- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Cigarette smoking is a risk factor for stroke and is linked to stroke severity. Previous studies have shown that cigarette smoke extract (CSE) triggers endothelial dysfunction _in
vitro_ by initiating oxidative stress and/or an inflammatory response. In addition, cerebral endothelial dysfunction (particularly at the level of the blood-brain barrier [BBB]) contributes
to stroke pathogenesis. Therefore, we hypothesized that cigarette smoking may influence stroke, at least in part, by exacerbating ischaemia-induced BBB disruption. To test this, we examined
the effect of CSE on the permeability of cerebral endothelial cells exposed to oxygen glucose deprivation and reoxygenation (OGD + RO). We found that the loss of BBB integrity following
ischaemic/reperfusion-like conditions was significantly worsened by CSE. Despite this being associated with increased mRNA expression of Nox catalytic subunits, reactive oxygen species (ROS)
levels were however markedly lower. Furthermore, this occurred in association with elevated expression of antioxidant enzymes (SOD1, SOD2, and Gpx-1), suggesting an antioxidant defence
response. Lastly, we found that CSE significantly upregulated mRNA expression of cytokines (IL-6 and TGF-β). Collectively, these results show that acute exposure to CSE worsens BBB
disruption caused by OGD + RO, however, this is not linked to elevated ROS levels but may involve inflammatory mechanisms. SIMILAR CONTENT BEING VIEWED BY OTHERS SALVIANOLIC ACID A PREVENTED
CEREBROVASCULAR ENDOTHELIAL INJURY CAUSED BY ACUTE ISCHEMIC STROKE THROUGH INHIBITING THE SRC SIGNALING PATHWAY Article 10 December 2020 MITOCHONDRIAL DNA IS A KEY DRIVER IN CIGARETTE SMOKE
EXTRACT-INDUCED IL-6 EXPRESSION Article 17 October 2023 NRF2 ACTIVATION AMELIORATES BLOOD–BRAIN BARRIER INJURY AFTER CEREBRAL ISCHEMIC STROKE BY REGULATING FERROPTOSIS AND INFLAMMATION
Article Open access 04 March 2024 INTRODUCTION Cigarette smoking is a major cause of morbidity and mortality worldwide1. In addition to causing various cancers2, cigarette smoking is linked
to the pathogenesis of a number of diseases that affect the brain. For example, smoking is a risk factor for ischaemic and haemorrhagic stroke3, and silent cerebral infarction4. Cigarette
smoking is also a major cause of chronic obstructive pulmonary disease (COPD), accounting for more than 95% of cases in industrialized countries5. Recent studies have shown that strokes are
more prevalent in COPD patients compared with the general population6,7,8, and clinical studies have revealed that COPD is linked to worse stroke outcomes including mortality9,10. Given
smoking is an established risk factor for stroke and is linked to worse stroke outcomes the association between COPD and stroke may be largely dependent on exposure to cigarette smoke6.
Cigarette smoke contains over 4,000 chemicals including nicotine and reactive oxygen species (ROS, e.g. superoxide, hydrogen peroxide)11. These oxidants give rise to secondary ROS by
inflammatory cells within the lung as part of an inflammatory-immune response towards a pathogen or irritant6. These inflammatory cells also have an impaired phagocytic function, resulting
in impairment in clearance of apoptotic cells, contributing to the chronic inflammatory state in the lungs and leading to an ongoing cycle of damage and remodelling in the airways and lung
tissue12,13. In addition to triggering local oxidative stress and inflammation in the lungs, the chemicals found in cigarette smoke can pass through the lung and initiate oxidative stress
and inflammation in cells remote from the primary site of exposure14,15. At the level of the cerebral vasculature this can in turn trigger cerebral endothelial dysfunction6,16. Indeed,
exposure of rats to cigarette smoke impairs endothelial-dependent vasodilator responses of cerebral arterioles _in vivo_ by activating the Nox-NADPH oxidases17, ROS generating enzymes that
are major contributors to cerebral endothelial dysfunction in numerous disease states including stroke16,18. Furthermore, using cigarette smoke extract (CSE) to mimic physiological
concentrations of heavy smokers, several studies have shown that cigarette smoking triggers blood-brain barrier (BBB) disruption _in vitro_ via oxidative and inflammatory mechanisms19,20.
Given cerebral endothelial dysfunction, particularly at the level of the BBB, is implicated in stroke pathogenesis21, it is conceivable that cigarette smoking may influence stroke, at least
in part, by exacerbating ischaemia-induced BBB disruption. Therefore, the aim of this study was to examine whether CSE worsens BBB disruption using a well-established _in vitro_ BBB stroke
model, and to determine whether this is associated with elevated ROS production and/or inflammation. METHODS CIGARETTE SMOKE EXTRACT (CSE) PREPARATION CSE was prepared as previously
described22. Briefly, this involved using one filtered Winfield Original Red cigarette (1.2 mg of nicotine, 16 mg of tar, 15 mg of CO). The cigarette was lit and using a 30 ml syringe
cigarette smoke was bubbled (flow rate of 3 mL/second) into 25 mL of culture media (Dulbecco’s modified Eagle’s medium [DMEM] media). This process was repeated until the cigarette had burned
through just prior to the filter. The resultant solution was defined as 100% CSE. 100% CSE was then filtered before being diluted in media. CSE was utilised within 15–30 minutes after
preparation. CULTURE OF MOUSE CEREBRAL MICROVASCULAR ENDOTHELIAL CELLS Mouse microvascular cerebral endothelial cells (bEnd.3 cells; ATCC CRL-2299) were grown in DMEM media (containing 10%
fetal bovine serum [FBS]) at 37 °C in a humidified 5% CO2 atmosphere23. Cells were passaged every 3–4 days. Culture media was changed after 24 h of passaging and every 2 days thereafter.
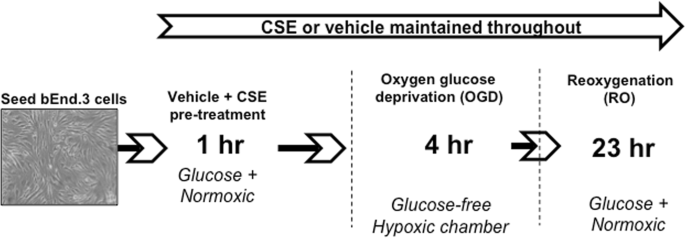
Experiments were performed with cells from passages 26 to 34. OXYGEN GLUCOSE DEPRIVATION (OGD) AND REOXYGENATION (RO) OF BEND.3 CELLS bEnd.3 cells were seeded at a density of 7 × 104
cells/cm2 in 96-well plates or T75 tissue culture flasks (Greiner Bio-One), or at 4 × 104 cells/well in tissue culture inserts (translucent polyethylene terephthalate [PET], 0.4 μm pore
size; Greiner Bio-One) and grown to confluence. Two-days post-confluent cells were washed twice with DMEM glucose-containing media to remove culture media, containing FBS, and replaced with
either CSE (5%, 10%, 20% or 40%) diluted in DMEM glucose-containing media or media alone (vehicle). Cells were incubated for 1 h at 37 °C (5% CO2 atmosphere), washed twice with DMEM
glucose-free media pre-equilibrated in OGD gas mixture for 5 mins (95% N2 and 5% CO2). Cells were then incubated for 4 h in a humidified hypoxia chamber (Biospherix, Lacona, USA; 95% N2, 5%
CO2) in either OGD media containing CSE (5, 10, 20 or 40%) or OGD media alone (vehicle) (Fig. 1)23. A digital oxygen controller maintained the oxygen level at 0.3% and CO2 at 5% for the
duration of the experiment. After 4 h of OGD, media was replaced with either vehicle or CSE (5%, 10%, 20% or 40%) diluted in glucose-containing, serum-free DMEM media (oxygenated with air)
for a further 23 h incubation at 37 °C (5% CO2 atmosphere) (Fig. 1). For each OGD + RO experiment, time-controlled normoxic controls were run alongside by incubating cells for 27 h in CSE
(5%, 10%, 20% or 40%) diluted in glucose-containing, serum-free DMEM media or media alone (vehicle) at 37 °C (5% CO2 atmosphere). MEASUREMENT OF CELL VIABILITY OF BEND.3 CELLS Morphological
changes in bEnd.3 cells were assessed using phase contrast imaging. Briefly, bEnd.3 cells were seeded into 12-well plates and exposed to either normoxic or OGD + RO conditions, as described
above, and treated with either vehicle or CSE (5, 10, 20 or 40%). Cells were then imaged using a microscope (Nikon Eclipse TS100, Japan) at ×100 magnification and image capture camera
(Infinity 3 Lumen_era_, Canada). To measure cell viability a 3-(4,5 dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) assay (Promega, Australia)
was performed as previously described23. bEnd.3 cells were seeded into clear 96-well plates (Greiner, Bio-One) and exposed to OGD + RO or normoxic conditions with either vehicle or CSE (5,
10, 20 or 40%) treatment, as described above. Following RO, bEnd.3 cells were washed twice with pre-warmed Krebs-HEPES buffer [composed of (in mmol/L) 99 NaCl, 4.7 KCl, 1.2 MgSO4, 1.0
KH2PO4, 19.6 NaHCO3, 11.2 Glucose 20 Na-HEPES and 2.5 CaCl2, pH = 7.4] prior to incubating with the MTS solution for 1 h at 37 °C (5% CO2). A BMG Clariostar plate reader (BMG Labtech,
Germany) was used to measure absorbance at a wavelength of 490 nm. MEASUREMENT OF PARACELLULAR PERMEABILITY OF BEND.3 CELLS Paracellular permeability was assessed by measuring the diffusion
of 70 kDa fluorescein isothiocynate (FITC)-dextran (Sigma) across monolayer cultures of bEnd.3 cells, as previously described23,24. Tissue culture inserts (0.4 μm pore size; Greiner Bio-One)
were fitted into 24-well culture plates (Greiner Bio-One) and then pre-soaked with culture media in both the upper ‘apical’ chamber and lower chamber 1 h prior to seeding. bEnd.3 cells were
subsequently seeded into the upper chamber in culture media as above. Culture media was replaced at this time and after 24 hours. Two-days post-confluent, bEnd.3 cells were exposed to OGD +
RO or normoxic conditions with either vehicle or CSE (10%) treatment, as described above. Following RO, pre-warmed Krebs-HEPES buffer was replaced in the lower chamber of fresh wells and
inserts transferred into these wells. Both the upper and lower chambers were then washed with pre-warmed Krebs-HEPES buffer twice and then FITC-dextran (1 mg/mL) was added to the upper
chamber and left to incubate for 1 h at 37 °C (5% CO2). Duplicate samples from the lower chamber were removed and transferred into a black 96-well plate (PerkinElmer, Australia). Using a BMG
Clariostar plate reader, fluorescence intensity was measured at an excitation wavelength of 492 nm and emission wavelength of 518 nm. MEASUREMENT OF SUPEROXIDE LEVELS IN BEND.3 CELLS bEnd.3
cells were seeded into a white 96-well plate (PerkinElmer) and exposed to OGD + RO or normoxic conditions with either vehicle or CSE (5, 10, 20 or 40%) treatment, as described above.
Following RO, L-012 (100 μmol/L)24,25 was added to each well and photon counts measured using a BMG Clariostar plate reader (45 cycles, 3 seconds per well). MEASUREMENT OF HYDROGEN PEROXIDE
PRODUCTION BY BEND.3 CELLS bEnd.3 cells were seeded into a black 96-well plate (PerkinElmer) and exposed to OGD + RO or normoxic conditions with either vehicle or CSE (10%) treatment, as
described above. Cells were treated with 15 μmol/L Amplex Red® reagent (10H-Phenoxazine-3,7-diol, 10-acetyl- 119171-73-2) and 0.1 U/mL horseradish peroxidase (HRP) (Thermofisher
Scientific)25. Amplex red and HRP were also added to hydrogen peroxide standards (0.000, 0.078, 0.156, 0.312, 0.625 and 1.25 μmol/L). Fluorescence was then measured using a BMG Clariostar
plate reader (121 cycles, 40 s per well) at emission filter 590 nm and excitation filter 530 nm. RT-PCR bEnd.3 cells were seeded into T75 cell culture flasks (Greiner Bio-One). Two-days
post-confluent bEnd.3 cells were exposed to OGD + RO or normoxic conditions with vehicle or CSE (10%) treatment, as described above. Following RO, flasks were washed with 0.01 M phosphate
buffered saline (PBS) twice and cells scraped off with 0.01 M PBS. RNA was extracted using the Qiagen RNase-free DNase kit (Qiagen) as per manufacturer’s instructions. Both the purity and
yield of RNA were quantified using a Nanodrop 2000 spectrophotometer (Thermofisher Scientific). cDNA was synthesised using a QuantiTect Reverse Transcription kit (Qiagen). QuantiFast SYBR®
Green primers were used to measure Nox1 (NM_172203), Nox2 (NM_007807) and Nox4 (NM_015760), SOD1 (NM_011434), SOD2 (NM_013671), SOD3 (NM_011435), Gpx-1 (NM_008160), and the housekeeping gene
18S (NR_003278). TaqMan® Universal Master Mix (Applied Biosystems) and mouse-specific TaqMan® Gene Expression assays (Applied Biosystems) were used to measure IL-4 (NM_021283.3), IL-1β
(NM_008361.3), TNF-α (NM_013693.3), IFN-γ (NM_008337.3), IL-6 (NM_031168.1), TGF-β (NM_011577.1) and the housekeeping gene 18S (NR_003278.3) via a QuantStudioTM 7 Flex Real-Time PCR System
(Applied Biosystems). Data were normalized to ribosomal 18s and represented relative to expression levels in normoxic vehicle control samples using the 2−ΔΔCt method26. DATA AND STATISTICAL
ANALYSES All statistical analysis was performed using GraphPad Prism 6 (GraphPad Software, Version 6.07, La Jolla, CA, USA). Results are represented as mean ± SEM and a _P_ < 0.05 was
considered to be statistically significant. Normoxic groups were compared using a Student’s unpaired t-test. A one-way ANOVA with a Bonferroni post-hoc test was performed for OGD + RO groups
for FITC-dextran passage, cell viability, superoxide production, hydrogen peroxide production, and mRNA expression. RESULTS EFFECT OF CSE ON CELL VIABILITY OF BEND.3 CELLS Treatment of
normoxic or OGD + RO bEnd.3 cells with either 5 or 10% CSE resulted in no obvious morphological changes compared with vehicle-treated cells (Fig. 2A). However, 20% or 40% CSE resulted in a
loss of their typical spindle-shape appearance as well as visible cell retraction (Fig. 2A). 5, 10, or 20% CSE had no significant effect on the viability of either normoxic or OGD + RO
bEnd.3 cells relative to vehicle-treated cells (Fig. 2B,C, _P_ > 0.05, n = 6–8), whereas 40% CSE decreased cell viability by ~50% (Fig. 2B,C, _P_ < 0.05). EFFECT OF CSE ON PARACELLULAR
PERMEABILITY OF BEND.3 CELLS Treatment of normoxic bEnd.3 cells with 10% CSE significantly increased FITC-dextran passage (~2 fold) and thus endothelial cell paracellular permeability,
relative to vehicle-treated normoxic cells (Fig. 3A, _P_ < 0.05, n = 6–8). Exposure of bEnd.3 cells to OGD + RO significantly increased paracellular permeability (~4-fold) relative to
normoxic controls (Fig. 3B, _P_ < 0.05, n = 6–8). Furthermore, pre-treatment of cells with 10% CSE exacerbated (~2-fold) this hyperpermeability relative to vehicle-treated OGD + RO cells
(Fig. 3B, _P_ < 0.05, n = 8). EFFECT OF CSE ON SUPEROXIDE AND HYDROGEN PEROXIDE LEVELS IN BEND.3 CELLS Using L-012 chemiluminescence and the Amplex Red fluorescence assay, we next
assessed whether the effect of CSE on BBB permeability is associated with alterations in the production of superoxide and/or hydrogen peroxide, which both influence BBB integrity. Treatment
of normoxic bEnd.3 cells with 5% CSE had no significant effect on superoxide levels (Fig. 4A, _P_ > 0.05, n = 5), whereas superoxide levels were significantly lower in cells treated with
10% or 20% CSE (Fig. 4A, _P_ < 0.05, n = 6). Treatment of normoxic bEnd.3 cells with 10% CSE had no significant effect on hydrogen peroxide levels (Fig. 4C, _P_ > 0.05, n = 5).
Consistent with our previous work24, superoxide levels in bEnd.3 cells were markedly elevated (~47%) after exposure to OGD + RO when compared with normoxic controls (Fig. 4B, _P_ < 0.05,
n = 6). Furthermore, there was a trend for hydrogen peroxide levels to also be elevated (Fig. 4D, _P_ > 0.05, n = 5). By contrast, superoxide levels were not elevated after OGD + RO in
cells treated with either 10% or 20%. In fact, superoxide levels were significantly lower compared with vehicle-treated OGD + RO cells (Fig. 4B, _P_ < 0.05, n = 6). Hydrogen peroxide
levels were also significantly lower in cells treated with 10% CSE relative to vehicle-treated OGD + RO cells (Fig. 4D, _P_ < 0.05, n = 5). EFFECT OF CSE ON MRNA EXPRESSION OF NOX
CATALYTIC SUBUNITS, SOD, AND GPX-1 Using RT-PCR, we next assessed whether the effect of CSE on ROS levels is associated with altered expression levels of Nox oxidase catalytic subunits
(Nox1, Nox2, and Nox4) and/or antioxidant enzymes (SOD1-3, Gpx-1). 10% CSE significantly increased Nox1, Nox4, SOD1, SOD2, and Gpx-1 mRNA expression in normoxic bEnd.3 cells relative to
vehicle-treated cells (Figs 5A,C, 6A,B,D, _P_ < 0.05, n = 8), whereas Nox2 and SOD3 expression levels were comparable between the two groups (Figs 5B & 6C, _P_ > 0.05, n = 8).
Exposure of bEnd.3 cells to OGD + RO increased Nox1 and Nox4 mRNA expression relative to normoxic controls (Fig. 5D,F, _P_ < 0.05, n = 8), but had no significant effect on expression
levels of the SODs and Gpx-1 (Fig. 6E–H, _P_ > 0.05, n = 8). 10% CSE significantly increased Nox1 and Nox4 expression in OGD + RO cells relative to normoxic controls, and there was a
trend for an increase in expression levels of the three Nox isoforms relative to vehicle-treated OGD + RO cells (Fig. 5D–F, _P_ > 0.05, n = 8). 10% CSE also significantly increased
expression of SOD1, SOD2, and Gpx-1 in OGD + RO cells relative to vehicle treated normoxic, and OGD + RO cells (Fig. 6E,F,H, _P_ < 0.05, n = 8). By contrast, Nox2 and SOD3 expression were
comparable between groups (Figs 5E and 6G, _P_ > 0.05, n = 8). EFFECT OF CSE ON MRNA EXPRESSION OF INFLAMMATORY CYTOKINES CSE (10%) caused a significant increase in TGF-β mRNA expression
(Fig. 7A) and a trend for an increase in IL-6 mRNA expression (Fig. 7B) in normoxic cells. However, CSE significantly increased TGF-β and IL-6 mRNA expression in OGD + RO cells (Fig. 7C,D,
_P_ < 0.05, n = 6). IL-4, IL-1β, TNF-α, and IFN-γ mRNA were undetectable in all normoxic and OGD + RO experimental groups (data not shown). DISCUSSION This study aimed to determine
whether CSE worsens ischaemia-induced BBB disruption _in vitro_ by triggering oxidative stress and/or inflammation. We found that the loss of BBB integrity following
ischaemic/reperfusion-like conditions (OGD + RO) was indeed significantly worsened by CSE; however, contrary to our hypothesis this was not associated with elevated ROS levels. In fact,
levels of superoxide and hydrogen peroxide were markedly lower in response to CSE, possibly because of upregulated antioxidant defence. Lastly, CSE increased the expression of
pro-inflammatory cytokines in cerebral endothelial cells exposed to ischaemic/reperfusion-like conditions. Collectively, these results suggest that acute cigarette smoke exposure worsens BBB
disruption after stroke potentially via inflammatory mechanisms. Whilst _in vivo_ studies are needed, these findings raise the possibility that the increased stroke risk and severity in
smokers (and potentially COPD patients) might relate, at least in part, to the deleterious effect of cigarette smoke on BBB integrity. Cigarette smoking is a risk factor for stroke and is
linked to worse stroke outcomes27. Findings from this _in vitro_ study and previously published work17,19,20 show that CSE, which contain the soluble components of cigarette smoke, causes
dysfunction of the cerebrovascular endothelium (in the absence of ischaemia-like conditions). Indeed, using a concentration of CSE that does not induce overt endothelial cell death, we show
here that acute exposure of monolayers of normoxic cerebral endothelial cells to CSE markedly increased paracellular permeability, indicative of BBB disruption. These findings are consistent
with studies showing that CSE causes a loss of BBB integrity in monolayers of human cerebral endothelial cells (hCMEC/D3) in association with altered expression of tight junction proteins
(e.g. ZO-1, occludin, VE-cadherin and claudin-5)17,19,20. Cerebral endothelial dysfunction (particularly at the level of the BBB) contributes to and exacerbates ischaemic brain injury28.
Therefore, the primary focus of this study was to test whether cigarette smoking influences stroke pathogenesis, at least in part, by worsening ischaemia-induced BBB disruption. We tested
this using a well characterised _in vitro_ BBB stroke model, which produces many of the key features of BBB disruption _in vivo_ including paracellular hyperpermeability, tight junction
protein degradation, and MMP-expression23,24. Indeed, in this study we found that exposure of cerebral endothelial cells to OGD + RO caused a marked loss of BBB integrity. The new finding
from these experiments however was that the BBB disruption following ischaemic/reperfusion-like conditions was significantly worsened by prior treatment with CSE. Together these results
strengthen evidence that acute cigarette smoking triggers BBB disruption _per se_, and show for the first time that this in turn might exacerbate the loss of BBB function and integrity after
stroke. Oxidative stress and inflammation both contribute to the complex mechanisms of BBB disruption after cerebral ischaemia and reperfusion. Cigarette smoke contains over 4000 chemical
compounds including ROS, reactive nitrogen species (RNS), nitric oxide (NO), and free radicals of organic compounds11. In addition to these relatively short-lived ROS/RNS, cigarette smoke
contains more stable substances that have the potential to increase ROS production including saturated and unsaturated aldehydes, unsaturated ketones, and nicotine29,30. Some of these
compounds have been shown to react with thiol groups involved in the regulation of enzymes such as the ROS generating Nox oxidases29,30. In addition to triggering oxidative stress and
inflammation in the lungs, oxidants and more stable compounds can pass into the circulation and initiate oxidative stress and inflammation in cells and tissues remote from the primary site
of exposure6. Indeed, previous studies have shown that CSE elevates ROS levels in various cell types (e.g. neurons, glial, and endothelial and vascular smooth muscle cells of systemic
origin) via Nox oxidase activation, enzymes that a major sources of ROS in cerebral vessels25,31,32. We therefore postulated that CSE worsens BBB disruption after OGD + RO by augmenting ROS
levels and/or triggering an inflammatory response. Contrary to our hypothesis, although superoxide levels were elevated in control cells after OGD + RO, we found that prior treatment with
CSE did not elevate levels further. In fact, despite evidence for increased gene expression of Nox1 and Nox4 catalytic subunits, superoxide levels in CSE treated cells after OGD + RO were in
fact ~50% lower than controls. Consistent with this lower level of superoxide, hydrogen peroxide (the downstream metabolite of superoxide) levels were similarly lower in CSE treated cells
after OGD + RO. Thus, these _in vitro_ findings suggest that cigarette smoke exposure does not trigger or worsen BBB disruption by elevating ROS levels in cerebral endothelial cells. Some
studies have shown that antioxidant defence mechanisms are upregulated in endothelial cells in response to exposure to CSE. For example, acute exposure of human cerebral endothelial cells to
CSE upregulates the expression of nuclear-factor (erythroid derived 2) related factor-2 (Nrf2)19. Nrf2 is one of the major transcription factors regulating the antioxidant defence response
to oxidative stress, including the upregulation of the enzymes responsible for ROS metabolism (SODs, catalase, and Gpx1)33. Consistent with this, we found evidence of increased expression of
SOD1, SOD2, and Gpx1 in response to CSE. We propose that upregulation of these antioxidant enzymes occurs in response to, and to counteract the oxidative stress caused by cigarette smoking.
It remains to be determined, however, if this protection is sustained over longer periods of cigarette smoke exposure or whether the oxidative stimuli in cigarette smoke overcomes the
protective antioxidant mechanisms of cerebral endothelial cells. We then went on to explore whether the worsening of CSE-induced BBB disruption was associated with increased inflammatory
gene expression. We found that IL-4, IL-1β, TNF-α, and IFN-γ mRNA were all undetectable. Similarly, a study found that IL-1β and TNF-α levels were below the level of detection (ELISA) in
hCMEC/D3 cells following exposure to CSE34. In contrast to these cytokines, we found that IL-6 and TGF-β were significantly greater in CSE-treated cerebral endothelial cells after OGD + RO.
However, whether this translates to increased cytokine levels remains to be determined. Furthermore, future experiments should be performed to determine whether elevated gene expression of
IL-6 and TGF-β in response to CSE leads to increased cytokine-mediated signaling, e.g. phosphorylation of TGF-β receptor-regulated Smad proteins. Our findings are however consistent with
previous studies showing that CSE promotes cytokine production (e.g. IL-6, IL-8, TGF-β) and the expression of adhesion molecules (e.g. vascular endothelial adhesion molecule-1, platelet
endothelial cell adhesion molecule-1) in endothelial cells19,34. The roles of IL-6 and TGF-β in ischaemic stroke and BBB disruption are however not fully elucidated. IL-6 and TGF-β have been
shown to increase the paracellular permeability of monolayers of cerebral endothelial cells _in vitro_ by triggering a loss of tight junction proteins35,36. Consistent with this, inhibiting
the effects of IL-6 with neutralizing antibodies attenuates ischaemia-induced BBB disruption in a hypoxic-ischaemic brain injury model37. By contrast, IL-6 has been reported to support the
integrity of the BBB in a rat model of cerebral ischaemia38. Thus, future studies are needed to fully determine whether these cytokines contribute to the deleterious effects of cigarette
smoke exposure on BBB integrity. This study used a well-characterised and utilized immortalised cerebral endothelial cell line which displays many of the key features of the BBB such as the
expression of key tight junction proteins, as well as the expression of key endothelial cell markers23. Monolayers of bEnd.3 cells are known to exhibit moderate paracellular permeability to
high molecular weight molecules and have TEER readings ranging from 100–140 Ω cm2 39,40, which is similar to that of primary cultures from several species (50–180 Ω cm2)41. Furthermore,
bEnd.3 cells have the advantage of being cost effective compared with immortalised human cell lines or primary cerebral endothelial cells. Nevertheless, primary cerebral endothelial cells
have the closest similarity to the BBB phenotype _in vivo_ at low passage numbers. Thus, future studies should be performed to validate our findings in monolayers of primary cerebral
endothelial cells, and/or using an immortalised human cerebral endothelial cell line. In summary, the study found that cigarette smoke exposure worsens ischaemia-induced BBB disruption.
Whilst cigarette smoke exposure increased the gene expression of Nox oxidases this did not translate to elevated levels of either superoxide or its downstream metabolite hydrogen peroxide,
possibly because of upregulated antioxidant defence. Future studies should examine whether this protection is sustained chronically or whether cigarette smoke exposure overcomes the
protective antioxidant mechanisms of cerebral endothelial cells. Lastly, we found evidence of increased proinflammatory gene expression in response to cigarette smoke exposure, suggesting
the involvement of inflammatory mechanisms. The deleterious effect of CSE on BBB integrity might be consistent with the occurrence of a biological process known as endothelial to mesenchymal
transition (EndoMT). Similar to airway epithelial to mesenchymal transition (EMT), EndoMT is a complex process whereby endothelial cells lose their specific markers such as the adheren
junction protein VE-cadherins42,43, which is important for maintaining BBB integrity. Under chronic oxidative damage and inflammation (notably TGF-β) EndoMT can be initiated42,44. EMT is an
active process in smokers and COPD patients45,46. Moreover, there is evidence that EndoMT might also be active in the pulmonary vasculature during COPD47. Thus, future studies could explore
whether CSE initiates EndoMT in the cerebral endothelium and whether such a process contributes to the deleterious effects of CSE on BBB integrity. In conclusion, although future _in vivo_
studies are needed, our findings raise the possibility that the deleterious effect of cigarette smoke on BBB integrity may contribute to stroke risk and severity in smokers and potentially
COPD patients. DATA AVAILABILITY All data generated or analysed during this study are included in this published article. REFERENCES * Britton, J. Death, disease, and tobacco. _Lancet_ 389,
1861–1862, https://doi.org/10.1016/S0140-6736(17)30867-X (2017). Article PubMed Google Scholar * Brown, K. F. _et al_. The fraction of cancer attributable to modifiable risk factors in
England, Wales, Scotland, Northern Ireland, and the United Kingdom in 2015. _Br J Cancer_ 118, 1130–1141, https://doi.org/10.1038/s41416-018-0029-6 (2018). Article PubMed PubMed Central
Google Scholar * Mannami, T. _et al_. Cigarette smoking and risk of stroke and its subtypes among middle-aged Japanese men and women: the JPHC Study Cohort I. _Stroke_ 35, 1248–1253,
https://doi.org/10.1161/01.STR.0000128794.30660.e8 (2004). Article PubMed Google Scholar * Howard, G. _et al_. Cigarette smoking and other risk factors for silent cerebral infarction in
the general population. _Stroke_ 29, 913–917 (1998). Article CAS PubMed Google Scholar * Barnes, P. J., Shapiro, S. D. & Pauwels, R. A. Chronic obstructive pulmonary disease:
molecular and cellular mechanisms. _Eur Respir J_ 22, 672–688 (2003). Article CAS PubMed Google Scholar * Austin, V., Crack, P. J., Bozinovski, S., Miller, A. A. & Vlahos, R. COPD
and stroke: are systemic inflammation and oxidative stress the missing links? _Clin Sci (Lond)_ 130, 1039–1050, https://doi.org/10.1042/CS20160043 (2016). Article Google Scholar *
Kunisaki, K. M. _et al_. Exacerbations of Chronic Obstructive Pulmonary Disease and Cardiac Events. A Post Hoc Cohort Analysis from the SUMMIT Randomized Clinical Trial. _Am J Respir Crit
Care Med_ 198, 51–57, https://doi.org/10.1164/rccm.201711-2239OC (2018). Article CAS PubMed PubMed Central Google Scholar * Portegies, M. L. _et al_. Chronic Obstructive Pulmonary
Disease and the Risk of Stroke. The Rotterdam Study. _Am J Respir Crit Care Med_ 193, 251–258, https://doi.org/10.1164/rccm.201505-0962OC (2016). Article CAS PubMed Google Scholar *
Lekoubou, A. & Ovbiagele, B. Prevalance and Influence of Chronic Obstructive Pulmonary Disease on Stroke Outcomes in Hospitalized Stroke Patients. _eNeurologicalSci_ 6, 21–24,
https://doi.org/10.1016/j.ensci.2016.11.007 (2017). Article PubMed Google Scholar * Lin, C. S. _et al_. Risk of Stroke and Post-Stroke Adverse Events in Patients with Exacerbations of
Chronic Obstructive Pulmonary Disease. _PLoS One_ 12, e0169429, https://doi.org/10.1371/journal.pone.0169429 (2017). Article CAS PubMed PubMed Central Google Scholar * Pryor, W. A.
& Stone, K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. _Ann N Y Acad Sci_ 686, 12–27; discussion 27–18 (1993). * Barnes, P. J. Cellular
and molecular mechanisms of chronic obstructive pulmonary disease. _Clin Chest Med_ 35, 71–86, https://doi.org/10.1016/j.ccm.2013.10.004 (2014). Article PubMed Google Scholar * Donnelly,
L. E. & Barnes, P. J. Defective phagocytosis in airways disease. _Chest_ 141, 1055–1062, https://doi.org/10.1378/chest.11-2348 (2012). Article PubMed Google Scholar * Wouters, E. F.
Local and systemic inflammation in chronic obstructive pulmonary disease. _Proc Am Thorac Soc_ 2, 26–33, https://doi.org/10.1513/pats.200408-039MS (2005). Article CAS PubMed Google
Scholar * Rahman, I., Morrison, D., Donaldson, K. & MacNee, W. Systemic oxidative stress in asthma, COPD, and smokers. _Am J Respir Crit Care Med_ 154, 1055–1060,
https://doi.org/10.1164/ajrccm.154.4.8887607 (1996). Article CAS PubMed Google Scholar * Miller, A. A., Budzyn, K. & Sobey, C. G. Vascular dysfunction in cerebrovascular disease:
mechanisms and therapeutic intervention. _Clin Sci (Lond)_ 119, 1–17, https://doi.org/10.1042/CS20090649 (2010). Article CAS Google Scholar * Iida, H., Iida, M., Takenaka, M., Fukuoka, N.
& Dohi, S. Rho-kinase inhibitor and nicotinamide adenine dinucleotide phosphate oxidase inhibitor prevent impairment of endothelium-dependent cerebral vasodilation by acute cigarette
smoking in rats. _J Renin Angiotensin Aldosterone Syst_ 9, 89–94, https://doi.org/10.3317/jraas.2008.012 (2008). Article CAS PubMed Google Scholar * Drummond, G. R., Selemidis, S.,
Griendling, K. K. & Sobey, C. G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. _Nat Rev Drug Discov_ 10, 453–471, https://doi.org/10.1038/nrd3403
(2011). Article CAS PubMed PubMed Central Google Scholar * Prasad, S. _et al_. Impact of cigarette smoke extract and hyperglycemic conditions on blood-brain barrier endothelial cells.
_Fluids Barriers CNS_ 12, 18, https://doi.org/10.1186/s12987-015-0014-x (2015). Article CAS PubMed PubMed Central Google Scholar * Kim, J. H. _et al_. Oxidative Stress Induced by
Cigarette Smoke Extracts in Human Brain Cells (T98G) and Human Brain Microvascular Endothelial Cells (HBMEC) in Mono- and Co-Culture. _J Toxicol Environ Health A_ 78, 1019–1027,
https://doi.org/10.1080/15287394.2015.1043607 (2015). Article CAS PubMed Google Scholar * Jackman, K. & Iadecola, C. Neurovascular regulation in the ischemic brain. _Antioxid Redox
Signal_ 22, 149–160, https://doi.org/10.1089/ars.2013.5669 (2015). Article CAS PubMed PubMed Central Google Scholar * Laan, M., Bozinovski, S. & Anderson, G. P. Cigarette smoke
inhibits lipopolysaccharide-induced production of inflammatory cytokines by suppressing the activation of activator protein-1 in bronchial epithelial cells. _J Immunol_ 173, 4164–4170
(2004). Article CAS PubMed Google Scholar * Ku, J. M. _et al_. Characterisation of a mouse cerebral microvascular endothelial cell line (bEnd.3) after oxygen glucose deprivation and
reoxygenation. _Clin Exp Pharmacol Physiol_ 43, 777–786, https://doi.org/10.1111/1440-1681.12587 (2016). Article CAS PubMed Google Scholar * Ku, J. M. _et al_. Protective actions of
des-acylated ghrelin on brain injury and blood-brain barrier disruption after stroke in mice. _Clin Sci (Lond)_ 130, 1545–1558, https://doi.org/10.1042/CS20160077 (2016). Article CAS
Google Scholar * De Silva, T. M., Broughton, B. R., Drummond, G. R., Sobey, C. G. & Miller, A. A. Gender influences cerebral vascular responses to angiotensin II through Nox2-derived
reactive oxygen species. _Stroke; a journal of cerebral circulation_ 40, 1091–1097, https://doi.org/10.1161/STROKEAHA.108.531707 (2009). Article CAS Google Scholar * Schmittgen, T. D.
& Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. _Nature protocols_ 3, 1101–1108 (2008). Article CAS PubMed Google Scholar * Allen, C. L. &
Bayraktutan, U. Risk factors for ischaemic stroke. _Int J Stroke_ 3, 105–116, https://doi.org/10.1111/j.1747-4949.2008.00187.x (2008). Article PubMed Google Scholar * Gursoy-Ozdemir, Y.,
Yemisci, M. & Dalkara, T. Microvascular protection is essential for successful neuroprotection in stroke. _Journal of neurochemistry_ 123(Suppl 2), 2–11,
https://doi.org/10.1111/j.1471-4159.2012.07938.x (2012). Article CAS PubMed Google Scholar * Stedman, R. L. The chemical composition of tobacco and tobacco smoke. _Chem Rev_ 68, 153–207
(1968). Article CAS PubMed Google Scholar * Jaimes, E. A., DeMaster, E. G., Tian, R. X. & Raij, L. Stable compounds of cigarette smoke induce endothelial superoxide anion production
via NADPH oxidase activation. _Arteriosclerosis, thrombosis, and vascular biology_ 24, 1031–1036, https://doi.org/10.1161/01.ATV.0000127083.88549.58 (2004). Article CAS PubMed Google
Scholar * Miller, A. A., Drummond, G. R., Schmidt, H. H. & Sobey, C. G. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. _Circulation
research_ 97, 1055–1062, https://doi.org/10.1161/01.RES.0000189301.10217.87 (2005). Article CAS PubMed Google Scholar * De Silva, T. M., Brait, V. H., Drummond, G. R., Sobey, C. G. &
Miller, A. A. Nox2 oxidase activity accounts for the oxidative stress and vasomotor dysfunction in mouse cerebral arteries following ischemic stroke. _PloS one_ 6, e28393,
https://doi.org/10.1371/journal.pone.0028393 (2011). Article CAS ADS PubMed PubMed Central Google Scholar * Chen, B., Lu, Y., Chen, Y. & Cheng, J. The role of Nrf2 in oxidative
stress-induced endothelial injuries. _J Endocrinol_ 225, R83–99, https://doi.org/10.1530/JOE-14-0662 (2015). Article CAS PubMed Google Scholar * Naik, P. _et al_. Oxidative and
pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: is smoking reduced or nicotine-free products really safe? _BMC Neurosci_ 15, 51,
https://doi.org/10.1186/1471-2202-15-51 (2014). Article CAS PubMed PubMed Central Google Scholar * Rochfort, K. D., Collins, L. E., Murphy, R. P. & Cummins, P. M. Downregulation of
blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. _PLoS One_ 9,
e101815, https://doi.org/10.1371/journal.pone.0101815 (2014). Article CAS ADS PubMed PubMed Central Google Scholar * Shen, W. _et al_. Tyrosine phosphorylation of VE-cadherin and
claudin-5 is associated with TGF-beta1-induced permeability of centrally derived vascular endothelium. _Eur J Cell Biol_ 90, 323–332, https://doi.org/10.1016/j.ejcb.2010.10.013 (2011).
Article CAS PubMed Google Scholar * Zhang, J. _et al_. Anti-IL-6 neutralizing antibody modulates blood-brain barrier function in the ovine fetus. _FASEB journal: official publication of
the Federation of American Societies for Experimental Biology_ 29, 1739–1753, https://doi.org/10.1096/fj.14-258822 (2015). Article CAS Google Scholar * Feng, Q., Wang, Y. I. & Yang,
Y. Neuroprotective effect of interleukin-6 in a rat model of cerebral ischemia. _Exp Ther Med_ 9, 1695–1701, https://doi.org/10.3892/etm.2015.2363 (2015). Article CAS PubMed PubMed
Central Google Scholar * Zehendner, C. M. _et al_. Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein
disruption. _PLoS One_ 8, e82823, https://doi.org/10.1371/journal.pone.0082823 (2013). Article CAS ADS PubMed PubMed Central Google Scholar * Brown, R. C., Morris, A. P. & O’Neil,
R. G. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. _Brain Res_ 1130, 17–30,
https://doi.org/10.1016/j.brainres.2006.10.083 (2007). Article CAS PubMed Google Scholar * de Vries, H. E. _et al_. The influence of cytokines on the integrity of the blood-brain barrier
_in vitro_. _J Neuroimmunol_ 64, 37–43 (1996). Article PubMed Google Scholar * Eapen, M. S. _et al_. sE-cadherin and sVE-cadherin indicate active epithelial/endothelial to mesenchymal
transition (EMT and EndoMT) in smokers and COPD: implications for new biomarkers and therapeutics. _Biomarkers_ 23, 709–711, https://doi.org/10.1080/1354750X.2018.1479772 (2018). Article
CAS PubMed Google Scholar * Sohal, S. S. Endothelial to mesenchymal transition (EndMT): an active process in Chronic Obstructive Pulmonary Disease (COPD)? _Respir Res_ 17, 20,
https://doi.org/10.1186/s12931-016-0337-4 (2016). Article CAS PubMed PubMed Central Google Scholar * Piera-Velazquez, S., Li, Z. & Jimenez, S. A. Role of endothelial-mesenchymal
transition (EndoMT) in the pathogenesis of fibrotic disorders. _Am J Pathol_ 179, 1074–1080, https://doi.org/10.1016/j.ajpath.2011.06.001 (2011). Article CAS PubMed PubMed Central Google
Scholar * Mahmood, M. Q. _et al_. Epithelial mesenchymal transition in smokers: large versus small airways and relation to airflow obstruction. _Int J Chron Obstruct Pulmon Dis_ 10,
1515–1524, https://doi.org/10.2147/COPD.S81032 (2015). Article CAS PubMed PubMed Central Google Scholar * Sohal, S. S. _et al_. Evaluation of epithelial mesenchymal transition in
patients with chronic obstructive pulmonary disease. _Respir Res_ 12, 130, https://doi.org/10.1186/1465-9921-12-130 (2011). Article CAS PubMed PubMed Central Google Scholar * Reimann,
S. _et al_. Increased S100A4 expression in the vasculature of human COPD lungs and murine model of smoke-induced emphysema. _Respir Res_ 16, 127, https://doi.org/10.1186/s12931-015-0284-5
(2015). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This study was supported was supported by the University of Glasgow Centre of Excellence
Award (RE/13/5/3017) and the National Health and Medical Research Council of Australia [Project Grant ID 1120522]. AUTHOR INFORMATION Author notes * These authors contributed equally: Ross
Vlahos and Alyson A. Miller. AUTHORS AND AFFILIATIONS * School of Health and Biomedical Sciences, RMIT University, PO Box 71, Bundoora, VIC, 3083, Australia Ashton Bernard, Jacqueline M. Ku,
Ross Vlahos & Alyson A. Miller * Institute of Cardiovascular & Medical Sciences, British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, G12
8TA, United Kingdom Ashton Bernard & Alyson A. Miller Authors * Ashton Bernard View author publications You can also search for this author inPubMed Google Scholar * Jacqueline M. Ku
View author publications You can also search for this author inPubMed Google Scholar * Ross Vlahos View author publications You can also search for this author inPubMed Google Scholar *
Alyson A. Miller View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS R.V. and A.A.M. conceived the study design. A.B., J.K., R.V. and A.A.M.
designed the experiments. A.B. and J.K. acquired the data. A.B., J.K., R.V. and A.A.M. analysed and interpreted the data. All authors drafted and reviewed the manuscript. CORRESPONDING
AUTHOR Correspondence to Alyson A. Miller. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Bernard, A., Ku, J.M., Vlahos, R. _et al._ Cigarette smoke extract exacerbates
hyperpermeability of cerebral endothelial cells after oxygen glucose deprivation and reoxygenation. _Sci Rep_ 9, 15573 (2019). https://doi.org/10.1038/s41598-019-51728-2 Download citation *
Received: 02 April 2019 * Accepted: 12 September 2019 * Published: 30 October 2019 * DOI: https://doi.org/10.1038/s41598-019-51728-2 SHARE THIS ARTICLE Anyone you share the following link
with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative










