- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Download PDF Article Open access Published: 16 July 2018 Copper/iron co-catalyzed alkoxycarbonylation of unactivated alkyl bromides Yahui Li1 & Xiao-Feng Wu1,2 Communications Chemistry
volume 1, Article number: 39 (2018) Cite this article
4806 Accesses
1 Altmetric
Metrics details
Subjects Homogeneous catalysisSynthetic chemistry methodology AbstractCarbonylative transformations of alkyl bromides have been explored less than those of aryl halides, in part because of the high barrier to activation of aryl bromides. Additionally,
alkyl-metal reagents formed in situ can tend to undergo β-hydride elimination. Here we describe a copper/iron co-catalyzed alkoxycarbonylation of unactivated alkyl bromides. In the presence
of catalytic quantities of iron and copper catalysts, primary, secondary, and tertiary alkyl bromides are carbonylatively transformed into the corresponding aliphatic esters in good yields.
A potential reaction mechanism is proposed based on control experiments.
Similar content being viewed by others The iron-catalysed Suzuki coupling of aryl chlorides Article Open access17 October 2024 Iron-catalyzed radical Markovnikov hydrohalogenation and hydroazidation of alkenes Article Open access 22 August 2024 Iron-catalyzed fluoroalkylative alkylsulfonylation of
alkenes via radical-anion relay Article Open access 17 February 2024 Introduction
Transition metal-catalyzed carbonylative transformation is one of the most potent methodologies in organic chemistry for the preparation of various carbonyl-containing compounds1,2. Through
carbonylation reactions, the carbon chain of the parent molecules can be easily increased, with carbon monoxide (CO) serving as one of the cheapest and most abundant C1 building blocks.
Since the pioneering achievements of Heck and co-workers in 19743,4, many transition-metal complexes, especially noble metals, became the catalysts of choice for the carbonylation reactions.
And numerous carbonylation procedures using aryl, vinyl, allyl, and benzyl halides as the starting materials to give the corresponding carboxylic acid derivatives have been developed and
applied during the past two decades (Fig. 1a)1,2,5.
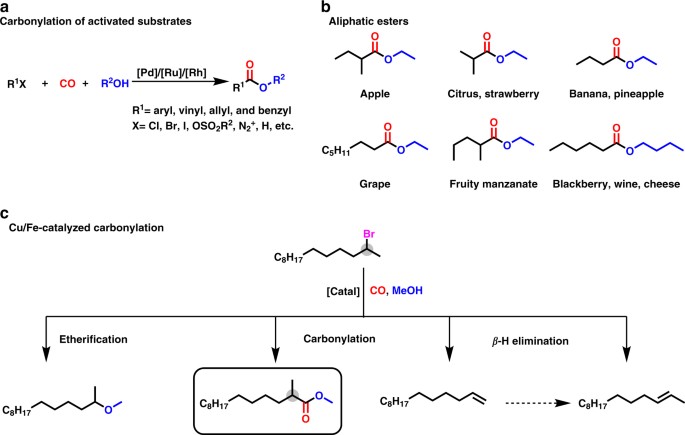
Fig. 1Challenges and application potentials for the Cu/Fe catalyzed alkoxycarbonylation of unactivated alkyl bromides. a Carbonylation of activated substrates. b Selected examples of aliphatic
esters and their organoleptic properties. c Products obtained from the alkoxycarbonylation of 2-bromotetradecane with methanol. The gray circles highlight the position of bromide
Fullsize image
Numerous cross-coupling reactions of alkyl halides have been reported6,7,8, but by contrast, their carbonylative transformations often encounter increased difficulties. This situation can
mainly be explained by the following three reasons: (i) further increased difficulty in the oxidative addition step due to the presence of π-acidic CO, which decreases the electron density
on the metal center; (ii) fast subsequent β-hydride elimination, and the produced metal-hydride will react with substrates and catalysis; (iii) facile nucleophilic substitution reaction
between alkyl halides and nucleophiles. Among the reported studies, alkyl iodides are more frequently applied and proceed through radical intermediates9. Only one exceptional example on
alkyl bromides was reported recently with palladium as the catalyst10. However, the substrates were limited to secondary bromides. Hence, challenges are still remaining to be resolved: (1)
concerning the high cost of palladium catalysts, it will be interesting to substitute them by more abundant and bio-relevant metals; (2) developing more general catalytic protocols for
unactivated alkyl halides. One solution could be the application of multi-catalyst systems. Most of the known carbonylation protocols generally rely on the interaction of a unique catalyst
with a certain substrate, and then lower the energetic barrier for new bond formation with another substrate. The cooperation of different catalytic centers could decrease the activation
energy parallelly or successively and will definitely provide a new solution for the above discussed challenges.
On the other hand, concerning the abundant and bio-relevant transitional metal catalysts, copper and iron salts are ideal choices. They are inexpensive, environmentally friendly, and
relatively non-toxic. However, few examples exist of their applications in carbonylative reactions11,12,13,14,15. The main reasons are: (i) the high affinity of CO with iron which make the
metal center saturately coordinated and then activity inhibited; (ii) the decreased ability of copper catalyst in the oxidative addition step and also the low stability of carbonyl-copper
intermediates. And the combination of iron and copper catalysts can have several potential advantages. Besides the sustainability and economic aspects, the interaction of copper and iron can
activate the added or in situ formed iron carbonyl complexes and also stabilize the carbonyl-copper intermediates. Motived by this state of the art and by our long-standing interest in
carbonylation reactions, we sought to develop a procedure for copper/iron co-catalyzed alkoxycarbonylation of alkyl halides.
Here we describe the copper/iron co-catalyzed carbonylative transformation of primary, secondary, and tertiary alkyl bromides into the corresponding esters using alcohols as the reaction
partner (Fig. 1c). Due to the widely natural occurrence of aliphatic esters and their importance as flavorings (Fig. 1b), we anticipate that this new method will find valuable
applications.
ResultsReaction optimizationAs the beginning of our study, 2-bromotetradecane and methanol were chosen as the benchmark substrates to establish the catalytic system. Upon the variation of reaction conditions, different
product yields could be obtained. Among the different metal catalyst precursors (Fig. 2), CuTc (copper(I)-thiophene-2-carboxylate) showed the best result (37% GC yield; 55% conversion).
Furthermore, catalytic experiments were carried out using Pd(OAc)2, PdCl2, and RhCl3 as well, no carbonylation product could be obtained in the three cases. It is well known that nitrogen
ligands can enhance the efficiency of copper catalyst16. Hence, the effects of different ligands were studied next to improve the reaction efficiency (Fig. 3). Apparently, the increased
steric hindrance of the ligand L2 caused by the ortho substitution led to lower yield than L1. The application of the other ligands, such as L3, L4, and L5, all gave higher catalytic
activity with more than 60% yield of the carbonylative product. However, L6 was less effective in this reaction due to its decreased electron density. In the presence of the
bathophenanthroline ligand L8, the desired product was obtained in 82% yield.
Fig. 2Catalyst effect for this alkoxycarbonylation of unactivated alkyl bromides. Reaction conditions: 2-bromotetradecane (0.5 mmol), Cu cat. (5 mol%), 1,10-Phen (5 mol%), Fe3(CO)12 (5 mol%),
NaOMe (0.75 mmol), CO (40 bar), MeOH/toluene (0.5/2 mL), 24 h. The yields (%) of esters are based on 2-bromotetradecane. The yields (%) of esters and conversion (%) of 2-bromotetradecane are
determined by GC analysis using hexadecane as the internal standard
Full size imageFig. 3Ligand effect for this alkoxycarbonylation of unactivated alkyl bromides. Reaction conditions: 2-bromotetradecane (0.5 mmol), CuTc. (5 mol%), L (5 mol%), Fe3(CO)12 (5 mol%), NaOMe (0.75
mmol), CO (40 bar), MeOH/toluene (0.5/2 mL), 24 h. The yields (%) of esters are based on 2-bromotetradecane. The yields (%) of esters and conversion (%) of 2-bromotetradecane are determined
by GC analysis using hexadecane as the internal standard
Full size imageSubsequently, with L8 as the ligand of choice, various iron salts were also studied. Selected results are shown in Fig. 4. Iron(II) and iron(III) salts were shown less effective compared
with iron carbonyl complexes. However, using iron(II) and iron(III) salts in carbonylation has been scarcely investigated. Additionally, the amount of catalysts and ligand were also checked
(Fig. 5). When decreasing the catalyst and ligand loading to 3 mol%, there was a slight drop in the yield (77% instead of 82%). Interestingly, even only 1 mol% of CuBr(Me2S), Fe3(CO)12, and
L8 ligand were used, the desired product was also obtained in 60%. Finally, the pressure of CO seemed to have less influence for this transformation, and 81% yield of the product could be
obtained when using 20 bar of CO (Fig. 6). Based on these preliminary studies, the following conditions were used for further substrates investigations: 5 mol% catalysts and ligand, NaOMe
(1.5 eq.), in 2/0.5 mL toluene/methanol, under CO (40 bar), at 80 °C for 24 h.
Fig. 4Iron catalyst effect for this alkoxycarbonylation of unactivated alkyl bromides. Reaction conditions: 2-bromotetradecane (0.5 mmol), CuTc (5 mol%), L8 (5 mol%), Fe.Cat (5 mol%), NaOMe (0.75
mmol), (40 bar CO), MeOH/toluene (0.5/2 mL), 24 h. The yields (%) of esters are based on 2-bromotetradecane. The yields (%) of esters and conversion (%) of 2-bromotetradecane are determined
by GC analysis using hexadecane as the internal standard. †1.5 mmol NaOMe
Full size imageFig. 5Other effect for this alkoxycarbonylation of unactivated alkyl bromides. Reaction conditions: 2-bromotetradecane (0.5 mmol), CuTc (5 mol%), L8 (5 mol%), Fe3(CO)12 (5 mol%), NaOMe (0.75
mmol), CO (40 bar), MeOH/Toluene (0.5/2 mL), 48 h. The yields (%) of esters are based on 2-bromotetradecane. The yields (%) of esters and conversion (%) of 2-bromotetradecane are determined
by GC analysis using hexadecane as the internal standard. †CuBr(Me2S) (1 mol%) instead of CuTc; ‡1.5 mmol NaOMe
Full size imageFig. 6Substrate scope for the alkoxycarbonylation of unactivated alkyl bromides. a Reaction studied, with the structure of L8 inset; b Table of substrates. All yields are isolated yields except
mentioned. The gray circles highlight the position of bromide. Reaction conditions: 2-bromotetradecane (0.5 mmol), CuTc (5 mol%), L (5 mol%), Fe3(CO)12 (5 mol%), NaOMe (0.75 mmol), CO (40
bar), MeOH/toluene (0.5/2 mL), 24 h. *GC yields. ‡2-bromotetradecane (0.5 mmol), CuTc (5 mol%), L (5 mol%), Fe3(CO)12 (5 mol%), NaOMe (1.5 mmol), CO (40 bar), alcohol (20 eq.), toluene (2
mL), 24 h. §NaOtBu instead of NaOMe
Full size imageSubstrate scopeWith the optimized reaction conditions in hand, different alkyl bromides were tested using either methanol or n-butanol (Fig. 6). Simple short- and long-chain alkyl bromides, for example
2-bromooctane or 2-bromotetradecane, can give good yields of the desired products (75% and 78% yield, respectively). Six- and twelve-membered carbocycles also reacted efficiently in our
approach. Other substrates including cyclohexyl, tetrahydropyranyl, and cyclododecyl bromides were examined as well and good yields can be achieved in general. Furthermore, a variety of
functional groups were tested. Alkyl bromides bearing -Ph, -OMe, -S, and -COOMe were shown to be compatible, and gave the corresponding esters in 61–81% yields. p-Toluenesulfonamide group
was compatible with the catalytic conditions and provided the desired ester in 64% yield. Further, alkyl bromide such as strained norbornene was well tolerated and afforded the corresponding
ester product in high yield. Remarkably, our catalytic reaction can also be extended to the efficient carbonylation of primary and tertiary alkyl bromides. The corresponding esters were
obtained in moderate to good yields. For example, 1-bromoadamantane, which has high steric bulk can, also be used and gave the desired product in 82% yield. We next surveyed a range of
alcohols with 2-bromotetradecane under the optimized conditions. Primary alcohols, such as ethanol, 1-propanol, and n-butanol, gave good yields of the desired esters. Similarly, using
more-bulky alcohols, for example, cyclobutylmethanol, led to a good result of 74% yield as well. Secondary alcohols were also effective in the alkoxycarbonylation. However, a decreased yield
23% (63% rsm) was obtained with CF3CH2OH as an example of less-basic alcohol. In the case of tert-butanol, very low yield of the desired product was obtained.
Mechanistic studyIn order to get some more insight into the reaction mechanism, some control experiments were carried out. Firstly, we conducted radical clock experiments, which are often used to probe the
radical reactivity of metal alkyl species. Under our reaction conditions, both primary and secondary alkyl bromides formed radical rearranged products in moderate to good yields (Fig. 1, 3t,
3u). Additionally, we studied the effect of catalyst, ligand, and radical inhibitors (Fig. 7a). In the absence of a catalyst or ligand, no desired product could be observed. Additionally,
no desired product could be detected by GC when 2 equivalents of BHT (butylhydroxytoluene) or TEMPO was added into our standard reaction conditions. These preliminary studies suggest against
a purely free radical pathway, and implicate the formation of either caged radical or organometallic intermediates. Then, designed experiments were performed to verify the roles of the
catalytic components (Fig. 7b). We reacted bromocyclohexane with copper catalyst or iron catalyst in the presence of TEMPO. From the obtained results, the following conclusions can be made:
(1) alkyl bromide was activated by copper catalyst; (2) CO is not only a carbonyl source, but also a ligand to produce the activate copper catalyst; (3) iron (II/III) salts can be reduced by
CO to iron(0); (4) iron carbonyl can act as a CO source to activate copper catalyst; (5) iron can be crucial in CO insertion stage. In addition, competition experiments between secondary
and tertiary alkyl bromides were conducted. The results clearly demonstrated that the carbonylation of secondary alkyl bromide is faster than that for tertiary alkyl bromide (Fig. 7c). This
result can be more useful when a substrate contains both secondary and tertiary bromides.
Fig. 7Mechanistic study. a The effect of catalyst, ligand, and radical inhibitors. b Experiments revealed the role of copper catalysts. c Kinetic experiments between secondary and tertiary alkyl
bromides
Full size imageOn the basis of our findings, a possible reaction mechanism is described in Fig. 8. Under the assistance of base, the copper complex 4 irreversibly abstracts a bromine atom from the alkyl
bromide generating a carbon-centered radical and a copper bromide intermediate 5. The analogous photochemical process has recently been demonstrated by Fu and co-workers17,18. We propose
that the key intermediates should be similar here, although further study is needed to substantiate this hypothesis given the different modes of activation. This step is then followed by
radical addition to copper complex 5 and form the new copper complex 6. Then complex 6 reacted with complex 7; the acylcarbonyl-iron complex 8 will be formed after transmetalation and CO
insertion steps. Subsequently, nucleophilic attack by the alcohol should lead to the desired ester product and regenerate the active iron species 7.
Fig. 8Proposed reaction mechanism. Iron/copper co-catalyzed carbonylative transformation of alkyl bromides
Full size imageDiscussionIn conclusion, we have developed an interesting copper/iron co-catalyzed alkoxycarbonylation of unactivated alkyl bromides. The general applicability is demonstrated by 28 examples,
including primary, secondary, and tertiary alkyl bromides. On the basis of control experiments, a reaction mechanism is proposed as well. Notably, besides the advantages, including
generality on substrates, non-expensive and environmental benign catalyst system, etc., this also represents, to our knowledge, the first example on non-noble metal-catalyzed
alkoxycarbonylation of unactivated alkyl bromides.
MethodsSynthesis and characterizationSee Supplementary Methods and Supplementary Figures 1–27.
General procedure AA 4-mL screw-cap vial was charged with CuTc (4.75 mg, 5 mol%), 4,7-diphenyl-1,10-phenanthroline (8.3 mg, 5 mol%), Fe3(CO)12 (12.57 mg, 5 mol%), NaOMe (40.5 mg, 1.5 eq.), and an oven-dried
stirring bar. The vial was closed by Teflon septum and phenolic cap and was connected with the atmosphere with a needle. The vial was flushed with argon three times. After 2-bromotetradecane
(0.5 mmol), MeOH (0.5 mL), and toluene (2 mL) were injected by a syringe, the vial was fixed in an alloy plate and put into Paar 4560 series autoclave (300 mL) under argon atmosphere. At
room temperature, the autoclave is flushed with CO three times and 40 bar of CO was charged. The autoclave was placed on a heating plate equipped with magnetic stirring and an aluminum
block. The reaction is allowed to be heated under 80 °C for 24 h. Afterward, the autoclave is cooled to room temperature and the pressure was carefully released. After the removal of the
solvent under reduced pressure, pure product was obtained by column chromatography on silica gel (eluent: pentane–pentane/ethyl acetate = 500–30:1).
General procedure BA 4-mL screw-cap vial was charged with CuTc (4.75 mg, 5 mol%), 4,7-diphenyl-1,10-phenanthroline (8.3 mg, 5 mol%), Fe3(CO)12 (12.57 mg, 5 mol%), NaOMe (40.5 mg, 1.5 eq.), and an oven-dried
stirring bar. The vial was closed by Teflon septum and phenolic cap and was connected with the atmosphere with a needle. The vial was flushed with argon three times. After 2-bromotetradecane
(0.5 mmol), alcohol (10 mmol), and toluene (2 mL) were injected by a syringe, the vial was fixed in an alloy plate and put into Paar 4560 series autoclave (300 mL) under argon atmosphere.
At room temperature, the autoclave is flushed with CO three times and 40 bar of CO was charged. The autoclave was placed on a heating plate equipped with magnetic stirring and an aluminum
block. The reaction is allowed to be heated under 80 °C for 24 h. Afterward, the autoclave is cooled to room temperature and the pressure was carefully released. After the removal of the
solvent under reduced pressure, pure product was obtained by column chromatography on silica gel (eluent: pentane–pentane/ethyl acetate = 500–30:1).
Data availabilityThe data sets generated and analyzed during the current study are included in the Supplementary Information file and also available from the corresponding authors on request.
References Beller, M. Applied Homogeneous Catalysis with Organometallic Compounds 2nd edn, Vol. 1 (Wiley-VCH, Weinheim, 2002).
Schiesser, C. H., Wille, U., Matsubara, H. & Ryu, I. Radicals masquerading as electrophiles: dual orbital effects in nitrogen-philic acyl radical cyclization and related addition reactions.
Acc. Chem. Res. 40, 303–313 (2007).
Article CAS PubMed Google Scholar
Schoenberg, A., Bartoletti, I. & Heck, R. F. Palladium-catalyzed carboalkoxylation of aryl, benzyl, and vinylic halides. J. Org. Chem. 39, 3318–3326 (1974).
Article CAS Google Scholar
Schoenberg, A., Bartoletti, I. & Heck, R. F. Palladium-catalyzed amidation of aryl, heterocyclic, and vinylic halides. J. Org. Chem. 39, 3327–3331 (1974).
Article CAS Google Scholar
Brennführer, A., Neumann, H. & Beller, M. Palladium-catalyzed carbonylation reactions of aryl halides and related compounds. Angew. Chem. Int. Ed. 48, 4114–4133 (2009).
Article CAS Google Scholar
Zultanski, S. L. & Fu, G. C. Nickel-catalyzed carbon−carbon bond-forming reactions of unactivated tertiary alkyl halides: suzuki arylations. J. Am. Chem. Soc. 135, 624–627 (2013).
Article CAS PubMed PubMed Central Google Scholar
Liu, Y., Cornella, J. & Martin, R. Ni-catalyzed carboxylation of unactivated primary alkyl bromides and sulfonates with CO2. J. Am. Chem. Soc. 136, 11212–11215 (2014).
Article CAS PubMed Google Scholar
Juliá-Hernández, F., Moragas, T., Cornella, J. & Martin, R. Remote carboxylation of halogenated aliphatic hydrocarbons with carbon dioxide. Nature 545, 84–88 (2017).
Article CAS PubMed Google Scholar
Sumino, S., Fusano, A., Fukuyama, T. & Ryu, I. Carbonylation reactions of alkyl iodides through the interplay of carbon radicals and Pd catalysts. Acc. Chem. Res. 47, 1563–1574 (2014).
Article CAS PubMed Google Scholar
Sargent, B. T. & Alexanian, E. J. Palladium-catalyzed alkoxycarbonylation of unactivated secondary alkyl bromides at low pressure. J. Am. Chem. Soc. 138, 7520–7523 (2016).
Article CAS PubMed PubMed Central Google Scholar
Kang, S.-K., Yamaguchi, T., Kim, T.-H. & Ho, P.-S. Copper-catalyzed cross-coupling and carbonylative cross-coupling of organostannanes and organoboranes with hypervalent iodine compounds. J.
Org. Chem. 61, 9082–9083 (1996).
Cheng, L.-J. & Mankad, N. P. Cu-catalyzed hydrocarbonylative C–C coupling of terminal alkynes with alkyl iodides. J. Am. Chem. Soc. 139, 10200–10203 (2017).
Article CAS PubMed Google Scholar
Li, Y., Dong, K., Zhu, F., Wang, Z. & Wu, X.-F. Copper-Catalyzed carbonylative coupling of cycloalkanes and amides. Angew. Chem. Int. Ed. 55, 7227–7230 (2016).
Article CAS Google Scholar
Devasagayaraj, A., Rao, M. L. N. & Periasamy, M. Carbonylation of R2BI in the presence of NaCo(CO)4 and Na2Fe(CO)4: a simple synthesis of dialkyl ketones. J. Organomet. Chem. 421, 147–150
(1991).
Article CAS Google Scholar
Driller, K. M., Prateeptongkum, S., Jackstell, R. & Beller, M. A general and selective iron-catalyzed aminocarbonylation of alkynes: synthesis of acryl- and cinnamides. Angew. Chem. Int. Ed.
50, 537–541 (2011).
Article CAS Google Scholar
Bhunia, S., Pawar, G. G., Kumar, S. V., Jiang, Y. & Ma, D. Selected copper-based reactions for C-N, C-O, C-S, and C-C Bond Formation. Angew. Chem. Int. Ed. 56, 16136–16179 (2017).
Article CAS Google Scholar
Ahn, J. M., Peters, J. C. & Fu, G. C. Design of a photoredox catalyst that enables the direct synthesis of carbamate-protected primary amines via photoinduced, copper-catalyzed N-alkylation
reactions of unactivated secondary halides. J. Am. Chem. Soc. 139, 18101–18106 (2017).
Article CAS PubMed Google Scholar
Fu, G. C. Transition-metal catalysis of nucleophilic substitution reactions: a radical alternative to SN1 and SN2 processes. ACS Cent. Sci. 3, 692–700 (2017).
Article CAS PubMed PubMed Central Google Scholar
Download references
AcknowledgementsThe authors thank the Chinese Scholarship Council for financial support. We also appreciate the general support from Professor Matthias Beller in LIKAT. The analytic supports of Dr. W.
Baumann, Dr. C. Fisher, S. Buchholz, and S. Schareina are gratefully acknowledged. The publication of this article was funded by the Open Access Fund of the Leibniz Association.
Author informationAuthors and Affiliations Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18059, Rostock, Germany
Yahui Li & Xiao-Feng Wu
Department of Chemistry, Zhejiang Sci-Tech University, Xiasha Campus, 310018, Hangzhou, People’s Republic of China
Xiao-Feng Wu
AuthorsYahui LiView author publications You can also search for this author inPubMed Google Scholar
Xiao-Feng WuView author publications You can also search for this author inPubMed Google Scholar
ContributionsX.F.W. and Y.L. conceived and designed the experiments. Y.L. performed the experiments and analyzed the data. X.F.W. and Y.L. co-wrote the paper. X.F.W. directed the project.
Corresponding author Correspondence to Xiao-Feng Wu.
Ethics declarations Competing interestsThe authors declare no competing interests.
Additional informationPublisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary materialSupplementaryInformationRights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and permissions
About this articleCite this article Li, Y., Wu, XF. Copper/iron co-catalyzed alkoxycarbonylation of unactivated alkyl bromides. Commun Chem 1, 39 (2018).
https://doi.org/10.1038/s42004-018-0039-6
Download citation
Received: 24 May 2018
Accepted: 19 June 2018
Published: 16 July 2018
DOI: https://doi.org/10.1038/s42004-018-0039-6
Share this article Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article.
Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative





