- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT RNA methyltransferases (MTases) have recently become increasingly important in drug discovery. Yet, most frequently utilized RNA MTase assays are limited in their throughput and
hamper this rapidly evolving field of medicinal chemistry. This study developed a microscale thermophoresis (MST)-based split aptamer assay for enzymatic MTase investigations, improving
current methodologies by offering a non-proprietary, cost-effective, and highly sensitive approach. Our findings demonstrate the assay’s effectiveness across different RNA MTases, including
inhibitor characterization of METTL3/14, DNMT2, NSUN2, and _S. aureus_ TrmD, enabling future drug discovery efforts. Using this concept, a pilot screening on the cancer drug target DNMT2
discovered several hit compounds with micromolar potency. SIMILAR CONTENT BEING VIEWED BY OTHERS TARGETED SYSTEMATIC EVOLUTION OF AN RNA PLATFORM NEUTRALIZING DNMT1 FUNCTION AND CONTROLLING
DNA METHYLATION Article Open access 06 January 2023 TIME-RESOLVED STRUCTURAL ANALYSIS OF AN RNA-CLEAVING DNA CATALYST Article 23 December 2021 POTENT AND SELECTIVE SETDB1 COVALENT NEGATIVE
ALLOSTERIC MODULATOR REDUCES METHYLTRANSFERASE ACTIVITY IN CELLS Article Open access 24 February 2025 INTRODUCTION RNA has recently emerged in modern drug discovery, serving not only as a
drug itself but also as a target for drugs, and notably, as a central component in cellular pathways linked to malignancies and infections1,2. Methylation of RNA, a metabolic process
observed across all organisms, has been validated for its association with numerous diseases3. In this regard, the introduction of STC-15, the first-in-class RNA methyltransferase (MTase)
inhibitor targeting METTL3/14, into clinical trials, marks a significant paradigm shift4. Thus, detecting and quantifying RNA modifications, including nucleobase methylation, has become a
focal point of research due to their physiological importance and implication in diseases. The current gold-standard bioanalytical techniques encompass mass spectrometry, radiometry, and
sequencing methods5. Yet, the detection of _S_-adenosylhomocysteine (SAH), a co-product of the MTase reactions, offers a simpler and universal assay methodology6. Enzymatic scintillation
assays using 3H-SAM and the LC/MS-based detection of the MTase reaction co-product SAH are state-of-the-art for screening drug candidates on RNA MTases such as METTL3/147, DNMT28, NSUN29,
and anti-infective targets like _H. influenzae_ TrmD or SARS-CoV2 nsp10/1610,11. Despite these advancements, existing RNA MTase assays face limitations in throughput, impeding the rapid
progress of medicinal chemistry in this domain. Efforts to overcome these limitations include the development of antibodies capable of discriminating between SAH and _S_-adenosylmethionine
(SAM)12. However, these antibodies lack the necessary affinity and specificity for robust SAH detection at low concentrations. Indirect SAH detection via enzyme-coupled assays is another
approach, yet none possess the requisite sensitivity for low nanomolar range detection13,14,15,16. Methyltransferases present numerous challenges in enzymatic assay development due to their
slow kinetic nature and substrate affinity in the sub-micromolar range17. This necessitates highly sensitive enzymatic assay methods, requiring the detection of SAH at very low
concentrations (<50 nM). Very recently, Pham et al. used an SAH-binding RNA aptamer to develop a time-resolved fluorescence energy transfer (TR-FRET)-based SAH sensor which is now sold
commercially as “AptaFluor™ SAH Methyltransferase Assay Kit”18. While this approach is superior to previous assay concepts in terms of sensitivity and robustness, it is marketed proprietary
(i.e., does not reveal the aptamer RNA sequences) and relatively high cost per treatment condition ($5/single assay reaction) still hampering high- or even medium-throughput assay campaigns
in academic settings. In this study, we followed a similar approach to develop an aptamer-based RNA MTase assay, aiming to address the major drawbacks of previous methods. We extend the
utility of the assay beyond TR-FRET readers to conventional FRET-compatible plate readers or microscale thermophoresis (MST) instruments. Hence, the development of an MST protocol for RNA
methylation quantification offers practical advantages over earlier technologies, potentially enabling RNA MTase drug discovery campaigns previously deemed unfeasible. Our protocol is
non-proprietary, revealing all necessary RNA aptamer sequences for independent assay setup and protocol modification at a moderate expense (<$0.05/single assay reaction). RESULTS AND
DISCUSSION DESIGN AND CALIBRATION OF A FRET- AND MST-CAPABLE SAH-SPLIT APTAMER To engineer a SAH-binding aptamer for RNA MTase enzyme assay development, we chose a naturally occurring
68-nucleotide RNA aptamer identified previously upstream of the metH gene in _D. aromatica_ that is part of a riboswitch, controlling metH expression depending on the SAH abundance in
bacteria19. RNA aptamers are highly structured polynucleotides that form selective binding pockets for specific ligands20. While the first aptamers were developed by in vitro selection
approaches, structures with similar properties were later discovered in nature, most often in bacteria, engineered aptamers can be developed to exhibit high affinity and selectivity for
their targets, and can have considerable potential for applications in biotechnology21. This metH aptamer represents the shortest consensus sequence within a group of sequences from related
organisms, as it lacks an optional structural element termed P3 stem. The apparent dissociation constant _K_D of this metH aptamer for SAH was determined by isothermal titration calorimetry
to be around 20 nM with at least 100-fold selectivity over other nucleosides (including SAM) highlighting the suitability for our assay development campaign22. In our study, we followed the
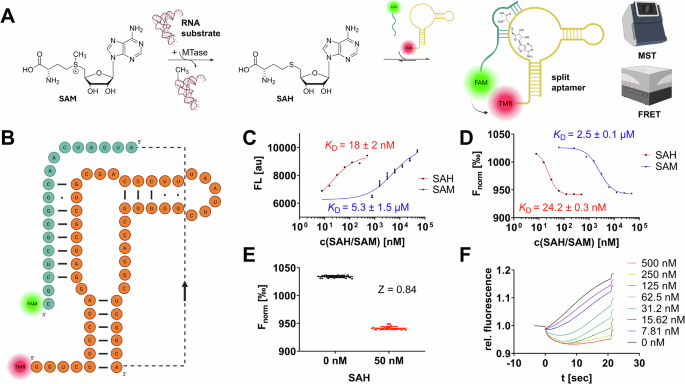
aim of developing a fluorescent split aptamer SAH biosensor (Fig. 1A) from the natural non-split metH RNA aptamer. The concept of fluorescent split aptamers relies on a series of two or more
independent fluorescently labeled aptamer oligonucleotides, able to assemble only in the presence of a specific target ligand. In a FRET-based approach, two fragments carry a FRET-pair of
communicating fluorescent dyes, such that, upon excitation of the donor dye, the acceptor dye will only emit fluorescence if the dyes are in spatial proximity in the ligand-bound complex,
thus indicating the presence of the analyte, i.e., SAH23. We hypothesized a suitable site for splitting the metH aptamer at adenine-50 from previously reported in-line probing and
crystallography experiments on SAH riboswitches19,22, hence, we will describe this specific design and sequences. Accordingly, we evaluated two split aptamer fragments labeled with
fluorescein (FAM) and tetramethylrhodamin (TAMRA) as a FRET-capable dye pair: aptaSAH1 and aptaSAH2 (Fig. 1B). To establish our experimental setup, we mixed both aptamer fragment
oligonucleotides at equimolar concentrations (final: 50 nM) and titrated series of SAH resp. SAM dilutions to this prototypical SAH biosensor. FRET experiments (λex = 485 nm; λem = 600 nm)
in 96-well plates yielded concentration-dependent fluorescence read-outs with apparent _K_D values for SAH and SAM of 18 nM and 5.3 µM in agreement with the native full-length metH aptamer
affinities (Fig. 1C)19. Hence, this 300-fold SAH over SAM selectivity and nanomolar sensitivity provides the necessary framework for the development of an enzymatic methyltransferase assay.
Yet, we tested the same experimental setup with an MST-based acquisition (Fig. 1D, F) which revealed comparable _K_D values (24 nM & 2.5 µM) but significantly improved reproducibility
and signal-to-noise properties (S/NMST = 41 dB vs. S/NFRET = 33 dB). Furthermore, MST assays require only 10 µL of sample volume per capillary compared to 30 µL per well in a 96-well plate
supporting high-throughput applications in material consumption. To assess the suitability for sensing SAH in a steady-state enzymatic assay, we examined statistical Z-values at 50 nM SAH
(which correspond to 5% SAM-to-SAH substrate conversion from 1 µM SAM substrate in a common RNA MTase reaction). By this, we determined an MST-based Z-value of 0.84 (FRET assay: _Z_ = 0.57)
highlighting the suitability of this framework for high-throughput screening (HTS) assay setups (Fig. 1E). For these reasons, we prefer the usage of the MST-based assay which the following
chapter will focus on, but it is noteworthy that plate reader-based FRET assays were also found equally functional. Furthermore, we performed comparative assays with the commercial AptaFluor
assay kit, which gave similar results to our assay in all respects (SI Fig. 2): Z-factor at 50 nM SAH = 0.84; SAH resp. SAM affinity _(K_D = 26 nM & 5.7 µM). MST-BASED ASSAYS FOR THE
CHARACTERIZATION OF RNA MTASE ACTIVITY AND FUNCTIONAL INHIBITORY AND ENZYMATIC PARAMETERS In this manuscript, we aimed to validate a split aptamer assay setup by reproducing functional
enzyme assay results using enzyme kinetics of RNA MTases and their known inhibitors. The enzyme kinetics were characterized by calculating the Michaelis-Menten constant (_K_M or _k__cat_)
and inhibitory constants (IC50 resp. _K_I). First, we focused on the characterization of human DNMT2, a well-characterized model of m5C methyltransferase, using its known enzyme kinetics and
inhibitors24. To assess DNMT2 activity, we incubated 250 nM DNMT2 with 5 µM tRNA, and 0.9 µM SAM for variable durations (0–120 min) and probed with the split aptamer and MST-based
evaluation for the SAH content formed during the reaction (Fig. 2A). The production of total SAH (in nM) could be calculated from the respective Fnorm values (Fig. 2B) and regression to the
calibration curve in Fig. 1D, indicating the enzymatic reaction reaches a plateau after 30 min when approximately 10% SAM consumption was reached (112 nM SAH from 900 nM SAM), consistent
with its intrinsic product inhibition reported in the literature25. The initial reaction velocity (_v__init_) was determined to be 0.104 nM s−1, and the turnover number (_k__cat_) was
calculated as 0.025 min−1. To evaluate the effect of inhibitors on DNMT2 activity, we used two known inhibitors, SHO108 and SH112. These SAH derivatives were selected based on their
documented efficacy for DNMT28,26. Before setting up an enzyme assay experiment, both inhibitors (without enzyme or RNA substrate) were titrated up to 100 µM to the split aptamer revealing
only minimal MST shifts compared to the SAH-positive control. This indicated that, although these inhibitors share structural features with the aptamer ligand SAH, the split aptamer provides
sufficient selectivity for SAH over such derivates to allow inhibition experiments with the present assay setup (Fig. 2C). Additionally, the natural _D. aromatica_ (non-split) SAH aptamer
has been tested for binding of 16 SAH-like derivatives in previous studies, showing excellent selectivity for SAH, as all derivatives exhibited binding affinities at least 3 orders of
magnitude weaker19. We followed this strategy, using our split aptamer construct, we tested 10 additional SAH derivatives at 10 µM among which only the SAH-mimetic and pan-MTase inhibitor
sinefungin showed a slight MST shift in the presence of the split aptamer, highlighting the excellent selectivity for SAH (SI Fig. 3). To further validate our findings on DNMT2, we compared
the split aptamer inhibition assay results of both inhibitors with those obtained from a previously published orthogonal assay method, the radiometric 3H incorporation assay8,26.
Dose-response experiments were conducted to measure inhibitory constants, demonstrating high reproducibility between the two methods (Fig. 2D–I). In summary, both SHO108 and SH112 inhibitors
showed dose-dependent inhibition of DNMT2 activity with comparable inhibitory constants between orthogonal methods, demonstrating that the split aptamer assay could reproduce previously
published results while circumventing the use of radioactivity. Using an additional non-enzymatic MTase assay technique, we also investigated the two DNMT2 inhibitors utilizing an orthogonal
MST method based on a fluorescent tracer displacement assay (SI Fig. 1)27,28. SHO108 and SH112 showed _K_D values of 4.7 and 7.8 µM, respectively, in good agreement with the inhibition data
presented in Fig. 2. These results validate the split aptamer assay setup for studying DNMT2 activity and inhibitor efficacy. Subsequently, we extended our investigation to include other
RNA MTases of clinical relevance, employing diverse inhibitor scaffolds as reported in the literature. Specifically, we studied the m6A MTase METTL3/14 and the m7G MTase _S. aureus_ TrmD.
For METTL3/14 (10 nM MTase, 500 nM SAM, 120 nM RNA substrate), we could successfully reproduce a reported _K__M_ value of 106 nM (Fig. 3A, B), demonstrating the assay’s capability in this
context29. To further illustrate the assay’s utility for drug discovery, we also performed inhibition experiments with the known inhibitor STM2457, yielding an apparent inhibition constant
_K_I of 5.1 nM (Fig. 3C). This result is consistent with both an orthogonal METTL3/14 assay utilizing an anti-m6A-antibody and the mass-spectrometric characterization reported in the
literature7,29, which confirms that our assay is also suitable for characterizing high-affinity inhibitors with low nanomolar potency. Additionally, we investigated the bacterial m7G MTase
_S. aureus_ TrmD, for which anti-infective inhibitors have been developed10. Time-course experiments with 50 nM TrmD, 2 µM SAM, and 1 µM tRNALeu indicated that our assay is also appropriate
for studying MTases with slow kinetics. TrmD exhibited a catalytic efficiency approximately one order of magnitude lower than DNMT2 and METTL3/14, with _v__init_ and _k__cat_ values of 4.16
nM min–1 and 0.083 min–1, respectively (Fig. 3D, E). Notably, the SAH production of TrmD does not reach saturation after 120 min. Furthermore, we could validate the assay’s applicability for
drug discovery by confirming a known TrmD inhibitor with an IC50 of 8.45 µM, closely matching the reported value of 11 µM (Fig. 3F)10. Moreover, we intended to use the split aptamer MST
assay for a stability assessment study of the m5C MTase NSUN2 in order to develop conditions suitable for stabilizing NSUN2 for several days to perform drug discovery screening applications
in the future30. To evaluate NSUN2 activity, we incubated 200 nM NSUN2 with 250 ng/µL tRNA and 1 µM SAM for varying durations (0–60 min) and measured the SAH content formed during the
reaction using the split aptamer and MST-based evaluation (Fig. 4A, B). NSUN2, a Rossmann-fold MTase, is prone to thermal unfolding31. In the storage buffer (25 mM HEPES pH 7.5, 300 mM NaCl,
10% glycerol, 0.04% Triton-X100, 0.5 mM TCEP) at 37 °C, we found NSUN2 has a catalytic t1/2 of ~4 h highlighting its intrinsic instability. We aimed to establish buffer and temperature
conditions that allow the maintenance of catalytic competency for at least 14 days. Because the literature suggested potential benefits from the addition of BSA on protein stability, we
conducted a series of incubation experiments at 4 °C and 20 °C in storage buffer, with and without 0.5% BSA (w/w) (Fig. 4C, D)32. After the designated incubation period, we initiated
enzymatic reactions with the appropriate tRNA substrate and SAM. Our findings revealed that, in contrast to the aforementioned expectations, only the buffer without BSA at 4 °C maintained
NSUN2’s catalytic competence for over 14 days (green curve, Fig. 4D). Finally, we utilized the newly identified MST assay for a pilot drug screening on the potential cancer drug target
DNMT2, which introduces the m5C modifications to various tRNAs24. Using an enzymatic tRNA methyltransferase assay screening setup, we have tested a commercial epigenetic target-focused
library including 160 drug-like compounds (Tocris) and an in-house covalent cysteine-focused warhead screening library (80 compounds) to identify novel DNMT2 binding scaffolds suitable for
subsequent lead optimization (Figs. 5, 6). The drug candidate library was added to a master mix of recombinantly expressed DNMT2 protein (250 nM) with 5 µM tRNA and 0.9 µM SAM. The final
drug compound concentration was adjusted to 500 µM for the epigenetic drug library (Fig. 5) resp. 100 µM for the covalent compound library (Fig. 6), and the enzymatic reaction was incubated
for 30 min at 25 °C. Subsequently, enzymatic reactions were terminated by the addition of 0.3% SDS, and the product SAH was quantified by the split aptamer MST assay (Figs. 5A, B, 6A).
Compounds that displayed Fnorm values > 1000‰ were considered primary screening hits (equals >80% inhibition). By this, we identified ten potential non-covalent hit compounds (Fig.
5G). Among those hits, alexidine (CPD 3) was found to be the most promising hit as this was the only compound that showed inhibition at concentrations <500 µM and was able to compete with
the fluorescent SAH-analog (5-FAM-triazolyl-adenosyl-diaminobutyric acid; FTAD) tracer during fluorescence polarization (FP) displacement experiments (Fig. 5C, E; _K_D = 364 µM)28. Of note,
the other nine compounds (Fig. 5G) were not able to displace the FTAD tracer during FP experiments, nor did they inhibit DNMT2 at decreased compound concentrations (200 µM) using the
enzymatic aptamer assay. We speculated these hits might bind very weakly to the tRNA-DNMT2 interaction site and by this inhibit DNMT2 activity. To test this hypothesis, we performed an
electrophoretic mobility shift assay (EMSA) using tRNAAsp (5 µM) and DNMT2 (10 µM) compared to the tRNA sample in absence of the DNMT2 protein (SI Fig. 4)33. Using this assay, the
heterodimeric DNMT2-tRNA complex resulted in a significant electrophoretic shift compared to the native tRNA sample. Subsequently, we treated the DNMT2-tRNA complex with all hit compounds at
500 µM and observed their potential to disrupt the complex. Interestingly, six out of ten compounds showed the potential to reverse the complex formation (namely CPDS 1, 2, 5, 6, 8, and 9)
providing first-in-class (yet low affinity) starting points for DNMT2-tRNA interaction inhibitors. Of note, CPD 3 (alexidine) did not influence DNMT2-tRNA complex formation indicating its
selective binding to the SAH binding pocket, and also, CPDS 4, 7, and 10 were found unable to disrupt the protein-tRNA complex, rendering these three compounds as DNMT2 inhibitors with an
unknown mode of action. Next, we investigated the target selectivity of all hit compounds by inhibition experiments using the MTases characterized in this study (DNMT2, METTL3/14, TrmD, and
NSUN2) as a selectivity panel. Of note, none of the identified hit compounds inhibited any other RNA MTases at ligand concentrations as high as 500 µM (Fig. 5D), rendering these compounds
DNMT2 selective hits. While CPD3 (alexidine) could be identified as a DNMT2 inhibitor during MST- and FP- experiments, the ligand’s hydrophobic nature raised the concern this compound might
be an aggregator of DNMT2. To exclude this possibility, we investigated the reversibility of alexidine binding using the FP displacement assay and separation of the alexidine ligand by
small-scale size-exclusion chromatography (SEC)34. Here, we found that the decrease in FP (92 to 5 mP) by alexidine displacing the fluorescent tracer FTAD is almost quantitatively reversed
after SEC removal of alexidine (Fig. 5F). Furthermore, the restored FP levels can be displaced again by alexidine, and hence, this compound is a fully reversible binder resp. inhibitor of
DNMT2 SAH pocket. Lastly, we used the DNMT2 aptamer assay for a pilot screening of a cysteine-focussed covalent inhibitor library, since DNMT2 Cys-79 was previously identified as a valid
target for covalent inhibitor development26. We screened this in-house library (_N_ = 80 compounds) analogously to the non-covalent compound library, but the final compound concentration was
adjusted to 100 µM. By this, we identified one single hit compound CPD 11 (adamantanyl-acryloylurea) which inhibited DNMT2 > 90% at 100 µM (Fig. 6A), displaced the FP-probe FTAD
effectively (Fig. 6B), and did not inhibit any of the other MTases (Fig. 5D). Dose-response experiments revealed a single-digit micromolar inhibition constant (Fig. 6C; IC50 = 4.1 µM) and
time-dependent displacement of the FTAD FP-tracer implying irreversible covalent inactivation of DNMT2 (Fig. 6D, E). To summarize this study, we successfully developed an MST-based split
aptamer assay for detecting RNA MTase activity, offering improved sensitivity and cost-effectiveness over existing methods. The assay was validated across multiple MTases, demonstrating high
reproducibility and potential for drug discovery and high-throughput screenings. METHODS RNA APTAMER The following two RNA aptamer sequences were synthesized and HPLC-purified by GenScript
on a 500 nmol and yielded 50 nmol of the final split aptamer oligonucleotides (total: $759; sufficient for 100k individual experiments in 10 µL MST capillaries; <$0.001/per assay
reaction). aptaSAH1: TAMRA-5′-CUGCCGAGGAGCGCUGCGACCCUUUAAUUCGGGGGCCAGGCUCGGCAAUGAUGCC-3′ aptaSAH2: 5′-AUGAUCAACGGCGCUCGC-3′-FAM MICROSCALE THERMOPHORESIS MTASE ASSAY MTase enzyme reaction
mixtures were prepared with buffer and substrate conditions as previously described8,10,29. Hence, the following enzyme-specific assay buffers and methylation conditions were used: DNMT2
& _S. aureus_ TrmD: 100 mM Tris-HCl, pH 8, 100 mM NH4OAc, 0.1 mM EDTA, 10 mM MgCl2, 10 mM DTT. For TrmD: 50 nM TrmD, 2 µM SAM, 1 µM tRNALeu. For DNMT2: 250 nM DNMT2, 900 nM SAM, 5 µM
tRNA METTL3/14: 20 mM Tris-HCl, pH 7.5, 20 mM KCl, 0.25 mM MgCl2, 1 mM DTT, 0.01% Triton-X100. 10 nM METTL3/14, 500 nM SAM, 120 nM RNA substrate. NSUN2: 50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 5
mM MgCl2, 1.5 mM DTT. 200 nM NSUN2, 1 µM SAM, 250 ng/µL tRNA (total from _E. coli_). CAVE: It is important to use high-quality SAM for assay preparations since frequent contamination in
commercial SAM is in fact SAH which shall be detected by this assay, and thus, can distort the final assay results. We found SAM from New England Biology (B9003S) to be sufficiently pure if
adequately stored (–20 °C, aliquot but do not dilute!) and used within the shelf life given by the manufacturer. When in doubt about a SAM stock solution, we found the quality can be easily
confirmed by LC/MS. For MTase assay detection by MST, enzyme reaction mixtures with a total volume of 10 µL per single treatment condition were prepared and incubated for the desired
reaction times. Inhibitors were added from corresponding DMSO stocks to a final DMSO concentration of max. 5%. Afterward, reactions were quenched at a given reaction time by the addition of
1 µL sodium dodecyl sulfate (SDS, solution, 3% in water) per 10 µL reaction mixture and mixed well by pipetting. Subsequently, 1 µL of a 10× aptamer-containing detection master mix (200 mM
Tris, pH 8.5, 200 mM MgCl2, 3 M NaCl, 10 mM urea, 0.2% Brij, 500 nM aptaSAH1, 500 nM aptaSAH2) was added to the quenched MTase reaction and incubated for 1 h at room temperature. Reaction
mixtures were loaded directly to MST capillaries without further purification or sample processing. MST experiments were carried out in at least technical triplicates using a Monolith NT.115
instrument (NanoTemper Technologies) using Monolith standard capillaries and the blue excitation laser setup. The instrument was calibrated according to the manufacturer’s instructions.
General settings were applied for all MST experiments as follows: manual temperature control: 25 °C, fluorescence measurement before MST: 4 s, MST (IR laser) on: 20 s, fluorescence after
MST: 2 s, delay: 25 s. The laser power was adjusted to optimize the signal-to-noise ratio and the fluorescence signal. LED and MST power settings were chosen individually for each sample by
adjusting the LED power to yield fluorescence signals of at least 300 units (blue laser) or 3000 units (red laser) and the MST power to achieve an appropriate thermophoretic response
(standard setting: medium MST power). MST measurements were analyzed using the NT analysis software (version 1.5.41) and exported for statistical analysis and plotting in GraphPad Prism
7.01. FRET-BASED MTASE ASSAY MTase reaction mixtures were prepared analogously to the MST-based detection approach but on a 30 µL scale (minimum volume for 96-well half-area plates).
Reactions were quenched at a given time by 0.3% SDS and supplemented with 3 µL of the 10× aptamer-containing detection master mix. Reaction mixtures were transferred in technical triplicates
to black Greiner 96-well half-area plate and incubated for 1 h at room temperature. Afterward, plates were measured using a Tecan Spark 10 M plate reader equipped with a monochromator setup
(λex = 485 nm; λem = 600 nm). Statistical analysis and plotting were performed in GraphPad Prism 7.01. ORTHOGONAL MTASE ENZYME ASSAYS Radiometric DNMT2 assays were carried out as described
previously26. HTRF MTase assays with the commercial AptaFluor SAH assay kit (BellBrook Labs) were performed as described in the manufacturer’s manual in 96-well format18. The inhibitors used
in this study include SHO108 (DNMT2 inhibitor, synthesized in-house, (_S_)-2-amino-4-((((2 _R_,3_S_,4 _R_,5
_R_)-5-(6-amino-9_H_-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(prop-2-yn-1-yl)amino)butanoic acid), SH112 (DNMT2 inhibitor, synthesized in house, (_S_)-2-amino-4-((((2 _R_,3_S_,4
_R_,5 _R_)-5-(6-amino-9_H_-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(4-chloro-3-(trifluoromethyl)benzyl)amino)butanoic acid), STM2457 (METTL3/14 inhibitor, commercial, biomol)
and 5-phenylthieno[2,3-_d_]pyrimidin-4(1_H_)-one (_S. aureus_ TrmD inhibitor, commercial, BLDpharm)7,8,10. A compound library for the DNMT2 pilot screening (including CPD 1–10) was
commercially obtained (Tocriscreen Epigenetics 3.0, Cat. No. 7578). CPD 11 (adamantanyl-acryloylurea) was obtained commercially from Enamine. _K_I values were calculated by correcting IC50
values to the zero-substrate concentration by the Cheng-Prusoff equation: _K_I = IC50/(1 + ([S]/_K__M_)). FLUORESCENT TRACER DISPLACEMENT ASSAY (FTAD-MST ASSAY) Fluorescent tracer
displacement MST assays for orthogonal ligand evaluation were conducted as described previously27. The samples contained 2 µM DNMT2, 100 nM FTAD probe, and 1.3% DMSO in MST buffer (50 mM
HEPES, 150 mM NaCl, 1 mM dithiothreitol (DTT), 0.1% PEG-8000, 0.05% polysorbate-20, pH 7.4) supplemented with variable concentrations of the screening compounds. All samples were measured in
triplicates in Monolith standard capillaries at 25 °C. The acquisition mode was set to “Nano-Blue” with an excitation power of 30% and a medium MST power. MST measurements were analyzed
using the NT analysis software (version 1.5.41) and exported for statistical analysis and plotting in GraphPad Prism 7.01. FLUORESCENCE POLARIZATION (FP-ASSAY) Fluorescence polarization
assays were conducted with a similar sample composition as described for FTAD-MST assays as described previously28. Triplicates (50 µL) were prepared in black 96-well half-area plates
(Greiner) and measured using a Spark 10 M plate reader (Tecan) equipped with polarization filters coupled to a monochromator setup (FTAD: λex = 480 nm, λem = 530 nm). Polarization values (in
mP) were calculated from polarization-specific parallel and orthogonal fluorescence intensities according to the Tecan in-built calculation routine. Reversibility of alexidine binding to
DNMT2 was evaluated with a modified FP-assay setup. Briefly, reaction mixtures containing alexidine were prepared in black 96-well half-area plates, and after FP measurement, the volume of
one well (50 µL) was collected and applied to a Zeba™ Spin Desalting Column (7 K MWCO) while 5 µL of a 10x DNMT2 buffer solution (500 mM HEPES, 1.5 M NaCl, 10 mM dithiothreitol (DTT), 1%
PEG-8000, 0.5% polysorbate-20, pH 7.4) was placed in the collection tube attached to the SEC column. This procedure separated alexidine and restored the initial composition of the DNMT2
assay solution. Kinetic FP assays were performed to assess the time-dependent inactivation of DNMT2 treated with covalent inhibitors. Briefly, samples contained 2 µM DNMT2, 100 nM FTAD
probe, and 1.3% DMSO in DNMT2 MST buffer. Inhibitors were added from DMSO stocks. Negative inhibition control was performed by mock treatment with DMSO. Reactions were initiated by the
addition of the CPD 11 (adamantanyl-acryloylurea) and monitored for 120 min at 25 °C in a Tecan Spark 10 M plate reader (FTAD: λex = 480 nm, λem = 530 nm) The irreversible inhibition
kinetics were analyzed as described previously35,36. ELECTROPHORETIC MOBILITY SHIFT ASSAY (EMSA) DNMT2 (10 µM) was mixed with tRNAAsp (5 µM) and treated with 500 µM of the respective
screening hit compound in DNMT2 assay buffer (100 mM Tris-HCl, pH 8, 100 mM NH4OAc, 0.1 mM EDTA, 10 mM MgCl2, 10 mM DTT). Subsequently, reaction samples (each 5 µL) were loaded to a native
10% polyacrylamide gel and separated electrophoretically (1 h, 180 V) in 1x TBE. RNA was stained in situ with 1x GelRed and PAGE analysis was conducted by fluorescence scanning at an
excitation wavelength of 532 nm (Amersham Typhoon 9400). Positive control: DNMT2+tRNA (band at high molecular weights); negative control: only tRNA (band at low molecular weights).
RECOMBINANT PROTEIN AND RNA PRODUCTION DNMT2, NSUN2, and _S. aureus_ TrmD were recombinantly expressed and purified as described previously8,37. For NSUN2, instead of the previously
described SEC buffer, a buffer containing 25 mM HEPES (pH 7.5), 300 mM NaCl, 10% Glycerol, 0.04% Triton X-100, and 0.5 mM TCEP was used for size-exclusion chromatography. Recombinant
METTL13/14 was obtained commercially (BPS Bioscience, BPS-100105). tRNA substrates for DNMT2 and NSUN2 were prepared as described previously8. RNA substrates for TrmD (synthetic tRNALeu:
5′-GCGAAGGUGGCGGAAUUGGUAGACGCGCUAGCUUCAGG UGUUAGUGUCCUUACGGACGUGGGGGUUCAAGUCCCCCCCCUCGCACCA-3′) and METTL3/14 (5′-AACUUAAUGUUGCAUUGGACUUGAGUUA-3′) were obtained commercially (GenScript).
REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Authors can confirm that all
relevant data are included in the paper and/or its supplementary information files. REFERENCES * Jonkhout, N. et al. The RNA modification landscape in human disease. _RNA_ 23, 1754–1769
(2017). Article CAS PubMed PubMed Central Google Scholar * Boriack-Sjodin, P. A., Ribich, S. & Copeland, R. A. RNA-modifying proteins as anticancer drug targets. _Nat. Rev. Drug
Discov._ 17, 435–453 (2018). Article CAS PubMed Google Scholar * Cayir, A. RNA modifications as emerging therapeutic targets. _WIREs RNA_ 13, e1702 (2022). Article CAS PubMed Google
Scholar * Ofir-Rosenfeld, Y. et al. STC-15, an oral small molecule inhibitor of the RNA methyltransferase METTL3, inhibits tumour growth through activation of anti-cancer immune responses
associated with increased interferon signalling, and synergises with T cell checkpoint blockade. _Eur. J. Cancer_ 174, S123 (2022). Article Google Scholar * Helm, M., Schmidt-Dengler, M.
C., Weber, M. & Motorin, Y. General principles for the detection of modified nucleotides in RNA by specific reagents. _Adv. Biol._ 5, 2100866 (2021). Article Google Scholar * Fischer,
T. R. et al. Chemical biology and medicinal chemistry of RNA methyltransferases. _Nucleic Acids Res._ 50, 4216–4245 (2022). Article CAS PubMed PubMed Central Google Scholar * Yankova,
E. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. _Nature_ 593, 597–601 (2021). Article CAS PubMed PubMed Central Google Scholar * Schwickert, M. et
al. Discovery of inhibitors of DNA methyltransferase 2, an epitranscriptomic modulator and potential target for cancer treatment. _J. Med. Chem._ 65, 9750–9788 (2022). Article CAS PubMed
Google Scholar * Tao, Y. et al. Chemical proteomic discovery of isotype-selective covalent inhibitors of the RNA methyltransferase NSUN2. _Angew. Chem., Int. Ed._ 62, e202311924 (2023).
Article CAS Google Scholar * Hill, P. J. et al. Selective inhibitors of bacterial t-RNA-(N 1 G37) methyltransferase (TrmD) that demonstrate novel ordering of the lid domain. _J. Med.
Chem._ 56, 7278–7288 (2013). Article CAS PubMed Google Scholar * Inniss, N. L. et al. Discovery of a druggable, cryptic pocket in SARS-CoV-2 nsp16 using allosteric inhibitors. _ACS
Infect. Dis._ 9, 1918–1931 (2023). Article CAS PubMed PubMed Central Google Scholar * Graves, T. L., Zhang, Y. & Scott, J. E. A universal competitive fluorescence polarization
activity assay for S-adenosylmethionine utilizing methyltransferases. _Anal. Biochem._ 373, 296–306 (2008). Article CAS PubMed Google Scholar * Hendricks, C. L., Ross, J. R., Pichersky,
E., Noel, J. P. & Zhou, Z. S. An enzyme-coupled colorimetric assay for S-adenosylmethionine-dependent methyltransferases. _Anal. Biochem._ 326, 100–105 (2004). Article CAS PubMed
Google Scholar * Wang, C., Leffler, S., Thompson, D. H. & Hrycyna, C. A. A general fluorescence-based coupled assay for S-adenosylmethionine-dependent methyltransferases. _Biochem.
Biophys. Res. Commun._ 331, 351–356 (2005). Article CAS PubMed Google Scholar * Dorgan, K. M. et al. An enzyme-coupled continuous spectrophotometric assay for
S-adenosylmethionine-dependent methyltransferases. _Anal. Biochem._ 350, 249–255 (2006). Article CAS PubMed Google Scholar * Hsiao, K., Zegzouti, H. & Goueli, S. A.
Methyltransferase-Glo: a universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. _Epigenomics_ 8, 321–339 (2016). Article CAS PubMed Google
Scholar * Janzen, W. P., Wigle, T. J., Jin, J. & Frye, S. V. Epigenetics: tools and technologies. _Drug Discov. Today_ 7, e59–e65 (2010). Article CAS Google Scholar * Pham, H.,
Kumar, M., Martinez, A. R., Ali, M. & Lowery, R. G. Development and validation of a generic methyltransferase enzymatic assay based on an SAH riboswitch. _SLAS Discov._ 29, 100161
(2024). Article CAS PubMed PubMed Central Google Scholar * Wang, J. X., Lee, E. R., Morales, D. R., Lim, J. & Breaker, R. R. Riboswitches that sense s-adenosylhomocysteine and
activate genes involved in coenzyme recycling. _Mol. Cell_ 29, 691–702 (2008). Article CAS PubMed PubMed Central Google Scholar * Breaker, R. R. The biochemical landscape of Riboswitch
ligands. _Biochemistry_ 61, 137–149 (2022). Article CAS PubMed Google Scholar * Breaker, R. R. Natural and engineered nucleic acids as tools to explore biology. _Nature_ 432, 838–845
(2004). Article CAS PubMed Google Scholar * Edwards, A. L., Reyes, F. E., Héroux, A. & Batey, R. T. Structural basis for recognition of S-adenosylhomocysteine by riboswitches. _RNA_
16, 2144–2155 (2010). Article CAS PubMed PubMed Central Google Scholar * Debiais, M., Lelievre, A., Smietana, M. & Müller, S. Splitting aptamers and nucleic acid enzymes for the
development of advanced biosensors. _Nucleic Acids Res._ 48, 3400–3422 (2020). Article CAS PubMed PubMed Central Google Scholar * Li, H. et al. Biological function molecular pathways
and druggability of DNMT2/TRDMT1. _Pharmacol. Res._ 205, 107222 (2024). Article CAS PubMed Google Scholar * Johannsson, S. et al. Structural insights into the stimulation of S. pombe
Dnmt2 catalytic efficiency by the tRNA nucleoside queuosine. _Sci. Rep._ 8, 8880 (2018). Article PubMed PubMed Central Google Scholar * Schwickert, M. et al. Covalent
S-adenosylhomocysteine-based DNA methyltransferase 2 inhibitors with a new type of aryl warhead. _ACS Med. Chem. Lett._ 14, 777–787 (2023). Article CAS PubMed PubMed Central Google
Scholar * Zimmermann, R. A. et al. an optimized microscale thermophoresis method for high-throughput screening of DNA methyltransferase 2 ligands. _ACS Pharmacol. Transl. Sci._ 5, 1079–1085
(2022). Article CAS PubMed PubMed Central Google Scholar * Meidner, J. L. et al. Nanomole scale screening of fluorescent RNA-methyltransferase probes enables the discovery of METTL1
inhibitors. _Angew. Chem. Int. Ed._ 63, e202403792 (2024). Article CAS Google Scholar * Kallert, E., Behrendt, M., Frey, A., Kersten, C. & Barthels, F. Non-covalent dyes in microscale
thermophoresis for studying RNA ligand interactions and modifications. _Chem. Sci._ 14, 9827–9837 (2023). Article CAS PubMed PubMed Central Google Scholar * Chellamuthu, A. & Gray,
S. G. The RNA methyltransferase NSUN2 and its potential roles in cancer. _Cells_ 9, 1758 (2020). Article CAS PubMed PubMed Central Google Scholar * Miao, W. et al. Glucose binds and
activates NSUN2 to promote translation and epidermal differentiation. _Nucleic Acids Res._ 52, 13577–13593 (2024). Article CAS PubMed PubMed Central Google Scholar * Shimizu, T.,
Korehisa, T., Imanaka, H., Ishida, N. & Imamura, K. Characteristics of proteinaceous additives in stabilizing enzymes during freeze-thawing and -drying. _Biosci. Biotechnol. Biochem._
81, 687–697 (2017). Article CAS PubMed Google Scholar * Müller, S. et al. Target recognition, RNA methylation activity and transcriptional regulation of the Dictyostelium discoideum
Dnmt2-homologue (DnmA). _Nucleic Acids Res._ 41, 8615–8627 (2013). Article PubMed PubMed Central Google Scholar * Winzor, D. J. Analytical exclusion chromatography. _J. Biochem.
Biopphys. Meth._ 56, 15–52 (2003). Article CAS Google Scholar * Barthels, F. et al. Asymmetric disulfanylbenzamides as irreversible and selective inhibitors of staphylococcus aureus
sortase A. _ChemMedChem_ 15, 839–850 (2020). Article CAS PubMed PubMed Central Google Scholar * Barthels, F. et al. 2-sulfonylpyrimidines as privileged warheads for the development of
S. aureus sortase A inhibitors. _Front. Mol. Biosci._ 8, 1284 (2022). Article Google Scholar * Zhang, Y. et al. Evolutionary adaptation of the essential tRNA methyltransferase TrmD to the
signaling molecule 3′,5′-cAMP in bacteria*. _J. Biol. Chem._ 292, 313–327 (2017). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS Financial support by the Deutsche
Forschungsgemeinschaft (DFG, German Research Foundation), TP C01 in TRR319, Project-ID 439669440 to M.H. is gratefully acknowledged. We would like to thank Angelika Gründling (Imperial
College London, United Kingdom) for the gift of the _S. aureus_ TrmD expression plasmid. FUNDING Open Access funding enabled and organized by Projekt DEAL. AUTHOR INFORMATION Author notes *
These authors contributed equally: Zarina Nidoieva, Mark O. Sabin. AUTHORS AND AFFILIATIONS * Institute of Pharmaceutical and Biomedical Sciences, Johannes Gutenberg-University, Mainz,
Germany Zarina Nidoieva, Mark O. Sabin, Tristan Dewald, Annabelle C. Weldert, Sabrina N. Hoba, Mark Helm & Fabian Barthels * Department of Molecular, Cellular, and Developmental Biology,
Yale University, New Haven, CT, USA Fabian Barthels Authors * Zarina Nidoieva View author publications You can also search for this author inPubMed Google Scholar * Mark O. Sabin View
author publications You can also search for this author inPubMed Google Scholar * Tristan Dewald View author publications You can also search for this author inPubMed Google Scholar *
Annabelle C. Weldert View author publications You can also search for this author inPubMed Google Scholar * Sabrina N. Hoba View author publications You can also search for this author
inPubMed Google Scholar * Mark Helm View author publications You can also search for this author inPubMed Google Scholar * Fabian Barthels View author publications You can also search for
this author inPubMed Google Scholar CONTRIBUTIONS Z.N.: formal analysis, investigation, validation. M.O.S.: formal analysis, investigation, validation. Z.N. and M.O.S. have contributed
equally. T.D.: formal analysis, investigation, validation. A.C.W.: investigation. S.N.H.: investigation. M.H.: conceptualization, writing—review & editing. F.B.: conceptualization,
visualization, writing— original draft. CORRESPONDING AUTHOR Correspondence to Fabian Barthels. ETHICS DECLARATIONS COMPETING INTERESTS Mark Helm is a consultant for Moderna Inc. All other
authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Communications Chemistry_ thanks the anonymous reviewers for their contribution to the peer review of this work.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION
SUPPLEMENTARY INFORMATION REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use,
sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative
Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Nidoieva, Z., Sabin, M.O., Dewald, T. _et al._ A microscale thermophoresis-based enzymatic RNA methyltransferase assay enables the discovery
of DNMT2 inhibitors. _Commun Chem_ 8, 32 (2025). https://doi.org/10.1038/s42004-025-01439-9 Download citation * Received: 19 September 2024 * Accepted: 29 January 2025 * Published: 03
February 2025 * DOI: https://doi.org/10.1038/s42004-025-01439-9 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative

:max_bytes(150000):strip_icc():focal(216x0:218x2)/benedict-cumberbatch-1-435-4-20cc736017b24435a3498a49d7c22b0e.jpg)