- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
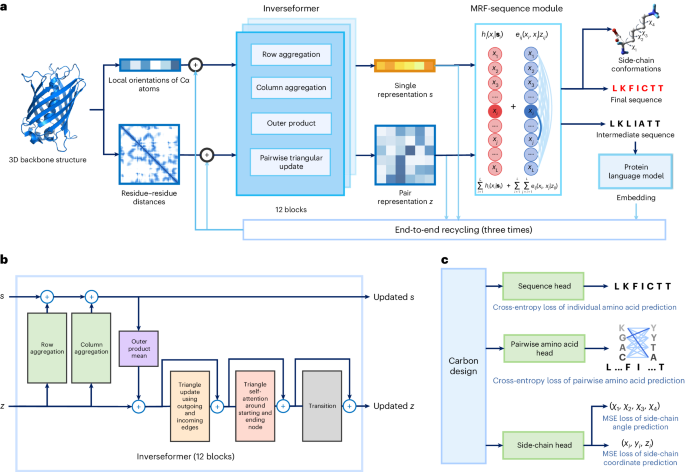
ABSTRACT Protein sequence design is critically important for protein engineering. Despite recent advancements in deep learning-based methods, achieving accurate and robust sequence design
remains a challenge. Here we present CarbonDesign, an approach that draws inspiration from successful ingredients of AlphaFold and which has been developed specifically for protein sequence
design. At its core, CarbonDesign introduces Inverseformer, which learns representations from backbone structures and an amortized Markov random fields model for sequence decoding. Moreover,
we incorporate other essential AlphaFold concepts into CarbonDesign: an end-to-end network recycling technique to leverage evolutionary constraints from protein language models and a
multitask learning technique for generating side-chain structures alongside designed sequences. CarbonDesign outperforms other methods on independent test sets including the 15th Critical
Assessment of protein Structure Prediction (CASP15) dataset, the Continuous Automated Model Evaluation (CAMEO) dataset and de novo proteins from RFDiffusion. Furthermore, it supports
zero-shot prediction of the functional effects of sequence variants, making it a promising tool for applications in bioengineering. Access through your institution Buy or subscribe This is a
preview of subscription content, access via your institution ACCESS OPTIONS Access through your institution Access Nature and 54 other Nature Portfolio journals Get Nature+, our best-value
online-access subscription $29.99 / 30 days cancel any time Learn more Subscribe to this journal Receive 12 digital issues and online access to articles $119.00 per year only $9.92 per issue
Learn more Buy this article * Purchase on SpringerLink * Instant access to full article PDF Buy now Prices may be subject to local taxes which are calculated during checkout ADDITIONAL
ACCESS OPTIONS: * Log in * Learn about institutional subscriptions * Read our FAQs * Contact customer support SIMILAR CONTENT BEING VIEWED BY OTHERS SPARKS OF FUNCTION BY DE NOVO PROTEIN
DESIGN Article 15 February 2024 COMPUTATIONAL PROTEIN DESIGN Article 27 February 2025 PROTEIN SEQUENCE DESIGN WITH A LEARNED POTENTIAL Article Open access 08 February 2022 DATA AVAILABILITY
The training data were obtained from the PDB website (http://www.rcsb.org/). The testing sets were acquired from CASP15 (https://predictioncenter.org/casp15/) and CAMEO
(https://www.cameo3d.org). Other datasets supporting the findings of this study are available in the paper and the Supplementary Information. Source data are provided with this paper. CODE
AVAILABILITY The CarbonDesign software is available on both GitHub (https://github.com/zhanghaicang/carbonmatrix_public) and Code Ocean (https://codeocean.com/capsule/5915382/tree)59.
REFERENCES * Cao, L. et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. _Science_ 370, 426–431 (2020). Article Google Scholar * Bryan, C. M. et al. Computational design
of a synthetic PD-1 agonist. _Proc. Natl Acad. Sci. USA_ 118, 2102164118 (2021). Article Google Scholar * Yeh, A. H.-W. et al. De novo design of luciferases using deep learning. _Nature_
614, 774–780 (2023). Article Google Scholar * Dou, J. et al. De novo design of a fluorescence-activating beta-barrel. _Nature_ 561, 485–491 (2018). Article Google Scholar * Vorobieva, A.
A. et al. De novo design of transmembrane beta barrels. _Science_ 371, 8182 (2021). Article Google Scholar * Kuhlman, B. et al. Design of a novel globular protein fold with atomic-level
accuracy. _Science_ 302, 1364–1368 (2003). Article Google Scholar * Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. _Nature_
https://doi.org/10.1038/s41586-023-06415-8 (2023). * Yim, J. et al. SE(3) diffusion model with application to protein backbone generation. In _Proc. of the 40th International Conference on
Machine Learning_ (eds Krause, A. et al.) 40001–40039 (PMLR, 2023). * Ingraham, J. et al. Illuminating protein space with a programmable generative model. _Nature_ 623, 1070–1078 (2023).
Article Google Scholar * Dauparas, J. et al. Robust deep learning-based protein sequence design using ProteinMPNN. _Science_ 378, 49–56 (2022). Article Google Scholar * Hsu, C. et al.
Learning inverse folding from millions of predicted structures. In _Proc. of the 39th International Conference on Machine Learning_ (eds Chaudhuri, K. et al.) 8946–8970 (PMLR, 2022). *
Anand, N. et al. Protein sequence design with a learned potential. _Nat. Commun._ 13, 746 (2022). Article Google Scholar * Liu, Y. et al. Rotamer-free protein sequence design based on deep
learning and self-consistency. _Nat. Comput. Sci._ 2, 451–462 (2022). Article Google Scholar * Huang, B. et al. Accurate and efficient protein sequence design through learning concise
local environment of residues. _Bioinformatics_ 39, 122 (2023). Article Google Scholar * Ingraham, J. et al. Generative models for graph-based protein design. In _Proc. of Advances in
Neural Information Processing Systems_ (eds Wallach, H. et al) 15820–15831 (NeurlPS, 2019). * Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. _Nature_ 596,
583–589 (2021). Article Google Scholar * Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. _Science_ 373, 871–876 (2021).
Article Google Scholar * Carreira, J. et al. Human pose estimation with iterative error feedback. In _Proc. of the IEEE Conference on Computer Vision and Pattern Recognition_ (eds Bajcsy,
R. et al.) 4733–4742 (IEEE, 2016). * Tu, Z. & Bai, X. Auto-context and its application to high-level vision tasks and 3D brain image segmentation. _IEEE Trans. Pattern Anal. Mach.
Intell._ 32, 1744–1757 (2010). Article Google Scholar * Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. _Science_ 379, 1123–1130
(2023). Article MathSciNet Google Scholar * Robin, X. et al. Continuous Automated Model EvaluatiOn (CAMEO)—perspectives on the future of fully automated evaluation of structure prediction
methods. _Proteins_ 89, 1977–1986 (2021). Article Google Scholar * _CASP15. Critical Assessment of Techniques for Protien Structure Prediction, 15th Round. Abstract Book_ (Protein
Structure Prediction Center, 2022); https://predictioncenter.org/casp15/doc/CASP15_Abstracts.pdf * Pearl, J. _Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference_
(Morgan Kaufmann, 1988). * Wainwright, M. J. & Jordan, M. I. Graphical models, exponential families, and variational inference. _Found. Trends Mach. Learn._ 1, 1–305 (2008). Article
Google Scholar * Zhang, H. et al. Predicting protein inter-residue contacts using composite likelihood maximization and deep learning. _BMC Bioinform._ 20, 537 (2019). Article Google
Scholar * Ekeberg, M., Lövkvist, C., Lan, Y., Weigt, M. & Aurell, E. Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models. _Phys. Rev. E_ 87, 012707
(2013). Article Google Scholar * Morcos, F. et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. _Proc. Natl Acad. Sci. USA_ 108,
1293–1301 (2011). Article Google Scholar * Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. _J. Chem. Theory Comput._ 13, 3031–3048 (2017).
Article Google Scholar * Henikoff, S. & Henikoff, J. G. Amino acid substitution matrices from protein blocks. _Proc. Natl Acad. Sci. USA_ 89, 10915–10919 (1992). Article Google
Scholar * Wang, W., Peng, Z. & Yang, J. Single-sequence protein structure prediction using supervised transformer protein language models. _Nat. Comput. Sci._ 2, 804–814 (2022). Article
Google Scholar * Chowdhury, R. et al. Single-sequence protein structure prediction using a language model and deep learning. _Nat. Biotechnol._ 40, 1617–1623 (2022). Article Google
Scholar * Sakuma, K., Koike, R. & Ota, M. Dual-wield NTPases: a novel protein family mined from AlphaFold DB. _Protein Science._ 33, e4934 (2024). Article Google Scholar * Varadi, M.
et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. _Nucleic Acids Res._ 50, 439–444 (2022). Article
Google Scholar * Yang, K. K., Wu, Z. & Arnold, F. H. Machine-learning-guided directed evolution for protein engineering. _Nat. Methods_ 16, 687–694 (2019). Article Google Scholar *
Shin, J.-E. et al. Protein design and variant prediction using autoregressive generative models. _Nat. Commun._ 12, 2403 (2021). Article Google Scholar * Lek, M. et al. Analysis of
protein-coding genetic variation in 60,706 humans. _Nature_ 536, 285–291 (2016). Article Google Scholar * Frazer, J. et al. Disease variant prediction with deep generative models of
evolutionary data. _Nature_ 599, 91–95 (2021). Article Google Scholar * Meier, J. et al. Language models enable zero-shot prediction of the effects of mutations on protein function. In
_Proc. of Advances in Neural Information Processing Systems_ (eds Ranzato, M. et al.) 29287–29303 (NeurlPS, 2021). * Notin, P. et al. Tranception: protein fitness prediction with
autoregressive transformers and inference-time retrieval. In _Proc. of the 39th International Conference on Machine Learning_ (eds Chaudhuri, K. et al.) 16990–17017 (PMLR, 2022). * Rao, R.
M. et al. MSA transformer. In _Proc. of the 38th International Conference on Machine Learning_ (eds Meila, M and Zhang, T.) 8844–8856 (PMLR, 2021). * Findlay, G. M. et al. Accurate
classification of BRCA1 variants with saturation genome editing. _Nature_ 562, 217–222 (2018). Article Google Scholar * Kotler, E. et al. A systematic p53 mutation library links
differential functional impact to cancer mutation pattern and evolutionary conservation. _Mol. Cell_ 71, 178–1908 (2018). Article Google Scholar * Mighell, T. L., Evans-Dutson, S. &
O’Roak, B. J. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. _Am. J. Hum. Genet._ 102, 943–955 (2018). Article
Google Scholar * Jia, X. et al. Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. _Am. J. Hum. Genet._ 108, 163–175 (2021). Article Google
Scholar * Pan, X. et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. _Science_ 362, 2486 (2018). Article Google Scholar * Hennig, M., Darimont, B.,
Sterner, R., Kirschner, K. & Jansonius, J. N. 2.0 Å structure of indole-3-glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: possible determinants of protein
stability. _Structure_ 3, 1295–1306 (1995). Article Google Scholar * Banerjee, S. et al. Protonation state of an important histidine from high resolution structures of lytic polysaccharide
monooxygenases. _Biomolecules_ https://doi.org/10.3390/biom12020194 (2022). * Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. _Nature_ 620, 1089–1100
(2023). Article Google Scholar * Leman, J. K. et al. Macromolecular modeling and design in rosetta: recent methods and frameworks. _Nat. Methods_ 17, 665–680 (2020). Article MathSciNet
Google Scholar * Madani, A. et al. Large language models generate functional protein sequences across diverse families. _Nat. Biotechnol._ https://doi.org/10.1038/s41587-022-01618-2 (2023).
* Hie, B. L. et al. Efficient evolution of human antibodies from general protein language models. _Nat. Biotechnol._ https://doi.org/10.1038/s41587-023-01763-2 (2023). * Suzek, B. E. et al.
UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. _Bioinformatics_ 31, 926–932 (2015). Article Google Scholar * Mitchell, A. L. et al.
MGnify: the microbiome analysis resource in 2020. _Nucleic Acids Res._ 48, 570–578 (2020). Google Scholar * Mirdita, M. et al. Uniclust databases of clustered and deeply annotated protein
sequences and alignments. _Nucleic Acids Res._ 45, 170–176 (2017). Article Google Scholar * Remmert, M., Biegert, A., Hauser, A. & Söding, J. HHblits: lightning-fast iterative protein
sequence searching by HMM-HMM alignment. _Nat. Methods_ 9, 173–175 (2012). Article Google Scholar * Johnson, L. S., Eddy, S. R. & Portugaly, E. Hidden Markov model speed heuristic and
iterative HMM search procedure. _BMC Bioinform._ 11, 431 (2010). Article Google Scholar * Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. In _Proc. of the
International Conference on Learning Representations_ (eds Bengio, Y. et al.) 210–219, (ICLR 2015). * Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library.
In _Proc. of Advances in Neural Information Processing Systems_ (eds Wallach, H. et al.) 8024–8035 (NeurlPS, 2019). * Ren, M., Yu, C., Bu, D. & Zhang, H. Accurate and robust protein
sequence design with Carbondesign. _Code Ocean_ https://doi.org/10.24433/CO.5915382.v2 (2024). Download references ACKNOWLEDGEMENTS We acknowledge the financial support from the National
Natural Science Foundation of China (grant no. 32370657) and the Project of Youth Innovation Promotion Association CAS to H.Z. We also acknowledge the financial support from the Development
Program of China (grant no. 2020YFA0907000) and the National Natural Science Foundation of China (grant nos. 32271297 and 62072435). We thank Beijing Paratera Co., Ltd and the ICT
Computing-X Center, Chinese Academy of Sciences, for providing computational resources. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * SKLP, Institute of Computing Technology, Chinese Academy
of Sciences, Beijing, China Milong Ren, Chungong Yu, Dongbo Bu & Haicang Zhang * University of Chinese Academy of Sciences, Beijing, China Milong Ren, Chungong Yu, Dongbo Bu &
Haicang Zhang * Central China Institute of Artificial Intelligence, Zhengzhou, China Chungong Yu, Dongbo Bu & Haicang Zhang Authors * Milong Ren View author publications You can also
search for this author inPubMed Google Scholar * Chungong Yu View author publications You can also search for this author inPubMed Google Scholar * Dongbo Bu View author publications You can
also search for this author inPubMed Google Scholar * Haicang Zhang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS H.Z. conceived the
ideas and implemented the CarbonDesign model and algorithms. H.Z. and M.R. designed the experiments, and M.R. conducted the main experiments and analysis. M.R. wrote the manuscript. H.Z.,
D.B. and C.Y. revised the manuscript. CORRESPONDING AUTHORS Correspondence to Dongbo Bu or Haicang Zhang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests.
PEER REVIEW PEER REVIEW INFORMATION _Nature Machine Intelligence_ thanks Haiyan Liu and Dong Xu for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S
NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary
Notes 1–4, Figs. 1–6 and Tables 1–17. REPORTING SUMMARY SUPPLEMENTARY DATA 1 Statistical Source Data for Supplementary Fig. 3. SUPPLEMENTARY DATA 2 Statistical Source Data for Supplementary
Fig. 4. SUPPLEMENTARY DATA 3 Statistical Source Data for Supplementary Fig. 5. SUPPLEMENTARY DATA 4 Statistical Source Data for Supplementary Fig. 6. SOURCE DATA SOURCE DATA FIG. 2
Statistical Source Data for Fig. 2. SOURCE DATA FIG. 3 Statistical Source Data for Fig. 3. SOURCE DATA FIG. 4 Statistical Source Data for Fig. 4. SOURCE DATA FIG. 5 Statistical Source Data
for Fig. 5. RIGHTS AND PERMISSIONS Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or
other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ren, M., Yu, C., Bu, D. _et al._ Accurate and robust protein sequence design with CarbonDesign. _Nat Mach Intell_ 6, 536–547 (2024).
https://doi.org/10.1038/s42256-024-00838-2 Download citation * Received: 10 August 2023 * Accepted: 10 April 2024 * Published: 23 May 2024 * Issue Date: May 2024 * DOI:
https://doi.org/10.1038/s42256-024-00838-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative









