, Engelman RW, Kerr WG et al. SHIP-1 deficiency in the myeloid compartment is insufficient to induce myeloid expansion or chronic inflammation. _Genes Immun_ 2014; 15: 233–240.
Article CAS Google Scholar * Li HL, Davis WW, Whiteman EL, Birnbaum MJ, Pure E . The tyrosine kinase Syk and Lyn expert opposing effects on the activation of protein kinase Akt/PKB in B
lymphocytes. _Proc Natl Acad Sci USA_ 1999; 96: 6890–6895. Article CAS Google Scholar * Avila M, Martinez-Juarez A, Ibarra-Sanchez A, Gonzalez-Espinosa C . Lyn kinase controls
TLR4-dependent IKK and MAPK activation modulating the activity of TRAF-6/TAK-1 protein complex in mast cells. _Innate immun_ 2012; 18: 648–660. Article CAS Google Scholar * Heid ME, Keyel
PA, Kamga C, Shiva S, Watkins SC, Salyer RD . Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. _J Immunol_ 2013; 191: 5230–5238.
Article CAS Google Scholar * Wu R, Li Y, Guo Z, Gong J, Zhu W, Li N et al. Triptolide ameliorates ileocolonic anastomosis inflammation in IL-10 deficient mice by mechanism involving
suppression of miR-155/SHIP-1 signaling pathway. _Mol Immunol_ 2013; 56: 340–346. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We sincerely acknowledge Dr O Miura of
Tokyo Medical and Dental University for kindly providing GST-Lyn constructs with different functional domains and also thank S Abrahamson (University of North Dakota Imaging Core Facility
supported by a COBRe grant: NIH 1P30GM103329) for help with imaging. This work was supported by National Institute of Health (AI109317-01A1 and AI109373-01). AUTHOR INFORMATION Author notes
* Rongpeng Li and Lizhu Fang: Co-first author. AUTHORS AND AFFILIATIONS * Department of Biomedical Sciences, University of North Dakota, Grand Forks, North Dakota, USA Rongpeng Li, Lizhu
Fang, Qinqin Pu, Ping Lin, Austin Hoggarth, Huang Huang, Xuefeng Li & Min Wu * Key Laboratory of Biotechnology for Medicinal Plants of Jiangsu Province, Jiangsu Normal University,
Xuzhou, Jiangsu 221116, P.R., China Rongpeng Li * State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, and Collaborative Innovation Center for
Biotherapy, Chengdu, China Qinqin Pu & Min Wu * Institute of Human Virology, Sun Yat-sen University, Guangzhou, China Xuefeng Li * Inflammation and Allergic Disease Research Unit, First
Affiliated Hospital of Southwest Medical University, Luzhou, China Guoping Li Authors * Rongpeng Li View author publications You can also search for this author inPubMed Google Scholar *
Lizhu Fang View author publications You can also search for this author inPubMed Google Scholar * Qinqin Pu View author publications You can also search for this author inPubMed Google
Scholar * Ping Lin View author publications You can also search for this author inPubMed Google Scholar * Austin Hoggarth View author publications You can also search for this author
inPubMed Google Scholar * Huang Huang View author publications You can also search for this author inPubMed Google Scholar * Xuefeng Li View author publications You can also search for this
author inPubMed Google Scholar * Guoping Li View author publications You can also search for this author inPubMed Google Scholar * Min Wu View author publications You can also search for
this author inPubMed Google Scholar CONTRIBUTIONS RL and MW designed research and wrote the manuscript. RL, LF, QP, CZ, PL and HH performed experiments. CORRESPONDING AUTHORS Correspondence
to Guoping Li or Min Wu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Supplementary Information accompanies the paper on the
_Signal Transduction and Targeted Therapy_ website SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION (DOC 206 KB) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons
Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the
credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of
this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Li, R., Fang, L., Pu, Q. _et al._ Lyn prevents aberrant
inflammatory responses to _Pseudomonas_ infection in mammalian systems by repressing a SHIP-1-associated signaling cluster. _Sig Transduct Target Ther_ 1, 16032 (2016).
https://doi.org/10.1038/sigtrans.2016.32 Download citation * Received: 10 October 2016 * Revised: 18 November 2016 * Accepted: 22 November 2016 * Published: 16 December 2016 * DOI:
https://doi.org/10.1038/sigtrans.2016.32 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative










