- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Coronary artery disease (CAD) is the leading cause of death, and genetic factors contribute significantly to risk of CAD. This study aims to identify new CAD genetic loci through a
large-scale linkage analysis of 24 large and multigenerational families with 433 family members (GeneQuest II). All family members were genotyped with markers spaced by every 10 cM and a
model-free nonparametric linkage (NPL-all) analysis was carried out. Two highly significant CAD loci were identified on chromosome 17q21.2 (NPL score of 6.20) and 7p22.2 (NPL score of 5.19).
We also identified four loci with significant NPL scores between 4.09 and 4.99 on 2q33.3, 3q29, 5q13.2 and 9q22.33. Similar analyses in individual families confirmed the six significant CAD
loci and identified seven new highly significant linkages on 9p24.2, 9q34.2, 12q13.13, 15q26.1, 17q22, 20p12.3, and 22q12.1, and two significant loci on 2q11.2 and 11q14.1. Two loci on 3q29
and 9q22.33 were also successfully replicated in our previous linkage analysis of 428 nuclear families. Moreover, two published risk variants, SNP rs46522 in _UBE2Z_ and SNP rs6725887 in
_WDR12_ by GWAS, were found within the 17q21.2 and 2q33.3 loci. These studies lay a foundation for future identification of causative variants and genes for CAD. SIMILAR CONTENT BEING VIEWED
BY OTHERS DISCOVERY AND SYSTEMATIC CHARACTERIZATION OF RISK VARIANTS AND GENES FOR CORONARY ARTERY DISEASE IN OVER A MILLION PARTICIPANTS Article Open access 06 December 2022 LARGE-SCALE
GENOME-WIDE ASSOCIATION STUDY OF CORONARY ARTERY DISEASE IN GENETICALLY DIVERSE POPULATIONS Article 01 August 2022 CHROMOSOME 1Q21.2 AND ADDITIONAL LOCI INFLUENCE RISK OF SPONTANEOUS
CORONARY ARTERY DISSECTION AND MYOCARDIAL INFARCTION Article Open access 04 September 2020 INTRODUCTION Genetic factors contribute to the risk of developing coronary artery disease (CAD) and
its major complication, myocardial infarction (MI), which is the result of the accumulation of atherosclerotic plaques in the walls of the coronary arteries1,2,3. Existing knowledge of
genetic components affecting the risk of CAD is largely based on results from genome-wide association studies (GWAS), a systematic, unbiased and powerful approach to identify
disease-associated variants using population samples. Although the majority of GWAS have focused on European ancestry populations4,5,6,7,8,9,10,11,12,13,14, several GWAS were also reported
in African Americans15, East Asians16,17,18,19,20,21 and South Asians9, 22. Due to newly developed SNP imputation methods23,24,25 based on the HapMap project
(https://www.ncbi.nlm.nih.gov/probe/docs/projhapmap/) and the 1000 Genome project (http://www.internationalgenome.org/), meta-GWAS is becoming a more popular strategy for CAD and other
complex diseases. The largest meta-GWAS recently analyzed 9.4 million imputed SNPs among >185,000 samples and identified 10 novel CAD loci14. To date, there have been 65 independent CAD
susceptibility loci reported at a genome-wide significance level (i.e., _P_ < 5.0 × 10−8). The heritability of CAD has been estimated from 40% to 60% by genetic-epidemiologic studies26.
However, recent studies strongly indicate that GWAS variants cannot fully explain the heritability of CAD, and all published risk variants explained only 10–20% of heritability13, 14, 27.
Genome-wide linkage analysis (GWLA) is another systematic and unbiased approach to identify genetic loci for human complex diseases and to search for evidence of major genetic effects. The
first GWLA for CAD, conducted in 2000, involved an analysis of 156 affected sibling pairs and revealed two genetic loci on chromosomes 2q21.1–22 and Xq23–26 that were linked to premature
CAD28. Since then, over ten GWLAs, including our own studies, have identified additional genetic loci for CAD or MI, including 1p34–36,1q25, 2q14.3, 2q36–37.3, 2q13, 3q13, 5q31, 7p14, 8p22,
13q12–13,14q32.3, 15q26.3, 16p13, and 17p11.2–17q2128,29,30,31,32,33,34,35,36,37,38,39. Recently, we have completed a genome-wide linkage scan in a well characterized U.S GeneQuest cohort
with 428 nuclear families and identified six novel CAD loci on chromosomes 3p25.1, 3p29, 9q22.3, 9p34.11, 17p12, and 21q22.340. In contrast to aforementioned GWASs, the number of genetic
loci identified by GWLA was much smaller and independent, suggesting that many linkage loci remain to be identified in new CAD or MI families40. Most GWLAs for CAD have been conducted in
either single large pedigrees or a large number of nuclear families. Increasing the number of family members within families can improve the power of linkage analysis36, 40, 41. In this
study, we performed a large scale GWLA in a well-characterized U.S. cohort of 24 large, multigenerational CAD families (mean pedigree size = 18). This cohort, referred to as GeneQuest II,
was independent from our previously reported GeneQuest cohort with 428 nuclear families40. The most attractive feature of the GeneQuest II cohort is the inclusion of extended family members
of affected siblings or trios. To our knowledge, this is the largest linkage analysis of multiple large pedigrees to identify genetic loci for CAD, and significant susceptibility loci were
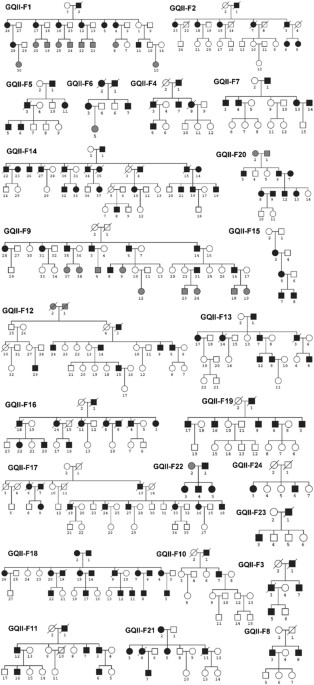
identified. RESULTS CHARACTERISTICS OF 24 LARGE GENEQUEST II FAMILIES The 24 large and multigenerational families with CAD and MI were genetically characterized (Table 1). The pedigrees of
the 24 GeneQuest II families are shown in Fig. 1. 433 family members from the 24 families were included in the linkage analysis. The pedigree size ranged from 5 to 38 members per family, and
the average age of onset of CAD was 51.3 ± 9.2 years in GeneQuest II. There were 162 patients affected with CAD and 247 family members without a diagnosis of CAD. Overall, there were 209
males and 224 females. However, among the CAD group, male patients (107 or 66.0%) were more predominant than female patients (55 or 34.0%). On the contrary, among the non-CAD group, 154
members were females (62.3%). These data are consistent with the notion that the male gender is an important risk factor for CAD. A full set of 410 microsatellite markers spanning the entire
human genome by every 10 cM were initially genotyped for all 433 family members in the 24 CAD families. 36 markers were excluded for further analysis, including 9 autosomal markers with
genotype and pedigree errors and 27 makers on X and Y chromosomes. Therefore, after quality control, 374 microsatellite markers on autosomes 1–22 from 433 family members were subjected to
subsequent statistical analysis. GENOME-WIDE LINKAGE SCANS As shown in Table 2, genome-wide two-point NPL linkage analysis identified two highly significant linkages at markers D7S3056
(7p22.2, NPL score = 5.19) and D17S1299 (17q21.2, NPL score = 6.20), respectively. Three significant linkages were also identified at markers D2S1384 (2q33.3, NPL score = 4.36), GATA138B05
(5q13.2, NPL score = 4.44) and D9S910 (9q22.33, NPL score = 4.54), respectively (Table 2). Multipoint NPL analysis was further performed. Multipoint NPL scores were plotted along the genetic
map for each of 22 chromosomes (Figs 2 and 3). Multipoint NPL analysis identified four significant genetic loci for CAD on chromosomes 17q21.1, 7p22.2, 2q33.3 and 3q29. The top CAD locus on
chromosome 17q21.2 identified by the two-point linkage analysis remained to be a highly significant linkage peak with a NPL score of 5.38 by multipoint NPL analysis. The CAD locus on
17q21.2 covered a genetic interval from 56.9 cM to 83.1 cM (Fig. 3). The second best CAD locus identified by multipoint NPL analysis was on 7p22.2 with a NPL score of 4.74, and the linkage
covered an interval between 1.4 cM and 11.0 cM (Fig. 2). Compared with two-point NPL scores, multipoint NPL scores of the six CAD loci slightly decreased except for the 3q29 locus with an
improved NPL score from 4.00 to 4.49. Moreover, both two-point and multipoint NPL analyses were carried out in individual families. Each of the 6 significant CAD loci was found to occur in
at least one individual family (Table 3). NPL scores in single families were, in general, higher than those in the combined families (Table 3). In addition, this analysis identified 15 new
linkages for CAD, including 7 highly significant linkages on chromosomes 12q13.13, 17q22, 20p12.3, 22q12.1, 15q26.1, 9q34.2, and 9p24.2, 2 significant linkages on 11q14.1 and 2q11.2, and 6
suggestive linkages on 10p15.3, 10q21.3, 2p16.3, 20q13.32, 12q23.1, and 4p16.3 (Table 4). POTENTIAL CAD-RELATED GENES UNDERLYING SIX SIGNIFICANT CAD LOCI To explore candidate genes for CAD
under the six significant genetic loci identified for CAD in the combined GeneQuest II families, we annotated all genes underlying each linkage. Genetic intervals of the six linkages were
converted to physical locations according to the genetic maps generated by the HapMap 2 project (lifted over to hg19). RefSeq genes located under the six linkages were retrieved from the
UCSC database (Tack: RefSeq Genes; Assembly, GRCh37/hg19), and then evaluated for potential relationship with cardiovascular diseases using the online program DisGeNET42, 43. Counts of
RefSeq genes and gene-disease pairs with score of >0.001 are summarized in Table 5. DISCUSSION Identification of new genetic loci for CAD is critical for addressing the important issue of
“missing heritability” in the field of genetics, and in fully elucidating the genetic basis of CAD. In this study, we report a unique genome-wide linkage scan for CAD in 24 large,
multigenerational families from a well-characterized U.S cohort (GeneQuest II). We carried out a model-free NPL-all scan and identified six susceptibility loci for CAD on chromosomes 2q33.3,
3q29, 5q13.2, 7p22.2, 9q22.33 and 17q21.2. It is interesting to note that the 3q29 and 9q22.33 loci were previously identified by us in a genome-wide linkage scan for CAD in 428 nuclear
families in the GeneQuest population40. Suggestive evidence of linkage to the 3q29 CAD locus (_P_ = 2.0 × 10−4) was also found in a meta-analysis of four GWLS in Finnish, Mauritan, Germany,
and Australian cohorts44. Therefore, the present study provides strong validation of the 3q29 and 9q22.33 linkages for CAD using an independent, large family-based linkage scan, suggesting
that these two loci can be prioritized for identifying the underlying causative genes for CAD. Candidate genes for CAD at the 3q29 and 9q22.33 loci are listed in Table 5. There are 31 unique
RefSeq genes annotated within the CAD locus on 3q29. DisGenNET analysis identified 5 genes related to cardiovascular diseases (Table 5). The _UTS2B_ gene encodes Urotensin IIB and was shown
to play a role in the acceleration of atherosclerosis development. Increased human Urotensin II levels were observed in hypertension, diabetes, atherosclerosis and CAD45. There are 20
unique genes within the 9q22.33 locus and three genes (_TGFBR1_, _NR4A3_, _and INVS_) were linked to cardiovascular diseases (Table 5). _TGFBR1_ encodes transforming growth factor beta
receptor 1 (TGFβ1) and an increase in active TGFβ1 levels were correlated with both the occurrence and severity of CAD46. The four other CAD loci on 2q33.3, 5q13.2, 7p22.2 and 17q21.2 are
all novel. The chromosome 17q21.2 linkage is the most significant locus for CAD identified in this study. The 17q21.2 CAD locus was initially identified at marker D17S1299 at the position of
62.01 cM with a two-point NPL score of 6.20 and a multipoint NPL score of 5.38 (Table 2). This CAD locus spans a large genetic interval of 26.2 cM (corresponding to 34.40–57.50 Mb) (Fig.
3). Within the 17q21.2 CAD locus, we found SNP rs46522, which is a CAD-risk variant identified by a large-scale GWAS for CAD in 201312 and located about 8 Mb away from D17S1299. SNP rs46522,
located in the _UBE2Z_-_GIP_-_ADTP5G_ gene cluster, exhibited a strong cis-eQTL (expression quantitative trait locus) to _UBE2Z_ in whole blood samples and to _ATP5G1_ in left ventricle
samples according to the GTEx database v647. On the other hand, we identified a set of 514 unique RefSeq genes within the 17q21.2 CAD locus; 77 of them were linked to cardiovascular diseases
based on data from DisGenNET (Table 5). In particular, _CCL3_ and _CCL4_ encoding small CC chemokines known as macrophage inflammatory protein 1α and 1β, respectively, were well-recognized
as key mediators of both diabetes and atherosclerotic cardiovascular disease48. Elevated expression levels of both _CCL3_ and _CCL4_ were found in atherosclerotic lesions in _ApoE_ −/−
mice49. Leukocyte-derived CCL3 can induce neutrophil chemotaxis toward the atherosclerotic plaque, causing accelerated lesion formation50. _CCL4_ was also upregulated in atherosclerotic
plaques in stroke patients51. _NR1D1_ is also a candidate gene for CAD. It is located 600 kb from marker D17S1299, encodes a member of the nuclear receptor superfamily and regulates genes
involved in triglyceride metabolism, inflammatory and the pathogenesis of atherosclerosis52. NR1D1 can regulate apolipoprotein APOC3 via binding to the proximal promoter53. Future studies
may focus on these strong candidate genes to identify causative genes that contribute to the risk of CAD in families. The second most significant linkage for CAD on 7p22.2 was identified
with marker D7S3056 at a position of 7.44 cM (physical position: 4.49 Mb) with two-point NPL score of 5.19 and a multipoint NPL score of 4.74 (Table 2). This is a novel locus for CAD. No
GWAS variants were found to be located within the 7p22.2 locus. The closest GWAS SNP for CAD was rs2023938 in _HDAC9_, which is located at 7p21.113. There are 87 unique RefSeq genes located
within the 7p22.2 locus (0.88 Mb to 7.22 Mb). DisGenNET analysis identified 9 genes related to cardiovascular diseases (Table 5). _SDK1_ was found to be associated with hypertension in the
Japanese population54. _GPER1_ encodes a multi-pass membrane protein that is localized to the endoplasmic reticulum and _Gper1_ knockout mice showed increased atherosclerosis progress and
vascular inflammation55, 56. The 2q33.3 locus, represented by marker D2S1384 at 200.43 cM (physical position: 205.23 Mb), covers a genomic region of 15.14 Mb (Table 2 and Fig. 2). GWAS found
that SNP rs6725887 in _WDR12_, which is only 1.48 Mb from marker D2S1384, was associated with early-onset MI and ischemic stroke7, 12, 57 at a genome-wide signifcance level. Moreover,
DisGenNET analysis identified 21 genes related to cardiovascular disease (Table 3). _PDE1A_ encodes a cyclic nucleotide phosphodiesterase and differential expression of _PDE1A_ was observed
in human epicardial adipose tissues from male patients affected with CAD58. _TFPI_ encodes a tissue factor (TF)-dependent pathway of blood coagulation59. An elevated plasma TFPI level was
significantly associated with the presence and severity of CAD60, 61. _TFPI_ expression can be regulated by _ADTRP_, a CAD susceptibility gene identified by our group17. The 5q13.2 locus was
mapped at marker GATA138B05 at 78.80 cM (or 71.40 Mb) and spanned an interval of 5.1 cM (4.95 Mb) (Fig. 2). This is a novel locus for CAD. DisGenNET analysis identified 8 genes linked to
cardiovascular diseases at the 5q13.2 locus (Table 5). _PIK3R1_ encodes Phosphoinositide-3-Kinase Regulatory Subunit 1 and was predicted to be a cardiovascular disease-related gene by a
network topology analysis62. _PIK3R1_is a target of miR-221, and a recent small RNA sequencing analysis revealed that the _miR_-_221_- _PIK3R1_pair was deregulated in late endothelial
progenitor cells (late EPCs) of CAD patients63. _CCNB1_ encodes a regulatory protein involved in mitosis and a recent study showed that genetic variants in _CCNB1_ contributed to risk of the
restenosis of intracoronary stents64. The compelling results above demonstrated that linkage analysis with fewer but larger pedigrees can achieve comparable performance with hundreds of
small nuclear families. As shown in Table 2, the 3q29 and 9q22.33 CAD loci were identified by both GWLS with 24 large families (GeneQuest II) and by a similar analysis with 428 nuclear
families in the GeneQuest population40. Our results also demonstrate that GWLA has a comparable power to GWAS. The 2q33.3 and 17q21.2 CAD loci, which were identified by the GWLS with 24
large families here (GeneQuest II) and represented by D2S1384 and D17S1299, respectively, contain CAD-risk SNPs identified by GWAS (rs6725887 at 2q33.3 and rs46522 at 17q21.2) (Table 2, Figs
2 and 3). Therefore, we conclude that increasing family members within individual families can markedly improve the power for identifying disease linkage and loci. These data also suggest
that our GeneQuest II database is a promising resource for identifying novel risk genes for CAD. Future studies on fine mapping and targeted sequencing will uncover causative variants or
genes for CAD at the CAD loci identified in this study. We also carried out genome-wide linkage analysis in each GeneQuest II family and found that each of the six significant CAD loci
identified in the combined family cohort (Table 1) were also identified in at least one individual family (Table 3). For example, the top two CAD loci on chromosomes 17q21.2 and 2p22.2 were
observed in two families (the best NPL score = 16.81) and three individual families (the best NPL score = 12.42), respectively. Moreover, individual family-based analyses also identified 15
new, significant linkages in 5 families that were not captured by joint linkage analyses of 24 GeneQuest II families, including 7 highly significant linkages, 2 significant linkages, and 6
suggestive significant linkages (Table 4). None of the 15 new genetic loci have been previously reported for CAD. Of interest, the two top ranked CAD locus on 12q13.13, represented by
D12S297 (multipoint NPL score = 6.76) and 17q22 represented by D17S1290 (multipoint NPL score = 6.51), were linked to CAD-associated traits of body mass index (BMI)65 and metabolic
factors66. Despite a list of significant CAD loci identified in this study, there were several limitations. First, the density of microsatellite markers in this study was low (10 cM per
marker). Future fine mapping studies may be carried out with additional markers surrounding the microsatellite polymorphisms used for linkage analysis or SNP microarrays with a much
increased marker density. Single SNPs may not as informative as microsatellite markers for linkage analysis due to their bi-allelic status, but haplotypes constructed using multiple SNPs may
be considered as multi-allelic markers67. Fine mapping will confirm that the linkage loci are overlapping in different families, shorten and narrow the linked regions (if shared) and
eventually reduce the number of candidate genes for some loci. Moreover, fine mapping with SNP arrays may allow us to compare the SNP linkage data with the top hits from previous GWAS and
identify new SNPs associated with CAD. Similarly, ongoing whole genome sequencing may be another powerful approach to capture SNPs or causal variants associated with CAD in the 24 GeneQuest
II families. Second, we highlighted 3–77 genes at each CAD locus based on the evidence from existing literature with a purpose to illustrate the relevance of each CAD locus to etiological
process of CAD. However, the CAD causal genes being responsible for each linkage were possibly overlooked in this study (Table 5). Third, the 24 GeneQuest II families were of European
descent, and it is likely that some significant CAD loci may not be expanded to other ethnic populations. In summary, we report the results of a genome-wide linkage scan of 24 large
GeneQuest II families and uncover six genetic loci for CAD on chromosomes 2q33.3, 3q29, 5q13.2, 7p22.2, 9q22.33 and 17q21.2. Our study identifies four novel CAD loci (2q33.3, 5q13.2, 7p22.2
and 17q21.2). Similar analysis in individual families confirmed the six significant CAD loci and also identified nine new significant linkages on 2q11.2, 9p24.2, 9q34.2, 11q14.1, 12q13.13,
15q26.1, 17q22, 20p12.3, and 22q12.1. Our study also independently confirms the 3q29 and 9q22.33 CAD loci identified by our earlier genome-wide linkage scan for CAD in 428 nuclear families.
Two loci on 2q33.3 and 17q21.2 contain GWAS risk variants identified from population samples. These studies may provide a new framework for uncovering causative variants, genes and
biological pathways involved in the pathogenesis of CAD. METHODS STUDY PARTICIPANTS Twenty-four large, extended, and multigenerational CAD families were recruited at the Center for
Cardiovascular Genetics of the Cleveland Clinic. The study was referred to as GeneQuset II to distinguish it from the original GeneQuest study which recruited more than 428 nuclear families,
mostly for sib-pair analysis. The GeneQuest II study started in the year of 2001 and is completely independent from the earlier GeneQuest study carried out between 1995 and 2000. This study
was reviewed and approved by the Cleveland Clinic Institutional Review Board (IRB) on Human Subject Research, and conformed to the guidelines set forth by the Declaration of Helsinki.
Written informed consent was obtained from all participants. Clinical phenotypic evaluation of study participants was carefully carried out by a panel of cardiologists. The presence or
absence of CAD was assessed according to coronary angiography with >70% stenosis, a history of revascularization procedures such as percutaneous coronary angioplasty (PCA) or coronary
artery bypass (CABG), and a previous diagnosis of myocardial infarction (MI) as described35, 68, 69. Families or patients with hypercholesterolemia, insulin-dependent diabetes, childhood
hypertension, and congenital heart disease were excluded from this study. Each family has at least four definitely diagnosed CAD patients; and the average pedigree size was 18. Clinical and
demographic features of the 24 GeneQuest II CAD families with 433 family members are summarized in Table 1. All recruited family members were Caucasians. The distinguishing features for the
GeneQuest II cohort are large families with three or more generations, 100% whites and a well-balanced male versus female ratio (209/224). A total of 398 sibling pairs were generated in this
cohort, including 154 sister/sister pairs, 105 brother/brother pairs and 139 brother/sister pairs. In contrast to sib-pair analysis of 428 nuclear families in our previous study40,
genome-wide linkage analysis was carried out using all family members instead of sibling pairs only, given the large pedigrees collected in GeneQuest II (Fig. 1). EXTRACTION OF HUMAN GENOMIC
DNA AND GENOTYPING Whole blood samples were drawn from each study participant. Genomic DNA was isolated using the Gentra Puregene blood (QIAGEN, Valencia, CA, USA). All DNA samples were
quantified using NanoDrop 2000 (Thermo Scientific, Wilmington, DE, USA) and inspected for quality by agarose gel electrophoresis. Genome-wide genotyping was performed by Mammalian Genotyping
Service of the National Heart, Lung, and Blood Institute directed by Dr. James L. Weber at Center for Medical Genetics at Marshfield Clinic
(http://research.marshfieldclinic.org/genetics/GeneticResearch/screeningsets.asp) using Screening Set 11. The screening set consists of 410 microsatellite markers spanning the whole human
genome by every 10 cM on average. LINKAGE ANALYSIS Prior to linkage analysis, raw genotyping data were cleaned as described in our previous studies35, 40. In brief, genotypes with
non-consensus calls were re-genotyped or deleted. Microsatellite markers on sex chromosomes were excluded. Missing parental genotypes were added and treated as missing values to complete
family pedigrees (Fig. 1) for linkage analysis. Mendelian inconsistencies were detected by using MARKERINFO built in software S.A.G.E (Statistical Analysis for Genetic Epidemiology)70.
Genotypes with Mendelian errors were excluded from further genome-wide linkage analysis by Genehunter version 2.1_r2 beta71. Relationship between family members (i.e., sibling pairs,
parents-offerings trios) within each family was verified by the RELTEST program included in the S.A.G.E software page70. The RELTEST program did not detect any inconsistent family
relationship. Allele frequencies for all microsatellite markers were estimated by module FREQ in S.A.G.E in the pooled samples containing all of our existing family studies. Program Mega272
was used to generate the input format required for Genehunter version 2.1_r2 beta71. Affected and unaffected individuals were coded as “2” and “1”, respectively, whereas individuals with
uncertain phenotype were coded as “0”. The principle of the Genehunter linkage analysis is to examine any excess of identity-by-decent73 allele-sharing between all affected subjects within a
family. We used the NPL-all statistic within Genehunter version 2.1_r2 beta for linkage analysis, which examines all individuals in the 24 GeneQuest II families simultaneously and provides
a more powerful test (www.broad.mit.edu/ftp/distribution/software/genehunter/). Without specifying the disease transmission model for all markers, non-parametric linkage (NPL) analysis was
carried out to jointly analyze genotype data of all 24 GeneQuest II families. The linkage between CAD and a genetic marker was evaluated by calculating NPL score Z, which is the summation of
standardized identity-by-descent allele-sharing scores across multiple families. Under a null hypothesis of no linkage, Z has mean 0 and variance 1 by choosing appropriate weighting
factors. Statistical significance of Z can be inferred by comparing the observed Z against to its null distribution. Two types of NPL scores were calculated for each marker: 1) A two-point
NPL score examined whether a single marker was linked to CAD; 2) A multipoint NPL score investigated whether a group of markers were linked to CAD. The advantage of the multipoint approach
is its capability of incorporating the information of adjacent markers into linkage analysis (making markers more informative). The NPL-all linkage analysis was also carried out individually
in each of the 24 GeneQuest II families. The larger a NPL score is, the stronger the linkage it indicates. As suggested by Lander and Kruglyak74, linkage peaks were defined in three
categories: (1) Highly significant linkage: NPL of 4.99 (or _P_ value of 3 × 10−7); (2) Significant linkage: NPL of 4.08 (or _P_ value of 2.2 × 10−5); (3) Suggestive Linkage: NPL of 3.18 (or
_P_ value of 7.4 × 10−4). PUBLIC RESOURCES UCSC database: http://genome.ucsc.edu/. DisGenNET42: http://www.disgenet.org/web/DisGeNET/menu/home. GTEx47 portal: http://gtexportal.org/home/.
Genetic map of microsatellite markers: http://research.marshfieldclinic.org/genetics/GeneticResearch/screeningsets.asp. Physical map based on hg19:
https://github.com/joepickrell/1000-genomes-genetic-maps. Genehunter version 2.1_r2 beta: www.broad.mit.edu/ftp/distribution/software/genehunter/. REFERENCES * McPherson, R. &
Tybjaerg-Hansen, A. Genetics of Coronary Artery Disease. _Circ Res_ 118, 564–578, doi:10.1161/CIRCRESAHA.115.306566 (2016). Article CAS PubMed Google Scholar * Lusis, A. J. Genetics of
atherosclerosis. _Trends Genet_ 28, 267–275, doi:10.1016/j.tig.2012.03.001 (2012). Article CAS PubMed PubMed Central Google Scholar * Lusis, A. J. Atherosclerosis. _Nature_ 407,
233–241, doi:10.1038/35025203 (2000). Article CAS PubMed PubMed Central Google Scholar * Clarke, R. _et al_. Genetic variants associated with Lp(a) lipoprotein level and coronary
disease. _N Engl J Med_ 361, 2518–2528, doi:10.1056/NEJMoa0902604 (2009). Article CAS PubMed Google Scholar * Erdmann, J. _et al_. New susceptibility locus for coronary artery disease on
chromosome 3q22.3. _Nat Genet_ 41, 280–282, doi:10.1038/ng.307 (2009). Article CAS PubMed PubMed Central Google Scholar * Gudbjartsson, D. F. _et al_. Sequence variants affecting
eosinophil numbers associate with asthma and myocardial infarction. _Nat Genet_ 41, 342–347, doi:10.1038/ng.323 (2009). Article CAS PubMed Google Scholar * Myocardial Infarction
Genetics, C. _et al_. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. _Nat Genet_ 41, 334–341, doi:10.1038/ng.327
(2009). Article Google Scholar * Trégouët, D.-A. _et al_. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease.
_Nature genetics_ 41, 283–285 (2009). Article PubMed Google Scholar * Consortium, I. K. C. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. _PLoS
Genet_ 7, e1002260, doi:10.1371/journal.pgen.1002260 (2011). Article Google Scholar * Erdmann, J. _et al_. Genome-wide association study identifies a new locus for coronary artery disease
on chromosome 10p11.23. _Eur Heart J_ 32, 158–168, doi:10.1093/eurheartj/ehq405 (2011). Article CAS PubMed Google Scholar * Reilly, M. P. _et al_. Identification of ADAMTS7 as a novel
locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. _The Lancet_ 377,
383–392 (2011). Article CAS Google Scholar * Schunkert, H. _et al_. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. _Nat Genet_ 43,
333–338, doi:10.1038/ng.784 (2011). Article CAS PubMed PubMed Central Google Scholar * Consortium, C. A. D. _et al_. Large-scale association analysis identifies new risk loci for
coronary artery disease. _Nat Genet_ 45, 25–33, doi:10.1038/ng.2480 (2013). Google Scholar * Nikpay, M. _et al_. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of
coronary artery disease. _Nat Genet_ 47, 1121–1130, doi:10.1038/ng.3396 (2015). Article CAS PubMed PubMed Central Google Scholar * Lettre, G. _et al_. Genome-wide association study of
coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. _PLoS Genet_ 7, e1001300, doi:10.1371/journal.pgen.1001300 (2011). Article CAS PubMed
PubMed Central Google Scholar * Aoki, A. _et al_. SNPs on chromosome 5p15.3 associated with myocardial infarction in Japanese population. _J Hum Genet_ 56, 47–51, doi:10.1038/jhg.2010.141
(2011). Article CAS PubMed Google Scholar * Wang, F. _et al_. Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. _Nat
Genet_ 43, 345–349, doi:10.1038/ng.783 (2011). Article CAS PubMed Google Scholar * Lu, X. _et al_. Genome-wide association study in Han Chinese identifies four new susceptibility loci
for coronary artery disease. _Nat Genet_ 44, 890–894, doi:10.1038/ng.2337 (2012). Article CAS PubMed PubMed Central Google Scholar * Takeuchi, F. _et al_. Genome-wide association study
of coronary artery disease in the Japanese. _Eur J Hum Genet_ 20, 333–340, doi:10.1038/ejhg.2011.184 (2012). Article CAS PubMed Google Scholar * Lee, J. Y. _et al_. A genome-wide
association study of a coronary artery disease risk variant. _J Hum Genet_ 58, 120–126, doi:10.1038/jhg.2012.124 (2013). Article CAS PubMed Google Scholar * Hirokawa, M. _et al_. A
genome-wide association study identifies PLCL2 and AP3D1-DOT1L-SF3A2 as new susceptibility loci for myocardial infarction in Japanese. _European Journal of Human Genetics_ 23, 374–380
(2015). Article CAS PubMed Google Scholar * Coronary Artery Disease Genetics, C. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary
artery disease. _Nat Genet_ 43, 339–344, doi:10.1038/ng.782 (2011). Article Google Scholar * Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate
genotype imputation in genome-wide association studies through pre-phasing. _Nat Genet_ 44, 955–959, doi:10.1038/ng.2354 (2012). Article CAS PubMed PubMed Central Google Scholar * van
Leeuwen, E. M. _et al_. Population-specific genotype imputations using minimac or IMPUTE2. _Nat Protoc_ 10, 1285–1296, doi:10.1038/nprot.2015.077 (2015). Article PubMed Google Scholar *
Browning, B. L. & Browning, S. R. Genotype Imputation with Millions of Reference Samples. _Am J Hum Genet_ 98, 116–126, doi:10.1016/j.ajhg.2015.11.020 (2016). Article CAS PubMed
PubMed Central Google Scholar * Chan, L. & Boerwinkle, E. Gene-environment interactions and gene therapy in atherosclerosis. _Cardiology in Review_ 2, 130–137 (1994). Article Google
Scholar * Ozaki, K. & Tanaka, T. Molecular genetics of coronary artery disease. _J Hum Genet_ 61, 71–77, doi:10.1038/jhg.2015.70 (2016). Article CAS PubMed Google Scholar *
Pajukanta, P. _et al_. Two loci on chromosomes 2 and X for premature coronary heart disease identified in early- and late-settlement populations of Finland. _Am J Hum Genet_ 67, 1481–1493,
doi:10.1086/316902 (2000). Article CAS PubMed PubMed Central Google Scholar * Francke, S. _et al_. A genome-wide scan for coronary heart disease suggests in Indo-Mauritians a
susceptibility locus on chromosome 16p13 and replicates linkage with the metabolic syndrome on 3q27. _Hum Mol Genet_ 10, 2751–2765 (2001). Article CAS PubMed Google Scholar * Broeckel,
U. _et al_. A comprehensive linkage analysis for myocardial infarction and its related risk factors. _Nat Genet_ 30, 210–214, doi:10.1038/ng827 (2002). Article CAS PubMed Google Scholar
* Harrap, S. B. _et al_. Genome-wide linkage analysis of the acute coronary syndrome suggests a locus on chromosome 2. _Arterioscler Thromb Vasc Biol_ 22, 874–878 (2002). Article CAS
PubMed Google Scholar * Wang, L., Fan, C., Topol, S. E., Topol, E. J. & Wang, Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. _Science_ 302,
1578–1581, doi:10.1126/science.1088477 (2003). Article ADS CAS PubMed PubMed Central Google Scholar * Hauser, E. R. _et al_. A genomewide scan for early-onset coronary artery disease
in 438 families: the GENECARD Study. _Am J Hum Genet_ 75, 436–447, doi:10.1086/423900 (2004). Article CAS PubMed PubMed Central Google Scholar * Helgadottir, A. _et al_. The gene
encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. _Nat Genet_ 36, 233–239, doi:10.1038/ng1311 (2004). Article CAS PubMed Google Scholar * Wang,
Q. _et al_. Premature myocardial infarction novel susceptibility locus on chromosome 1P34-36 identified by genomewide linkage analysis. _Am J Hum Genet_ 74, 262–271, doi:10.1086/381560
(2004). Article CAS PubMed PubMed Central Google Scholar * Samani, N. J. _et al_. A genomewide linkage study of 1,933 families affected by premature coronary artery disease: The British
Heart Foundation (BHF) Family Heart Study. _Am J Hum Genet_ 77, 1011–1020, doi:10.1086/498653 (2005). Article PubMed Google Scholar * Farrall, M. _et al_. Genome-wide mapping of
susceptibility to coronary artery disease identifies a novel replicated locus on chromosome 17. _PLoS Genet_ 2, e72, doi:10.1371/journal.pgen.0020072 (2006). Article PubMed PubMed Central
Google Scholar * Mani, A. _et al_. LRP6 mutation in a family with early coronary disease and metabolic risk factors. _Science_ 315, 1278–1282, doi:10.1126/science.1136370 (2007). Article
ADS CAS PubMed PubMed Central Google Scholar * Engert, J. C. _et al_. Identification of a chromosome 8p locus for early-onset coronary heart disease in a French Canadian population.
_Eur J Hum Genet_ 16, 105–114, doi:10.1038/sj.ejhg.5201920 (2008). Article CAS PubMed Google Scholar * Gao, H. _et al_. Genome-wide linkage scan identifies two novel genetic loci for
coronary artery disease: in GeneQuest families. _PLoS One_ 9, e113935, doi:10.1371/journal.pone.0113935 (2014). Article ADS PubMed PubMed Central Google Scholar * Ogut, F., Bian, Y.,
Bradbury, P. J. & Holland, J. B. Joint-multiple family linkage analysis predicts within-family variation better than single-family analysis of the maize nested association mapping
population. _Heredity (Edinb)_ 114, 552–563, doi:10.1038/hdy.2014.123 (2015). Article CAS Google Scholar * Bauer-Mehren, A., Rautschka, M., Sanz, F. & Furlong, L. I. DisGeNET: a
Cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. _Bioinformatics_ 26, 2924–2926, doi:10.1093/bioinformatics/btq538 (2010). Article CAS PubMed Google
Scholar * Pinero, J. _et al_. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. _Database (Oxford)_ 2015, bav028, doi:10.1093/database/bav028
(2015). Article Google Scholar * Chiodini, B. D. & Lewis, C. M. Meta-analysis of 4 coronary heart disease genome-wide linkage studies confirms a susceptibility locus on chromosome 3q.
_Arterioscler Thromb Vasc Biol_ 23, 1863–1868, doi:10.1161/01.ATV.0000093281.10213.DB (2003). Article CAS PubMed Google Scholar * Watanabe, T., Kanome, T., Miyazaki, A. & Katagiri,
T. Human urotensin II as a link between hypertension and coronary artery disease. _Hypertens Res_ 29, 375–387, doi:10.1291/hypres.29.375 (2006). Article CAS PubMed Google Scholar * Wang,
X. L., Liu, S. X. & Wilcken, D. E. Circulating transforming growth factor beta 1 and coronary artery disease. _Cardiovasc Res_ 34, 404–410 (1997). Article CAS PubMed Google Scholar
* Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. _Nat Genet_ 45, 580–585, doi:10.1038/ng.2653 (2013). Article Google Scholar * Chang, T. T. & Chen, J. W. Emerging
role of chemokine CC motif ligand 4 related mechanisms in diabetes mellitus and cardiovascular disease: friends or foes? _Cardiovasc Diabetol_ 15, doi:10.1186/s12933-016-0439-9 (2016). *
Moos, M. P. _et al_. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E-deficient mice. _Arterioscler Thromb Vasc Biol_ 25, 2386–2391,
doi:10.1161/01.ATV.0000187470.31662.fe (2005). Article CAS PubMed Google Scholar * de Jager, S. C. A. _et al_. Leukocyte-Specific CCL3 Deficiency Inhibits Atherosclerotic Lesion
Development by Affecting Neutrophil Accumulation. _Arterioscl Throm Vas_ 33, E75–+, doi:10.1161/Atvbaha.112.300857 (2013). Article Google Scholar * Montecucco, F. _et al_. Systemic and
Intraplaque Mediators of Inflammation Are Increased in Patients Symptomatic for Ischemic Stroke. _Stroke_ 41, 1394–1404, doi:10.1161/Strokeaha.110.578369 (2010). Article PubMed Google
Scholar * Ma, H. _et al_. Increased atherosclerotic lesions in LDL receptor deficient mice with hematopoietic nuclear receptor Rev-erbalpha knock- down. _J Am Heart Assoc_ 2, e000235,
doi:10.1161/JAHA.113.000235 (2013). Article PubMed PubMed Central Google Scholar * Coste, H. & Rodriguez, J. C. Orphan nuclear hormone receptor Rev-erbalpha regulates the human
apolipoprotein CIII promoter. _J Biol Chem_ 277, 27120–27129, doi:10.1074/jbc.M203421200 (2002). Article CAS PubMed Google Scholar * Oguri, M. _et al_. Assessment of a polymorphism of
SDK1 with hypertension in Japanese Individuals. _Am J Hypertens_ 23, 70–77, doi:10.1038/ajh.2009.190 (2010). Article PubMed Google Scholar * Meyer, M. R. _et al_. Erratum: G
Protein-coupled Estrogen Receptor Protects from Atherosclerosis. _Sci Rep_ 5, 13510, doi:10.1038/srep13510 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Meyer, M. R.
_et al_. G protein-coupled estrogen receptor protects from atherosclerosis. _Sci Rep_ 4, 7564, doi:10.1038/srep07564 (2014). Article CAS PubMed PubMed Central Google Scholar * Dichgans,
M. _et al_. Shared genetic susceptibility to ischemic stroke and coronary artery disease: a genome-wide analysis of common variants. _Stroke_ 45, 24–36, doi:10.1161/STROKEAHA.113.002707
(2014). Article CAS PubMed Google Scholar * Guauque-Olarte, S. _et al_. The transcriptome of human epicardial, mediastinal and subcutaneous adipose tissues in men with coronary artery
disease. _PLoS One_ 6, e19908, doi:10.1371/journal.pone.0019908 (2011). Article ADS CAS PubMed PubMed Central Google Scholar * Mukherjee, M. _et al_. Anticoagulant versus amidolytic
activity of tissue factor pathway inhibitor in coronary artery disease. _Blood Coagul Fibrinolysis_ 11, 285–291 (2000). CAS PubMed Google Scholar * Lima, L. M. _et al_. Tissue factor and
tissue factor pathway inhibitor levels in coronary artery disease: correlation with the severity of atheromatosis. _Thromb Res_ 121, 283–287, doi:10.1016/j.thromres.2007.04.013 (2007).
Article CAS PubMed Google Scholar * Morange, P. E. _et al_. Prognostic value of plasma tissue factor and tissue factor pathway inhibitor for cardiovascular death in patients with
coronary artery disease: the AtheroGene study. _J Thromb Haemost_ 5, 475–482, doi:10.1111/j.1538-7836.2006.02372.x (2007). Article CAS PubMed Google Scholar * Sarajlic, A., Janjic, V.,
Stojkovic, N., Radak, D. & Przulj, N. Network topology reveals key cardiovascular disease genes. _PLoS One_ 8, e71537, doi:10.1371/journal.pone.0071537 (2013). Article ADS CAS PubMed
PubMed Central Google Scholar * Chang, T. Y. _et al_. miRNome traits analysis on endothelial lineage cells discloses biomarker potential circulating microRNAs which affect progenitor
activities. _BMC Genomics_ 15, 802, doi:10.1186/1471-2164-15-802 (2014). Article PubMed PubMed Central Google Scholar * Silvestre-Roig, C. _et al_. Genetic variants in CCNB1 associated
with differential gene transcription and risk of coronary in-stent restenosis. _Circ Cardiovasc Genet_ 7, 59–70, doi:10.1161/CIRCGENETICS.113.000305 (2014). Article CAS PubMed Google
Scholar * Norris, J. M. _et al_. Quantitative trait loci for abdominal fat and BMI in Hispanic-Americans and African-Americans: the IRAS Family study. _Int J Obes (Lond)_ 29, 67–77,
doi:10.1038/sj.ijo.0802793 (2005). Article CAS Google Scholar * Cheng, C. Y. _et al_. Genome-wide linkage analysis of multiple metabolic factors: evidence of genetic heterogeneity.
_Obesity (Silver Spring)_ 18, 146–152, doi:10.1038/oby.2009.142 (2010). Article ADS CAS Google Scholar * Flaquer, A. _et al_. Genome-wide linkage analysis of congenital heart defects
using MOD score analysis identifies two novel loci. _BMC Genet_ 14, 44, doi:10.1186/1471-2156-14-44 (2013). Article CAS PubMed PubMed Central Google Scholar * McCarthy, J. J. _et al_.
Large scale association analysis for identification of genes underlying premature coronary heart disease: cumulative perspective from analysis of 111 candidate genes. _J Med Genet_ 41,
334–341 (2004). Article CAS PubMed PubMed Central Google Scholar * Shen, G. Q. _et al_. An LRP8 variant is associated with familial and premature coronary artery disease and myocardial
infarction. _Am J Hum Genet_ 81, 780–791, doi:10.1086/521581 (2007). Article CAS PubMed PubMed Central Google Scholar * Kong, X. _et al_. A combined linkage-physical map of the human
genome. _Am J Hum Genet_ 75, 1143–1148, doi:10.1086/426405 (2004). Article CAS PubMed PubMed Central Google Scholar * Li, H. & Schaid, D. J. GENEHUNTER: application to analysis of
bipolar pedigrees and some extensions. _Genet Epidemiol_ 14, 659–663, doi:10.1002/(SICI)1098-2272 (1997). Article CAS PubMed Google Scholar * Baron, R. V., Kollar, C., Mukhopadhyay, N.
& Weeks, D. E. Mega2: validated data-reformatting for linkage and association analyses. _Source Code Biol Med_ 9, 26, doi:10.1186/s13029-014-0026-y (2014). Article PubMed PubMed
Central Google Scholar * Beaudoin, M. _et al_. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. _PLoS Genet_
9, e1003723, doi:10.1371/journal.pgen.1003723 (2013). Article CAS PubMed PubMed Central Google Scholar * Lander, E. & Kruglyak, L. Genetic dissection of complex traits: guidelines
for interpreting and reporting linkage results. _Nat Genet_ 11, 241–247, doi:10.1038/ng1195-241 (1995). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank
the patients and their relatives for their enthusiastic participation in the study. We thank Annabel Z. Wang for proofreading the manuscript and corrections of spelling and grammar errors.
This study was supported by NIH/NHLBI grants R01 HL121358 and R01 HL126729, the China National Natural Science Foundation grants (91439129 and 31430047), the Chinese National Basic Research
Programs (973 Programs 2013CB531101 and 2012CB517801), Hubei Province Natural Science Key Program (2014CFA074), Hubei Province’s Outstanding Medical Academic Leader Program, and Specialized
Research Fund for the Doctoral Program of Higher Education from the Ministry of Education. We thank J. Weber and the NHLBI Mammalian Genotyping Service for genotyping. AUTHOR INFORMATION
Author notes * Yang Guo, Fan Wang, Lin Li and Hanxiang Gao contributed equally to this work. AUTHORS AND AFFILIATIONS * Center for Cardiovascular Genetics, Department of Molecular
Cardiology, Lerner Research Institute, Cleveland Clinic, Cleveland, OH, 44195, USA Yang Guo, Fan Wang, Lin Li, Hanxiang Gao, Stephen Arckacki, Isabel Z. Wang, Qiuyun Chen & Qing K. Wang
* Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH, 44195, USA Yang Guo, Fan Wang, Lin Li, Stephen Arckacki,
Qiuyun Chen & Qing K. Wang * Heart Center, the First Affiliated Hospital, Lanzhou University, Lanzhou, Gansu, 730000, P. R. China Hanxiang Gao * Shaker Heights High School, Shaker
Heights, OH, 44120, USA Isabel Z. Wang * Department of Quantitative Health Sciences, Cleveland Clinic, Cleveland, OH, 44195, USA John Barnard * Department of Cardiovascular Medicine, Sydell
& Arnold Miller Family Heart & Vascular Institute, Cleveland Clinic, Cleveland, OH, 44195, USA Stephen Ellis, Carlos Hubbard & Qing K. Wang * Scripps Translational Science
Institute, Scripps Research Institute, Scripps Clinic, La Jolla, CA, 92037, USA Eric J. Topol * Department of Genetics and Genome Sciences, Case Western Reserve University School of
Medicine, Cleveland, OH, 44106, USA Qing K. Wang Authors * Yang Guo View author publications You can also search for this author inPubMed Google Scholar * Fan Wang View author publications
You can also search for this author inPubMed Google Scholar * Lin Li View author publications You can also search for this author inPubMed Google Scholar * Hanxiang Gao View author
publications You can also search for this author inPubMed Google Scholar * Stephen Arckacki View author publications You can also search for this author inPubMed Google Scholar * Isabel Z.
Wang View author publications You can also search for this author inPubMed Google Scholar * John Barnard View author publications You can also search for this author inPubMed Google Scholar
* Stephen Ellis View author publications You can also search for this author inPubMed Google Scholar * Carlos Hubbard View author publications You can also search for this author inPubMed
Google Scholar * Eric J. Topol View author publications You can also search for this author inPubMed Google Scholar * Qiuyun Chen View author publications You can also search for this author
inPubMed Google Scholar * Qing K. Wang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Q.C. E.J.T. and Q.K.W. conceived and designed the
experiments. Y.G., F.W., L.L., H.Z., H.X. performed all data analysis, reports and interpretation. S.E., C.H., E.J.T., Q.C. and Q.K.W. participated in sample collection. Y.G., F.W., L.L.,
H.Z., H.X., Q.C. and Q.K.W. drafted the manuscript. S.A., I.Z.W., J.B., S.E., C.H., E.J.T. Q.C., and Q.K.W. contributed critical comments and revised the manuscript. All authors have
reviewed and approved the final version of the manuscript. CORRESPONDING AUTHORS Correspondence to Qiuyun Chen or Qing K. Wang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare
that they have no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution
and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if
changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the
material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to
obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Guo, Y., Wang, F., Li, L. _et al._ Genome-Wide Linkage Analysis of Large Multiple Multigenerational Families Identifies Novel Genetic Loci for Coronary Artery Disease. _Sci Rep_ 7,
5472 (2017). https://doi.org/10.1038/s41598-017-05381-2 Download citation * Received: 24 November 2016 * Accepted: 30 May 2017 * Published: 14 July 2017 * DOI:
https://doi.org/10.1038/s41598-017-05381-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative