- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Whole-body physical exercise has been shown to promote retinal structure and function preservation in animal models of retinal degeneration. It is currently unknown how exercise modulates
retinal inflammatory responses. In this study, we investigated cytokine alterations associated with retinal neuroprotection induced by voluntary running wheel exercise in a retinal
degeneration mouse model of class B1 autosomal dominant retinitis pigmentosa, I307N Rho. I307N Rho mice undergo rod photoreceptor degeneration when exposed to bright light (induced). Our
data show, active induced mice exhibited significant preservation of retinal and visual function compared to inactive induced mice after 4 weeks of exercise. Retinal cytokine expression
revealed significant reductions of proinflammatory chemokines, keratinocyte-derived chemokine (KC) and interferon gamma inducible protein-10 (IP-10) expression in active groups compared to
inactive groups. Through immunofluorescence, we found KC and IP-10 labeling localized to retinal vasculature marker, collagen IV. These data show that whole-body exercise lowers specific
retinal cytokine expression associated with retinal vasculature. Future studies should determine whether suppression of inflammatory responses is requisite for exercise-induced retinal
protection.
Mutations in genes encoding retinal trafficking proteins often manifest as blinding diseases, such as retinitis pigmentosa (RP). RP is a heterogeneous group of hereditary disorders that lead
to the progressive loss of retinal function. RP is one of the most inherited blinding diseases, affecting about 1.5 million people globally, with roughly 25% of these cases being autosomal
dominant retinitis pigmentosa (adRP)1,2,3,4. To date, 24 genes have been associated with adRP, (RetNet; final version, dated October 7, 2022) and over 1000 mutations have been reported in
these genes5. Clinical features include a reduction in the peripheral visual field, leading to tunnel vision and eventual progression to total blindness. Disease onset typically begins in
the early teenage years, and severe visual impairment occurs between the ages of 45 and 606. Although therapeutic strategies targeting specific mutations are being developed2,7,8, clinical
trials and retrospective studies suggest that exercise may have preventative and rehabilitative effects in patients with retinal degenerative diseases, including inherited retinal
degenerations9,10,11,12.
Recent studies have shown physical exercise to be an effective, non-invasive strategy to prevent and halt neurodegenerative disease progression11,13,14,15. For the past decade, our group has
investigated the neuroprotective effects of exercise in several animal models of retinal disease. We have found exercise to be associated with reduced photoreceptor cell death, reduced
retinal pigment epithelium (RPE) stress, conserved retinal structure, preserved visual function, and increased neuroplasticity in retinal astrocytes16,17,18,19,20,21,22. Recently, we
determined voluntary running wheel exercise preserved retinal function, morphology and retinal pigmented epithelial (RPE) integrity in aged (10–20-month-old) I307N Rho mice18. This model has
high translational relevance, because it specifically mimics several phenotypes associated with class B1 adRP mutations, such as slower disease progression following induction, focal rod
degeneration, and the initiation of innate and acquired retinal immune responses23,24. Immune dysregulation is one of the key mechanisms underlying retinal degenerative disease
pathogenesis25. Using cytokine knockout animal models, studies have shown that chronic stress in a deficient innate immune system may tip the balance of the cytokine network towards an
exacerbated immune response, resulting in tissue and cellular damage26,27,28. It is currently unknown how whole-body exercise affects retinal immune responses and cytokine signaling
networks, specifically in adRP. Our study provides insight for future methods and treatments to halt the progression of inherited retinal degenerations. In this study, we sought to
investigate the effects of voluntary exercise on retinal structure, function, and retinal cytokine response in younger (3–4 month-old) I307N Rho mice.
All animal procedures were approved by the Atlanta VA Institutional Animal Care and Use Committee, conform to the Association for Research in Vision and Ophthalmology (ARVO) Statement for
the Use of Animals in Ophthalmic and Vision Research, and all methods are reported in accordance with ARRIVE guidelines. A colony of I307N Rho mice was established and maintained at Emory
University by breeding homozygous I307N Rho mice (gift of Dr. Patsy Nishina of the Jackson Laboratory, Bar Harbor, ME; RRID: IMSR_JAX:030638; MGI: J:159523) to produce heterozygous progeny
for use in experiments29,30. (N.B.: The mice from Dr. Nishina originated from a two-generation backcross mating scheme using chemically mutated F1 hybrid 129/SvJae × C57BL/6J embryonic stem
cells, followed by a minimum of five backcrosses onto the C57BL/6J background)29. Adult male and female (3–4 months-old, n = 11–16 per group) heterozygous I307N Rho mice were then
transferred to the Atlanta VA and housed under a 12-h light/dark cycle (06:00–18:00 h). During the light cycle, light levels measured at the bottom of the mouse cages approximately 50 lx.
Mice had access to standard mouse chow (Teklad Global 18% Protein Rodent Diet 2918, Irradiated, Rockville, MD) and water ad libitum. Mice were placed in single housing cages with low-profile
running wheels (Med-Associates, Inc.; St. Albans, VT) that were either functional (active) or locked (inactive; see schematic of experimental timeline in Fig. 1a). Mice had continuous
access to wheels for the entirety of the experiment, with the exception of when retinal and visual functional testing was performed and during light induction (detailed below). Through use
of voluntary running wheels, we are able to model running patterns similar to natural running behavior in mice, providing a non-stress environment comparatively to forced exercise such as
treadmill running31. When given access to running wheels, mice have been reported to run a range between 4 and 20 km per day with a total activity time of roughly 3 to 7 h a day31. Following
testing at baseline, light exposure, and testing at 1- and 2-weeks post-light induction, mice were then transferred back to maintenance housing with active or inactive running wheels. At
the end of 4-weeks, mice were euthanized via cervical dislocation, eyes were enucleated for histology and immunohistochemistry, and retinal extract was collected for a multiplexed cytokine
immunoassay.
Animals were dark adapted overnight and atropine eye drops (0.2% diluted from 1% atropine ophthalmic solution, Akorn Inc., Lake Forest, IL; diluted with Refresh Tears (Allergan, Irvine, TX))
were administered twice, with the last eye drops applied 30 min before toxic light exposure. To induce I307N Rho degeneration, animals were individually housed in shoebox containers with a
white light emitting diode (LED) light panel (LED500A; Fancierstudio, Hayward, CA) fitted to a standard mouse cage as previously described19. Mice were exposed to either bright light (6000
lx, induced) to induce retinal degeneration or dim light (50 lx, uninduced) for 5 min. Room and light box temperatures were closely monitored to ensure animal welfare. Light induction
occurred between 10 and 11 AM. Following light induction, mice were returned to their home cages under normal lighting conditions for the remainder of the experiment.
Retinal function (n = 11–13 per group) was measured with a commercial electroretinography (ERG) system (Bigshot; LKC Technologies, Gaithersburg, MD) as previously described by our
group16,19. After overnight dark adaptation, mice were anesthetized (ketamine [80 mg/kg]/xylazine [16 mg/kg]). All procedures were performed under dim red light. The pupils were dilated (1%
tropicamide; Alcon Laboratories, Ft. Worth, TX) and corneas were anesthetized (1% tetracaine; Alcon Laboratories, Ft. Worth, TX). Body temperature was maintained with a heating pad (ATC
1000; World Precision Instruments, Sarasota, FL) for the duration of the session. The ERG protocol consisted of a ten-step series of full-field flash stimuli produced by a Ganzfeld dome
under both dark-adapted scotopic conditions (− 3.0 to 2.1 log cd s/m2) to test rod and rod/cone pathways and light-adapted photopic conditions to test cone pathway function. (2.0 log cd s/m2
presented as 6.1 Hz flicker with a 30 cd/m2 background light). Custom gold-loop wire electrodes were placed on the center of each eye through a layer of 1% methylcellulose to measure the
electrical response of the eye to each flash. Both reference and ground platinum needle reference electrodes (1 cm; Natus Medical Incorporated, Pleasanton, CA) were inserted subcutaneously
in each cheek and the tail, respectively. Once ERG was completed, mice were given an IP injection of atipamezole (1 mg/kg; Antisedan, Zoetis, Parsippany, NJ) to counteract the effects of
xylazine32, administered saline eye drops and allowed to recover on a heating pad (37 °C) before being returned to housing. ERG responses from both eyes were averaged together.
A virtual OMR tracking system was used to measure visual function in all experimental groups (n = 11–16 per group; OMR; OptoMotry®; Cerebral Mechanics Inc., Lethbridge, AB, Canada) as
previously described by our group and others33,34,35,36. Mice were placed on a circular platform in the center of a virtual reality chamber composed of four computer monitors, which present
vertical sine wave gratings revolving at a speed of 12 deg/s. A video camera monitored the animals’ behavior in real time throughout the experiment. The presence or absence of reflexive
movements by the mouse’s head were noted as it tracked the rotating gratings moving either in a clockwise or counter-clockwise direction. When mice became distracted, their attention was
regained by gently tapping the instrument. OptoMotry® uses a staircase paradigm to automatically calculate spatial frequency and contrast sensitivity thresholds. To evaluate spatial
frequency, contrast was held constant at 100% while spatial frequency gratings started with a frequency of 0.042 cyc/deg and adjusted over time with the presence or absence of head
movements. To assess contrast sensitivity, spatial frequency was held constant at the peak of the contrast sensitivity curve at 0.064 cycles per deg (c/d), while contrast began at 100% and
adjusted over time. The contrast sensitivity reported here was calculated as a reciprocal of the Michelson contrast from the screen’s luminance (max + min/max − min), as previously
described37. The OMR was assessed by a masked observer. The spatial frequency and contrast sensitivity thresholds were determined after three positive responses and calculated as the average
of the reported values for both eyes and were compared across time points.
For histological analyses, eyes were enucleated and then immersion-fixed in 4% paraformaldehyde for 2 h at room temperature and rinsed with 0.1 M phosphate buffer. Posterior eyecups were
processed through a graded alcohol series and embedded in plastic resin (Embed 812/DER 736, Electron Microscopy Science). Using an ultramicrotome (Reichert Ultracut, Leica, Inc., Buffalo
Grove, IL) with a histo-diamond knife, sections (0.5 μm) were cut through the superior to inferior retina bisecting the optic nerve. Plastic embedded retinal sections were stained with 1%
aqueous toluidine blue (Millipore Sigma, Burlington, MA) and imaged using a Leica DMLB microscope with a 40 × N Plan brightfield objective (Leica, Inc., Buffalo Grove, IL). Retinal
spidergrams were constructed by plotting the number of photoreceptor nuclei present in the outer nuclear layer (ONL) and cone photoreceptor quantifications as a function of position in the
retina relative to the optic nerve. ONL and cone nuclei were manually counted using Adobe Photoshop (24.0.0 release) within 100-μm-wide segments spaced at 250, 500, 750, 1000 and 1250 μm
from the optic nerve head in the superior and inferior directions within whole retinal sections (n = 8 per group, 3 retinal sections quantified per animal).
Eyes were fixed in methanol acetic acid, dehydrated, embedded in paraffin, and sectioned through the sagittal plane on a microtome (5 μm). Sections containing the optic nerve were selected
for staining to ensure that consistent regions were examined between animals. The slides were deparaffinized across five Coplin jars with 100 mL of xylene for 2 min each, consecutively. Then
the slides were rehydrated in a series of 100 mL ethanol solutions for 2 min each: 100%, 90%, 80%, 70%, 60%, and 50%. Following rehydration, retinal sections were blocked for 30 min at room
temperature in 5% normal donkey serum in PBS with 0.01% sodium azide and 0.3% Triton X-100. Primary antibodies were diluted in 5% normal donkey serum in PBS with 0.01% sodium azide and
incubations were performed overnight at 4 °C using KC monoclonal antibody (48415, Invitrogen, Waltham, MA, 1:100 dilution), IP-10 monoclonal antibody (sc-374092, Santa Cruz Biotechnology,
Dallas, TX, 1:100 dilution), Gt X Collagen Type IV antibody (AB769, Millipore Sigma, Burlington, MA, 1:100 dilution) and cone arrestin (15282, Millipore Sigma, Burlington, MA, 1:100
dilution). Secondary antibodies were diluted in PBS and incubations were performed for 1 h at room temperature in the dark (Alexa Fluor® 647 donkey anti-mouse; 1:500, A32787, ThermoFisher
Scientific, Waltham, MA; Alex Fluor® 568 donkey anti-goat; 1:500, A11057, ThermoFisher Scientific, Waltham, MA). Retinal sections were mounted with ProLong®Gold Antifade Reagent with DAPI
(#8961, Cell Signaling Technology Inc., Danvers, MA). Retinal sections were imaged using a Nikon A1R HD25 confocal microscope with a Plano Apo 20 × NA 0.75 objective and compiled and
quantified using ImageJ software. KC and IP-10 labeling quantifications are the result of positive KC and IP-10 labeling within the superior, inner and deep vascular retinal plexi (n = 6
animals per group, each symbol in the plots represents the average of three retinal sections per animal). Cone arrestin labeling quantification is the result of positive cone arrestin
labeling within the outer/inner segments and outer nuclear layer (n = 8 per group, 3 retinal sections quantified per animal).
Whole retinal extract (excluding RPE, n = 4–9 animals per group) was flash frozen and lysed using a Bio-Plex cell lysis kit (Bio-Rad, Fort Worth, TX) according to manufacturer’s protocol,
with the addition of one cOmplete mini (Roche, San Jose, CA) protease inhibitor per 5 mL of buffer. Lysates were placed on an end-over-end rotator for 20 min and centrifuged at 13.2kRPM.
Protein concentrations were determined using a Pierce BCA Protein Assay (Thermo Fisher, Waltham, MA). Samples were normalized to a concentration of 0.75 μg in 37.5 μL of Milliplex® MAP Assay
Buffer (EMD Millipore, Burlington, MA) because this loading fell within a linear range of detectable analytes38. Multiplexed cytokine quantification was conducted using the Milliplex® MAP
Mouse Cytokine/Chemokine 32-Plex Kit (Eotaxin*, G-CSF, GM-CSF*, IFN-γ*, IL-1α, IL-1β*, IL-2, IL-3*, IL-4*, IL-5*, IL-6*, IL-7*, IL-9*, IL-10*, IL-12p40*, IL-12p70*, IL-13, IL-15*, IL-17*,
IP-10, KC, LIF*, LIX*, MCP-1*, M-CSF*, MIG, MIP-1α, MIP-1β*, MIP-2, RANTES, TNF-α, and VEGF) (EMD Millipore) and read on a MAGPIX® system (Luminex, Austin, TX). Cytokines marked with an
asterisk did not fall within a linear range of fluorescent intensity vs. protein concentration and were therefore excluded from the analysis.
Sample size was determined based on our previously reported data16,17,19. The observers who analyzed the data were blinded to the experimental procedures and were masked to the specific
treatment group from which sampling arose. This included semi-automated marking of ERG a- and b-waves (Matlab 2021b, Mathworks, Natick, MA), image acquisition, semi-automated quantification
of KC and IP-10 fluorescence labeling in retinal sagittal sections (ImageJ software). Statistical analyses were performed using Graphpad Prism 9.0.0 (San Diego, CA). Two-way ANOVAs dividing
groups by exercise and light exposure to test the interactive effects of the independent variables and Tukey’s multiple comparison tests were performed on all visual and retinal functional
data, ONL counts, cone photoreceptor counts, and fluorescence quantification and are presented as mean ± standard error of the mean (SEM). A Pearson correlation coefficient was computed to
assess the linear relationship between individual animals KC and IP-10 expression, retinal function (scotopic a- and b-wave function and photopic b-wave function) and visual function
(spatial frequency and contrast sensitivity). Multiplex cytokine data were analyzed using both univariate and multivariate techniques. For univariate analysis, active vs inactive samples
were compared using an unpaired t-test with Welch’s correction to account for differing sample sizes among the groups. To account for co-variance among cytokines, we also conducted a partial
least squares discriminant analysis (PLSDA) using the ropls package in R (Bioconductor39,40). To ensure the results of the PLSDA were robust to a subset of the samples (mice), we conducted
a leave-n-out cross validation computing 100 iterations of PLSDA while each time leaving out four random samples (~ 15% of samples) then computed the standard deviation of loadings across
all iterations.
All p-values lower than 0.05 were considered statistically significant. The ROUT method (with Q set to 1%) was used to detect outliers, of which none were detected.
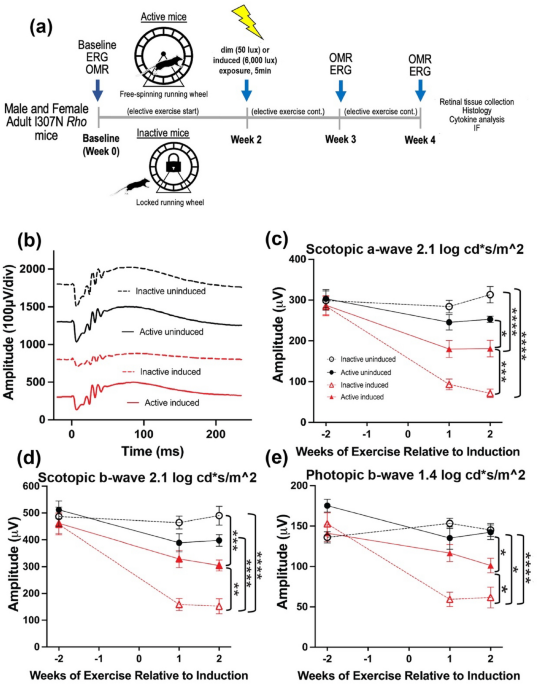
Electroretinography (ERG) was performed on all experimental groups to assess the effect of voluntary exercise on retinal function preservation in younger I307N Rho mice (Fig. 1a–e, Table 1).
Similar to our previous work, active induced mice undergoing retinal degeneration had statistically significant preservation of retinal function with 1.44 × greater scotopic a-wave
amplitudes (Fig. 1c, two-way ANOVA, F(3, 126) = 32.12, p










