- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
The appearance of multiple new SARS-CoV-2 variants during the COVID-19 pandemic is a matter of grave concern. Some of these variants, such as B.1.617.2, B.1.1.7, and B.1.351, manifest higher
infectivity and virulence than the earlier SARS-CoV-2 variants, with potential dramatic effects on the course of the pandemic. So far, analysis of new SARS-CoV-2 variants focused primarily
on nucleotide substitutions and short deletions that are readily identifiable by comparison to consensus genome sequences. In contrast, insertions have largely escaped the attention of
researchers although the furin site insert in the Spike (S) protein is thought to be a determinant of SARS-CoV-2 virulence. Here, we identify 346 unique inserts of different lengths in
SARS-CoV-2 genomes and present evidence that these inserts reflect actual virus variance rather than sequencing artifacts. Two principal mechanisms appear to account for the inserts in the
SARS-CoV-2 genomes, polymerase slippage and template switch that might be associated with the synthesis of subgenomic RNAs. At least three inserts in the N-terminal domain of the S protein
are predicted to lead to escape from neutralizing antibodies, whereas other inserts might result in escape from T-cell immunity. Thus, inserts in the S protein can affect its antigenic
properties and merit monitoring.
The first SARS-CoV-2 genome was sequenced in January 2020. Since then, more than a million virus genomes have been collected and sequenced. Comparative analysis of SARS-CoV-2 variants has
provided for the identification of the routes of virus transmission1,2,3,4, the selective pressure on different genes5, and the discovery of new variants associated with higher
infectivity6,7,8. In many cases, genome analysis only included search for point mutations, but some deletions also have been identified, such as del69-70, one of the characteristic mutations
of B.1.1.7 and Cluster 52,3 or del157-158 in B.1.617.2 (delta)9. Moreover, recently, recurrent deletions have been shown to drive antibody escape10. However, insertions are mostly ignored,
both during variant calling step and in the downstream analysis.
Although insufficiently studied, insertions appear to be crucial for beta-coronavirus evolution. Three insertions in the spike (S) glycoprotein and in the nucleoprotein (N), that occurred
early in sarbecovirus evolution, have been shown to differentiate highly pathogenic beta-coronaviruses (SARS-CoV-1, SARS-CoV-2 and MERS) from mildly pathogenic and non-pathogenic strains,
and suggested to be key determinants of human coronaviruses pathogenicity11. The best characterized insert in SARS-CoV-2 is the PRRA tetrapeptide that so far is unique to SARS-CoV-2 and
introduces a polybasic furin cleavage site into the S protein, enhancing its binding to the receptor12,13,14.
Inserts in the SARS-CoV-2 genome are categorized in the CoV-GLUE database15, and the preliminary results on systematic characterization of the structural variation and inserts in particular
have been reported16. Forty structural variants including three inserts, three nucleotides long each, were discovered and shown to occur in specific regions of the SARS-CoV-2 genome. These
variants have been further demonstrated to be enriched near the 5’ and 3’ breakpoints of the non-canonical (nc) subgenomic (sg) RNAs of coronaviruses. In addition, indels have been shown to
occur in arms of the folded SARS-CoV-2 genomic RNA16. However, longer inserts that might have been introduced into the virus genome during SARS-CoV-2 evolution, to our knowledge, have not
been systematically analyzed.
The mechanisms of sequence insertion in the genomes of RNA viruses, and coronaviruses in particular, are poorly understood. One potential route is recombination. Homologous recombination is
common among coronaviruses, and in particular, in the sarbecovirus lineage, and is likely to be a major evolutionary route producing coronavirus strains with changed properties17,18.
Specifically, the entire receptor-binding motif (RBM) domain of the S protein can be replaced by homologous recombination as it probably happened in RaTG13 and some other
sarbecoviruses17,19,20. In contrast, non-homologous recombination in RNA viruses appears to be rare, and its molecular mechanisms remains poorly understood21.
In infected cells, beta-coronaviruses produce 5–8 major sgRNAs22,23. Eight canonical sgRNAs are required for the expression of all encoded proteins of SARS-CoV-2. These sgRNAs are produced
by joining the transcript of the 5′ end of the genome (TRS site) with the beginning of the transcripts of the respective open reading frames (ORFs)24
In addition, SARS-CoV-2 has been reported to produce multiple nc sgRNAs, some of which include the TRS at 5′ end, whereas others are TRS-independent25,26; apparently, the ncRNAs are spurious
products of errors of transcription initiation.
Here we report the comprehensive census of the inserts that were incorporated into virus genomes during the evolution of SARS-CoV-2 over the course of the pandemic and show that they
occurred during virus evolution rather than resulting from experimental errors. These inserts are non-randomly distributed along the genome, most being located in the 3′terminal half of the
genome and co-localizing with 3′ breakpoints of nc-sgRNAs. We show that at least some long insertions occur either as a result of the formation of nc-sgRNAs or by duplication of adjacent
sequences. We analyze in detail the inserts in the S glycoprotein and show that at least three of these are located in a close proximity to the antibody-binding site in the N-terminal domain
(NTD), whereas others are also located in NTD loops and might lead to antibody escape, and/or T cell evasion.
To compile a reliable catalogue of inserts in SARS-CoV-2 genome, we analyzed all the 1,785,103 sequences present in the GISAID multiple genome alignment (compiled on June 17, 2021). From
this alignment, we extracted all sequences that contained insertions in comparison with the reference genome (1354 unique events in 2159 unique genomes). After the initial filtering
(Methods), insertions were identified in 752 unique genomes, with 544 unique events detected in total. We evaluated all regions around insertions in alignments and removed all inserts that
appeared due to misalignment. To the remaining inserts, we added four long inserts obtained from the GISAID metadata description (see Methods), resulting in a set of 354 unique inserts
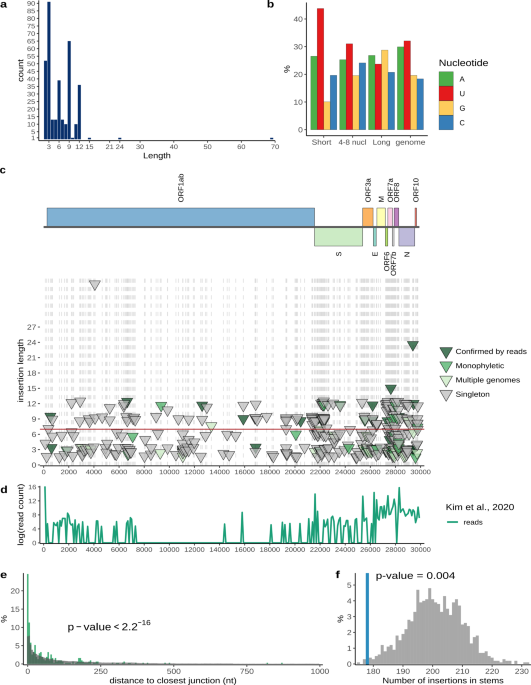
ranging in length from 2 to 69 nucleotides in 746 genomes (including identical sequences) (Fig. 1a; Supplementary Data 1; Supplementary Data 2).
a Distribution of insert lengths. b Nucleotide composition of inserts of different lengths and full SARS-CoV-2 genome. c Distribution of inserts along the genome. Each triangle represents
one insertion event. The level of confidence in each variant is represented by color: dark green, confirmed by sequencing read analysis; green, monophyletic in the tree, no read data
available; light green, observed multiple times, but not monophyletic; gray, singletons (Supplementary Data 2). The positions of inserts are marked with gray dashed lines. d Experimental
data on SARS-CoV-2 transcriptome26 showing template switch hotspots during the formation of sgRNAs, showing the distribution of junction reads connecting recombination hotspots along the
genome. e Distance from inserts to closest template switching hotspot site (green) compared with random expectation (gray). Wilcoxon rank sum test p value is provided. f The number of
inserts that occur in structured regions of SARS-CoV-2 genomic RNA (blue) compared with random expectation (gray). Permutation test p value is provided. The data on SARS-CoV-2 structure was
obtained from28. The code to reproduce the figure is provided in repository (see Code availability).
To further minimize the number of inserts that appeared due to sequencing errors, we screened the Sequence Read Archive (SRA) database for the corresponding raw read data. We were able to
obtain raw reads for 43 inserts, of which 40 were multiples of three by length, and one more was four nucleotides long but occurred in an intergenic region. We validated 35 insertions with
raw read data; only one of these was not a multiple of three in length and occurred within a gene (Supplementary Data 1; Supplementary Data 2). Among the inserts that were not validated by
raw reads 6 were singletons, whereas two others were short duplets and occurred in a polyU tract. We removed these unconfirmed events from our dataset, resulting in 346 unique inserts.
Assuming that the fraction of true positives is the same among all inserts as it is among those with available reads, 282 of these inserts are expected to reflect actual evolutionary events.
Among the inserts in our dataset, 234 (67%) were multiples of three, and of the remaining ones, 16 (5% of the total) were located in intergenic regions, 39 (11%) in orf1ab, and 57 (17%) in
other genes. It appears likely that most if not all frameshifting inserts are sequencing artifacts, but some of such inserts in other genes could be real events reflecting the dispensability
of these genes for virus reproduction. For example, we identified four frameshifting inserts in ORF6, for which deletion variants have been described earlier27





