- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT We describe a novel ERBB1/EGFR somatic mutation (p. C329R; c.985 T > C) identified in a patient with JAK2V617F Polycythaemia Vera (PV). This substitution affects a conserved
cysteine residue in EGFR domain 2 and leads to the formation of a ligand-independent covalent receptor dimer, associated with increased transforming potential. Aberrant signalling from the
EGFRC329R receptor is cell type-dependent and in the TF1.8 erythroid cell line expression of this mutant suppresses EPO-induced differentiation. Clonal analysis shows that the dominant
JAK2V617F-positive clone in this PV patient harbors EGFRC329R, thus this mutation may contribute to clonal expansion. Somatic mutations affecting other ERBB and related receptor tyrosine
kinases are observed in myeloproliferative neoplasms (MPN), and we show elevated EGFR levels in MPN samples, consistent with previous reports. Thus activation of this group of receptors, via
multiple mechanisms, may contribute to clonal growth and survival of the JAK2V617F disease clone in MPN. SIMILAR CONTENT BEING VIEWED BY OTHERS A NOVEL SUBCLONAL REARRANGEMENT OF THE
_STRN3::PDGFRB_ GENE IN DE NOVO ACUTE MYELOID LEUKEMIA WITH _NPM1_ MUTATION AND ITS LEUKEMOGENIC EFFECTS Article 07 August 2023 A NOVEL TRKB-ACTIVATING INTERNAL TANDEM DUPLICATION
CHARACTERIZES A NEW MECHANISM OF RECEPTOR TYROSINE KINASE ACTIVATION Article Open access 10 May 2025 CLINICOPATHOLOGIC SPECTRUM OF MYELOID NEOPLASMS WITH CONCURRENT MYELOPROLIFERATIVE
NEOPLASM DRIVER MUTATIONS AND _SRSF2_ MUTATIONS Article 11 June 2022 INTRODUCTION The ErbB receptor family, including epidermal growth factor receptor (ERBB1/EGFR), represents a group of
receptor tyrosine kinases (RTKs) with established roles in cancer. While the role of this family has not been investigated in detail in haematopoietic malignancies (HM), the activation of
RTKs by mutation has a well-characterised role in HM; in particular, activating mutations in FLT3 and KIT are well described in acute myeloid leukaemia (AML)1. Cancer-associated mutations
affecting the E3 Ubiquitin ligase, CBL, which is required for ubiquitination and degradation of multiple RTKs, are also recurrent across HM, including the BCR-ABL-negative myeloproliferative
neoplasms (MPN)2. Increased activity of the receptor-associated tyrosine kinase JAK2 by activating mutation (most frequently JAK2V617F) is a feature of this group of MPN, including
essential thrombocythaemia (ET), Polycythaemia Vera (PV) and Primary Myelofibrosis (PMF)3. Here we describe a novel somatic, transforming, EGFR variant with an extracellular cysteine
substitution in domain 2 (EGFRC329R), identified in a PV patient. PV is characterized by clonal erythroid hyperplasia and high frequency of JAK2 activating mutations (in >98% of cases)3.
PV is associated with clinical features including thrombosis, constitutional symptoms, and risk of transformation to myelofibrosis (MF) and AML. Loss of heterozygosity (LOH) for JAK2 occurs
frequently in PV and is associated with expansion of the erythroid lineage; however, JAK2V617F LOH alone is insufficient to sustain disease4. Co-operating mutations may contribute to the
disease phenotype. Hence, there is significant interest in the contribution of JAK2-independent signalling in MPN, particularly given that the same JAK2 mutation can lead to diverse disease
phenotypes, and since JAK inhibitor therapy typically does not lead to eradication of the MPN clone. Thus, acquisition of additional JAK2-independent events in the MPN clone is presumably
important for disease. Our characterization of EGFRC329R shows that the mutation induces ligand-independent covalent receptor dimerization and is associated with increased transforming
potential. This mechanism of activation is similar to that reported for recurrent mutations that affect the extracellular domain of EGFR in glioblastoma, EGFRvIII, as well as non-synonymous
mutations affecting residues in a conserved cysteine-rich region critical for the formation of intra-molecular disulphide bonds in domains 2 and 4 of the receptor5, 6. Consistent with a role
in clonal expansion and MPN pathogenesis, we show that the EGFRC329R mutant leads to loss of erythroid lineage markers, and reduced EPO-induced differentiation in an erythroid
differentiation model (TF-1.8 human erythroleukaemia cell line). A comprehensive survey of somatic mutations affecting ERBB genes, and the genes encoding the related RTKs, MET and ALK, shows
rare mutations across published MPN cohorts. Thus, we suggest that aberrant activation of these receptors by mutation is a rare event but likely cooperates with JAK2 signalling to influence
the growth and lineage properties of the MPN disease clone. RESULTS AND DISCUSSION IDENTIFICATION OF A SOMATIC EGFRC329R MUTATION IN PV Targeted exon capture and massively parallel
sequencing of 657 cancer-related genes in peripheral blood mononuclear cells (PBMNC) or granulocyte DNA from 15 JAK2V617F-positive PV patients (Suppl. Tables S1 and S2) identified a somatic
_EGFR_ variant (p. C329R; c.985 T > C) in one patient (PV17) (Suppl. Figure S1a). The screen included genes encoding multiple growth factor and cytokine receptors. Additional variants
identified are shown in Suppl. Table S3, and include a previously reported activating variant7 affecting MET, in another PV patient (METY1248H; Suppl. Figure S1a and Suppl. Table S5). The
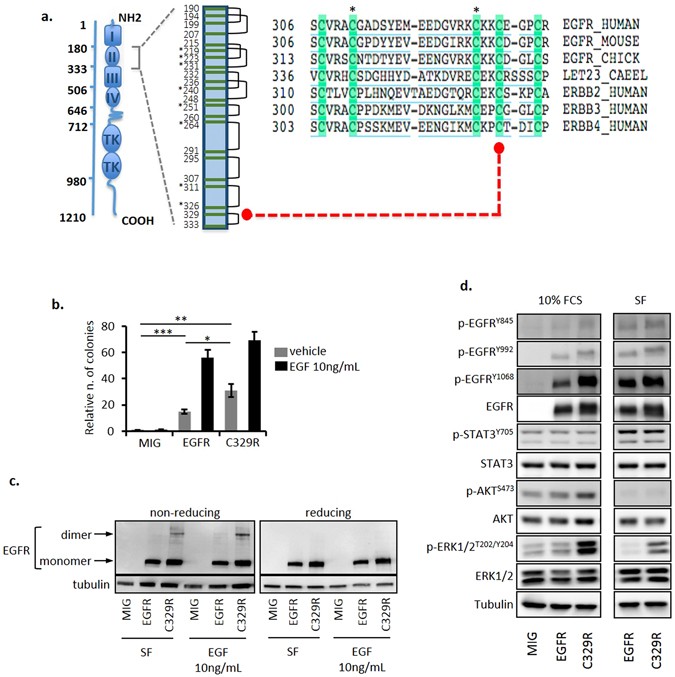
human EGFR C329 residue resides in the cysteine-rich region in extracellular domain 2, which promotes receptor dimerization (Fig. 1a) and is the target of recurrent somatic mutations in
glioblastoma6, 8. Moreover, it aligns with the C359 residue of the orthologous _C. elegans_ receptor, _let-23_, that is the target of a known gain-of-function mutation9, and with the
cysteine residue affected in the transforming human ERBB2/HER2 mutation, C334S10. THE EGFRC329R MUTANT DISPLAYS ONCOGENIC POTENTIAL AND CELL-TYPE SPECIFIC SIGNALLING We first investigated
the oncogenic potential of this novel EGFR variant by measuring anchorage-independent growth of transduced NIH3T3 cells expressing similar levels of either wild type (WT) EGFR (NIH3T3/EGFR)
or EGFRC329R (NIH3T3/C329R) (Suppl. Figure S2a). Colony formation in the absence of exogenous EGF was increased in NIH3T3/C329R cells compared to NIH3T3/EGFR cells (Fig. 1b and Suppl. Figure
S2b, respectively 31 ± 4.9 vs. 15 ± 1.6, _P_ < 0.05); however, in the presence of EGF (10 ng/mL), colony numbers were similar for both cell populations. Several mutations affecting EGFR
and ERBB2 extracellular cysteine residues lead to the formation of activated dimeric receptor through an intermolecular disulphide bond which involves the remaining free cysteine residue5,
10, 11. Based on the receptor modelling (Suppl. Figure S1b) we proposed a similar mechanism of dimerization and activation for EGFRC329R. We investigated this by western blot using reducing
and non-reducing conditions. The presence of the high molecular weight EGFR dimer was observed constitutively under non-reducing conditions in NIH3T3/C329R cells, but not in NIH3T3/EGFR
cells, and its abundance increased after treatment with EGF (10 ng/mL, Fig. 1c and Suppl. Figure S3). The presence of the EGFR dimer correlated with altered signalling responses.
NIH3T3/C329R cells displayed increased levels of phosphorylated p-EGFRTyr1068 compared to NIH3T3/EGFR in serum-containing media, and a profound increase of p-ERK1/2Thr202/Tyr204 in both the
presence and absence of serum (Fig. 1d and Suppl. Figure S4). These data confirm that the C329R domain 2 mutation promotes covalent, ligand-independent receptor dimerization and altered
signalling. We next investigated the effect of the C329R substitution on EGF sensitivity and EGF-induced growth and survival of transduced haematopoietic BaF3 cells expressing similar levels
of WT human EGFR or EGFRC329R (Suppl. Figure S2c). Even though both BaF3/EGFR and BaF3/C329R cells responded to increasing doses of EGF, which induced cell growth in the absence of mIL-3,
BaF3/C329R cells displayed hypersensitivity to the growth factor (Fig. 2a, EC50 BaF3/C329R 0.312 ± 0.082 ng/mL vs. BaF3/EGFR 0.732 ± 0.091 ng/mL, _P_ < 0.05). Furthermore, this difference
in sensitivity to low doses of EGF was associated with significantly increased output of viable BaF3/C329R cells over a 5 day period (Fig. 2b). Since we observed no difference in activation
of caspases 3 and 7 or in annexin V/7AAD staining (Suppl. Figure S5a,b) between these populations, the above results are consistent with the EGF-induced increase in cell numbers being
predominantly from increased proliferation and not decreased apoptosis. Analysis of signalling events associated with the mutant receptor in BaF3 and NIH3T3 cells revealed cell-type specific
differences. In contrast to NIH3T3 cells, there was no significant increase in basal p-ERK1/2 in response to expression of the mutant receptor in BaF3 (Fig. 2c, Suppl. Figure S6d). In the
presence of EGF (10 ng/mL), we observed phosphorylation of specific tyrosine residues selectively in the cells expressing WT receptor (Fig. 2c and Suppl. Figure S6a–c). In contrast, similar
levels of p-AKTS473, p-ERK1/2T202/Y204 and p-STAT5Y694 were observed following EGF stimulation in both cells expressing EGFR WT or C329R (Fig. 2c and Suppl. Figure S6b–d). Treatment of the
transduced cells with the selective EGFR inhibitor, gefitinib, blocked EGF-induced phosphorylation of EGFR, AKT, ERK1/2 and STAT5 in both EGFR WT and C329R expressing cells (Fig. 2c and
Suppl. Figure S6a–d). STAT3 was not phosphorylated in the presence or absence of EGF treatment in BaF3/EGFR and BaF3/C329R cells (Suppl. Figure S6a). These results are consistent with an
altered conformation of the EGFRC329R dimer which may prevent receptor trans-phosphorylation of specific tyrosine residues in response to EGF stimulation, but still permit downstream
activation of signalling intermediates. Further investigations, including detailed determination of the dimer configuration and receptor phosphorylation on tyrosine, serine and threonine
residues, are needed to establish the mechanism by which the C329R mutation selectively affects signalling responses induced by EGF. Distinct signalling and functional differences have been
described for different classes of EGFR mutations12 and it will be of interest to establish whether the signalling pattern that we identified is unique to EGFR extracellular mutants.
EXPRESSION OF EGFRC329R IN A HAEMATOPOIETIC CELL LINE MODEL OF ERYTHROID DIFFERENTIATION The human TF1.8 cell line is an erythroleukaemic, GM-CSF-dependent cell line, which displays
multi-lineage differentiation potential. To investigate the effects of EGFR signalling on haematopoietic lineage, we transduced TF1.8 cells with WT EGFR (TF1.8/EGFR), EGFRC329R (TF1.8/C329R)
or vector alone (TF1.8/MIG), and confirmed that TF1.8/EGFR and TF1.8/C329R expressed similar levels of receptor (Suppl. Figure S2d). We analysed myeloid/erythroid differentiation by
measuring surface expression of the granulocytic marker CD13, and the erythroid marker CD235a (glycophorin A). In the presence of GM-CSF, we observed a small increase in the percentage of
CD235a-negative cells in TF1.8/C329R cells compared to TF1.8/EGFR and TF1.8/MIG, with no significant change in CD13 (Fig. 3a, gate a, 3b, top panel). This result is consistent with the
mutant receptor signalling in the absence of exogenous EGF, and with such signalling being associated with a reduction in erythroid differentiation. To explore this further, we cultured the
TF1.8 transduced cells in EPO-containing medium, which induces an erythroid cell population defined by increased CD235a and reduced CD13 surface expression (Fig. 3a, gate c) at the expense
of the CD235a-negative, CD13Hi population (Fig. 3a, gate a). The erythroid shift with EPO (gate a to gate c) was comparable for TF1.8/MIG and TF1.8/EGFR; however, this response was
significantly reduced in the presence of EGFRC329R (Fig. 3a, EPO panels, gates a–c; Fig. 3b, panel EPO gate a). This observation suggests that signalling from the mutant receptor in the
absence of exogenous EGF impairs EPO-induced differentiation. To examine the EGF response of the transduced TF1.8 cells, they were treated with two doses of EGF (0.5 ng/mL and 10 ng/mL).
EGF-induced signalling suppressed erythroid differentiation as we observed a time-dependent increase in the CD235a-negative population (gate a) for both TF1.8/EGFR and TF1.8/C329R cells, but
this increase was much more pronounced at the higher dose of EGF (Fig. 3a, EGF panels, Fig. 3b, bottom panel). To investigate the mechanism by which EGFRC329R interferes with TF1.8
differentiation induced by EPO, we analysed EGFR signalling in these cells treated with GM-CSF or EGF (0.5 ng/mL and 10 ng/mL). We observed constitutive phosphorylation of tyrosine residues
selectively in WT EGFR. EGF induced phosphorylation of both EGFR WT and C329R, with reduced levels of phosphorylation observed for C329R (Suppl. Figure S7a–c). We did not observe significant
differences in signalling downstream from the receptors in any of the conditions tested (Suppl. Figure S7a–c). Overall these results are consistent with signalling from EGFRC329R limiting
EPO-induced erythroid differentiation in the absence of exogenous EGF. The mechanism for the receptor-induced suppression of erythroid differentiation is unclear, although the phenotype
resembles that reported for constitutive Ras signaling which blocks Epo-induced erythroid differentiation of progenitors, and promotes Epo-independent proliferation of erythroblasts13. We
note also that similar results have been observed in the same TF1.8 model following expression of a germline _ERBB3_ mutant (p. A1337T) identified in affected individuals from a family with
MDS/AML associated with erythroid hyperplasia14 (Suppl. Table S5). RELATIONSHIP OF EGFRC329R AND JAK2V617F IN PV PATIENT ERYTHROID COLONIES EGFR mRNA shows lineage-specific expression in
haematopoietic cells from healthy individuals15, and is significantly upregulated in MPN and AML patient samples16, 17. Therefore, we next analysed EGFR protein levels in healthy controls
(HC) and PV patients. We detected, by flow cytometry, increased surface EGFR levels on BMMNC from three PV patients compared to HC (Suppl. Figure S8a), and for patient PV17 we could detect
EGFR expression by Western blot using PBMNC lysate (Suppl. Figure S8b). These observations raise the possibility that EGFR protein level may be selectively increased in PV patients,
contributing to growth and survival of the disease clone. To test this, we treated patient BMMNC with gefitinib (25 nM) and measured the formation of EPO–independent (i.e. endogenous) BFU-E,
which are characteristic of PV. As shown in Suppl. Figure S9, we observed inhibition of EPO-independent BFU-E, consistent with EGFR-signaling contributing to growth and survival of clonal
cells derived from PV patients. We next determined the clonal evolution of the disease in patient PV17 by genotyping of individual BFU-E grown from PBMNC. We determined EGFRC329R and
JAK2V617F genotype using a quantitative single nucleotide primer extension assay with mass array (Sequenom). As shown in Fig. 3c and Suppl. Table S4, we identified heterozygous JAK2V617F
colonies that were WT for EGFR (13% of the total JAK2V617F-positive colonies), or heterozygous for EGFRC329R (5% of total JAK2V617F-positive), but no JAK2WT-EGFRC329R colonies were
identified. BFU-E with JAK2 LOH were predominantly heterozygous for EGFRC329R (77% of total JAK2-mutant colonies). Thus, in this patient, it is likely that there was an initial
JAK2V617F-heterozygous clone, and that LOH for JAK2V617F occurred secondarily in both JAK2V617F-single mutant (JAK2V617F-EGFRWT/WT) and double mutant (JAK2V617F-EGFRC329R/WT) derivatives
(Fig. 3c). The dominance of the JAK2V617F-EGFRC329R/WT clone over the JAK2V617F-EGFRWT/WT clone suggests that the aberrant RTK activity associated with the EGFRC329R mutant provides a clonal
advantage. A previous study in two PV patients with multiple LOH clones (identified by the extent of uniparental disomy) did not identify somatic mutations unique to the dominant clone
which might have explained its dominance18. They concluded that stochastic processes determine which LOH clone predominates. The mechanism by which activated EGFR may cooperate with
JAK2V617F signalling requires further study; nonetheless, a role in clonal expansion would be consistent with other studies indicating an important role for ERBB receptors in the expansion
of progenitor cells (or “transit amplifying cells” in solid tumours) associated with de-differentiation and stem cell characteristics19. Such a mechanism may be a common driver of clonal
amplification and accumulation of additional mutations in tumours. ROLE OF ABERRANT ERBB, MET AND ALK RECEPTOR SIGNALLING IN MPN While whole exome and targeted sequencing of MPN sample
cohorts have been undertaken by several groups, somatic mutations in EGFR have not previously been identified. We screened for recurrence of this _EGFR_ mutation in our Australian MPN cohort
(n = 160) using a custom Sequenom assay and did not identify any additional patients with this specific mutation. While mutations (most frequently in the tyrosine kinase domain) represent
the most common EGFR-activating event in solid tumours (reviewed in ref. 20), it is possible that aberrant EGFR signalling also occurs via non-mutational mechanisms in MPN. Other reported
mechanisms of oncogenic activation of EGFR include gene amplification, over-expression, and structural rearrangements (e.g. gene fusions) of the receptor, as well as tumour or stromal
over-expression of EGF–family ligands (reviewed in ref. 20). Recently, an acquired mutation in MET (N375S) has been reported as recurrent in AML21. Mutations affecting ERBB2 and ERBB3, or
the related MET and ALK receptors, can result in activation of overlapping signalling responses. Two separate studies have suggested a functional role for HGF/MET in PV22, 23, and a previous
exome sequencing study of MPN patient samples identified acquired mutations in MET (V1247A, adjacent to the somatic mutation identified in our screen) and in ERBB2 (L494F)24. Overall,
mutations in ERBB receptors, MET and ALK are rare in MPN (those described to date are summarized in Suppl. Table S5); however, recurrent mutations in genes that affect the signalling of this
group of receptors (e.g. CBL and RAS) have been reported in MPN2, 24. In summary, the characterization of the novel extracellular EGFRC329R mutation has revealed a potential role for
increased RTK signalling in parallel with, but independent of, JAK2 in MPN. Cross-talk between JAK2 and EGFR oncogenic signalling has recently been reported in lung adenocarcinoma where JAK2
activity has been shown to promote receptor turnover via the ubiquitin-mediated degradation pathway25. Since EGFR has been shown to promote HSC survival under conditions of increased HSC
proliferation26, we speculate that in the context of aberrant JAK2 activity in HSPC there may be a requirement for mechanisms that activate or sensitize RTK signalling. Aberrant activation
of signalling pathways by JAK2-independent mechanisms may also provide an explanation for the observed resistance of MPNs to JAK inhibitor therapy, which commonly has limited effect on JAK2
allelic burden, even in patients with a positive clinical response27. Furthermore, as inhibition of JAK2 has been shown to increase abundance of EGFR on the cell surface25, this raises the
possibility that combining JAK2 inhibitors with EGFR and/or MEK inhibition may be an effective strategy in selected MPN patients where the aim of treatment is to reduce the
disease-initiating clone. Such a dual-targeting approach has been reported to be effective in lung cancer patients resistant to EGFR inhibition28. MATERIAL AND METHODS MPN PATIENTS MPN
patients (92 PV, 39 ET, 17 MF, and 12 unclassified MPN) were recruited from 3 major South Australian hospitals (The Royal Adelaide Hospital – RAH, The Queen Elizabeth Hospital – TQEH, and
Flinders Medical Centre – FMC). Clinical information and samples were collected for research with informed consent from all subjects, and with approval from the following Human Research
Ethics Committees (HREC): RAH HREC, TQEH HREC, FMC HREC, and University of South Australia HREC. All experiments were performed in accordance with relevant guidelines and regulations with
approval from the RAH and University of South Australia HREC. EXON CAPTURE SEQUENCING Genomic DNA was prepared from granulocyte or peripheral blood mononuclear cells (PBMNC) from 15 PV
patients (all confirmed positive for JAK2V617F) and used in an initial exon capture experiment. 657 cancer-related genes were captured (Suppl. Table S1), including genes with mutations in
COSMIC (v48) and genes involved in AML, CML, ALL, CLL and lymphoma, using NimbleGen solid phase capture (2009) on genome build hg18 (Roche-NimbleGen). For capture, the 15 DNA samples were
combined into 5 pools without barcoding (Suppl. Table S2). Captured DNA was sequenced on a SOLiD (3.5) platform and base calling and alignment performed using SOLiD™ BioScope™ software 1.3
(Applied Biosystems). A total of 8 958 exons were captured and 88.3% of these produced efficient sequence. SeattleSeq Annotation was used to annotate sequence variants. To generate the list
of variants in Suppl. Table S3 we filtered for exonic Tier 1 variants and removed variants that were present in 1000 genomes or ExAC database. For the ESP database we filtered to remove
variants with a frequency in the Caucasian population of >1%. Finally, variants with variant allele frequency (VAF) of between 6% and 32% in any pool were included, and all variants that
were present at a frequency of >5% across all pools were excluded. For variants of interest we used Sanger sequencing of matched granulocyte and buccal DNA to determine the affected
patient within the pool, and germline/somatic status. HAEMATOPOIETIC CELL LINES The murine BaF3 cell line was cultured as previously described29. The human TF-1.8 cell line30 was cultured in
recombinant human GM-CSF (5 ng/mL; Peprotech, Rocky Hill, NJ, USA) as described31. EGFR RETROVIRAL EXPRESSION CONSTRUCTS The complete EGFR ORF was obtained from the EST Image Clone 4483665
(ThermoFisher Scientific, Waltham, MA, USA). EGFR was cloned into the retroviral vector, pMSCV-IRES-eGFP (MIG)32 and the C329R mutation introduced using the Phusion Site-directed Mutagenesis
kit (ThermoFisher Scientific). Retroviral transduction of cell lines was performed as previously described32. Cells were sorted for eGFP expression with a BD FACSAria II (BD Biosciences,
Franklin Lakes, NJ, USA). CASPASE ACTIVITY, VIABILITY AND PROLIFERATION ASSAYS To measure growth and survival of BaF3 cell populations expressing EGFR WT or EGFRC329R, cells were washed 3
times with PBS and incubated with growth factor (mIL-3 or rhEGF; R&D Systems, Minneapolis, MN, USA) at the indicated doses. Cell growth was assessed using the CellTiter 96 AQueous One
Solution Cell Proliferation Assay (MTS) (Promega, Madison, WI, USA) following the manufacturer’s instructions. Caspase activity was measured with Caspase-Glo 3/7 Assay (Promega) as per the
manufacturer’s instructions. Viability was measured by Annexin V (BD Biosciences) and 7AAD (BD Biosciences) staining, following the manufacturer’s protocol. SOFT-AGAR COLONY FORMATION Colony
formation assay with NIH3T3 transduced cells was conducted as described33 using a 6-well format. FLOW CYTOMETRY ANALYSIS For EGFR detection, 106 cells were fixed with 1.6% formaldehyde
(Sigma-Aldrich, St. Louis, MO, USA) for 10 min at room temperature (RT), pelleted and washed with PBS twice. For total EGFR expression, cells were permeabilised with 80% ethanol for 20 min
on ice followed by two washes with PBS. For total or surface EGFR expression, cells were then washed with 1% BSA (Sigma-Aldrich) in PBS, and finally pelleted and re-suspended in 100 µL of
anti-EGFR antibody (clone D38B1, cat #4267, Cell Signaling Technology, Danvers, MA, USA) diluted in 1% BSA in PBS. Incubation was for 1 hour at RT followed by one wash with PBS. Cells were
then re-suspended in 100 µL of anti-rabbit Alexa Fluor® 647 conjugate secondary antibody (cat #4414, Cell Signaling Technology) diluted in 1% BSA in PBS and incubated for 30 min at RT in the
dark. Finally, cells were washed twice with PBS, re-suspended in 250 µL of PBS and analysed on a GalliosTM Flow Cytometer (Beckman Coulter, Brea, CA, USA). For detection of erythroid
differentiation surface markers in TF1.8 transduced cell populations, staining was performed as above with the following antibodies: BV421 mouse anti-human CD235a (clone GA-R2-HIR2, cat
#562938, BD Biosciences) and PE anti-human CD13 (clone WM15, cat #301704, BioLegend, San Diego, CA, USA). Detection was performed on a BD LSRFortessa (BD Biosciences). Flow cytometry data
were analysed with FCS Express4 Flow Research Edition (DeNovo Software, Glendale, CA, USA). WESTERN BLOTTING For analysis of signalling responses and EGFR dimer, cells were washed with PBS
containing 10 mM iodoacetamide (Sigma-Aldrich) and then lysed with NP40 Cell Lysis Buffer (ThermoFisher Scientific) containing cOmplete Protease Inhibitor Cocktail (Roche Diagnostics,
Mannheim, Germany), PhosStop (Roche Diagnostics), Pefabloc (Roche Diagnostics) and iodoacetamide (Sigma-Aldrich). Western blot was performed as described32. Primary antibodies used were:
EGFR (clone 15F8, cat #4405, to detect EGFR dimer), EGFR (clone D38B1, cat #4267), p-EGFR Tyr845 (clone D63B4, cat #6963), p-EGFR Tyr992 (cat #2235), p-EGFR Tyr1068 (clone D7A5, cat #3777),
p44/42 MAPK (cat #9102), p-p44/42 MAPK Thr202/Tyr204 (cat #9101), STAT5 (clone 3H7, cat #9358), p-STAT5 Tyr694 (cat #9351), STAT3 (clone 79D7, cat #4904), p-STAT3 Tyr705 (clone D3A7, cat
#9145), AKT (cat #9272) and p-AKT Ser473 (cat #9271), all from Cell Signaling Technology, and anti-tubulin (clone E-19, sc-12462-R, Santa Cruz Biotechnology, Dallas, TX, USA). Secondary
antibody used was anti-rabbit-AP conjugate (Santa Cruz Biotechnology). Total protein levels were determined after stripping the blots probed with the phosphoprotein antibody and re-probing
with respective total protein antibody. Tubulin was used as loading control; blots were stripped after probing with total protein antibody and re-probed with anti-tubulin antibody. Blots
were imaged using the Typhoon FLA 9000 (GE Healthcare Life Sciences, Uppsala, Sweden) and images analysed using ImageQuant TL (GE Healthcare Life Sciences). GENOTYPING FOR EGFRC329R AND
JAK2V617F We used previously established methods based on Sequenom mass array34 for sensitive detection of EGFRC329R and JAK2V617F mutations. A multiplex assay was designed and performed on
PBMNC or granulocyte DNA from a cohort of 160 MPN patients with clinical diagnosis of PV (n = 92); ET (n = 39); MF (primary or post-PV/post-ET; n = 17); or uncharacterized MPN (n = 12).
REFERENCES * Coombs, C. C., Tallman, M. S. & Levine, R. L. Molecular therapy for acute myeloid leukaemia. _Nat Rev Clin Oncol_ 13, 305–318, doi:10.1038/nrclinonc.2015.210 (2016). Article
CAS PubMed Google Scholar * Grand, F. H. _et al_. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. _Blood_ 113, 6182–6192,
doi:10.1182/blood-2008-12-194548 (2009). Article CAS PubMed Google Scholar * Skoda, R. C., Duek, A. & Grisouard, J. Pathogenesis of myeloproliferative neoplasms. _Exp Hematol_ 43,
599–608, doi:10.1016/j.exphem.2015.06.007 (2015). Article CAS PubMed Google Scholar * Li, J. _et al_. JAK2V617F homozygosity drives a phenotypic switch in myeloproliferative neoplasms,
but is insufficient to sustain disease. _Blood_ 123, 3139–3151, doi:10.1182/blood-2013-06-510222 (2014). Article CAS PubMed Google Scholar * Greenall, S. A. _et al_. EGFRvIII-mediated
transactivation of receptor tyrosine kinases in glioma: mechanism and therapeutic implications. _Oncogene_ 34, 5277–5287, doi:10.1038/onc.2014.448 (2015). Article CAS PubMed Google
Scholar * Ymer, S. I. _et al_. Glioma Specific Extracellular Missense Mutations in the First Cysteine Rich Region of Epidermal Growth Factor Receptor (EGFR) Initiate Ligand Independent
Activation. _Cancers_ 3, 2032–2049, doi:10.3390/cancers3022032 (2011). Article CAS PubMed PubMed Central Google Scholar * Schmidt, L. _et al_. Germline and somatic mutations in the
tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. _Nat Genet_ 16, 68–73, doi:10.1038/ng0597-68 (1997). Article CAS PubMed Google Scholar * Freed, D. M.,
Alvarado, D. & Lemmon, M. A. Ligand regulation of a constitutively dimeric EGF receptor. _Nat Commun_ 6, 7380, doi:10.1038/ncomms8380 (2015). Article ADS PubMed PubMed Central Google
Scholar * Katz, W. S. _et al_. A point mutation in the extracellular domain activates LET-23, the Caenorhabditis elegans epidermal growth factor receptor homolog. _Mol Cell Biol_ 16,
529–537, doi:10.1128/MCB.16.2.529 (1996). Article CAS PubMed PubMed Central Google Scholar * Greulich, H. _et al_. Functional analysis of receptor tyrosine kinase mutations in lung
cancer identifies oncogenic extracellular domain mutations of ERBB2. _Proc Natl Acad Sci USA_ 109, 14476–14481, doi:10.1073/pnas.1203201109 (2012). Article ADS CAS PubMed PubMed Central
Google Scholar * Greenall, S. A., Donoghue, J. F., Gottardo, N. G., Johns, T. G. & Adams, T. E. Glioma-specific Domain IV EGFR cysteine mutations promote ligand-induced covalent
receptor dimerization and display enhanced sensitivity to dacomitinib _in vivo_. _Oncogene_ 34, 1658–1666, doi:10.1038/onc.2014.106 (2015). Article CAS PubMed Google Scholar *
Erdem-Eraslan, L. _et al_. Mutation specific functions of EGFR result in a mutation-specific downstream pathway activation. _Eur J Cancer_ 51, 893–903, doi:10.1016/j.ejca.2015.02.006 (2015).
Article CAS PubMed Google Scholar * Zhang, J., Socolovsky, M., Gross, A. W. & Lodish, H. F. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional
analysis by a flow cytometry-based novel culture system. _Blood_ 102, 3938–3946, doi:10.1182/blood-2003-05-1479 (2003). Article CAS PubMed Google Scholar * Braunstein, E. M. _et al_. A
Germline Mutation in ERBB3 Predisposes to Inherited Erythroid Myelodysplasia/Erythroleukemia. _Blood_ 126, 4105 (2015). Google Scholar * Bagger, F. O. _et al_. BloodSpot: a database of gene
expression profiles and transcriptional programs for healthy and malignant haematopoiesis. _Nucleic Acids Res_ 44, D917–924, doi:10.1093/nar/gkv1101 (2016). Article PubMed Google Scholar
* Skov, V. _et al_. Gene expression profiling with principal component analysis depicts the biological continuum from essential thrombocythemia over polycythemia vera to myelofibrosis.
_Exp Hematol_ 40, 771–780 e719, doi:10.1016/j.exphem.2012.05.011 (2012). Article CAS PubMed Google Scholar * Sun, J. Z. _et al_. Epidermal growth factor receptor expression in acute
myelogenous leukaemia is associated with clinical prognosis. _Hematol Oncol_ 30, 89–97, doi:10.1002/hon.1002 (2012). Article CAS PubMed Google Scholar * Godfrey, A. L. _et al_.
Nongenetic stochastic expansion of JAK2V617F-homozygous subclones in polycythemia vera? _Blood_ 124, 3332–3334, doi:10.1182/blood-2014-09-603043 (2014). Article CAS PubMed PubMed Central
Google Scholar * Schneider, M. R. & Yarden, Y. The EGFR-HER2 module: a stem cell approach to understanding a prime target and driver of solid tumors. _Oncogene_ 35, 2949–2960,
doi:10.1038/onc.2015.372 (2016). Article CAS PubMed Google Scholar * Hynes, N. E. & MacDonald, G. ErbB receptors and signaling pathways in cancer. _Curr Opin Cell Biol_ 21, 177–184,
doi:10.1016/j.ceb.2008.12.010 (2009). Article CAS PubMed Google Scholar * Marjanovic, I. _et al_. Parallel targeted next generation sequencing of childhood and adult acute myeloid
leukemia patients reveals uniform genomic profile of the disease. _Tumour Biol_ 37, 13391–13401, doi:10.1007/s13277-016-5142-7 (2016). Article CAS PubMed Google Scholar * Boissinot, M.
_et al_. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of
JAK2V617F. _Oncogene_ 30, 990–1001, doi:10.1038/onc.2010.479 (2011). Article CAS PubMed Google Scholar * Hoermann, G. _et al_. Identification of oncostatin M as a JAK2 V617F-dependent
amplifier of cytokine production and bone marrow remodeling in myeloproliferative neoplasms. _FASEB J_ 26, 894–906, doi:10.1096/fj.11-193078 (2012). Article CAS PubMed Google Scholar *
Tenedini, E. _et al_. Targeted cancer exome sequencing reveals recurrent mutations in myeloproliferative neoplasms. _Leukemia_ 28, 1052–1059, doi:10.1038/leu.2013.302 (2014). Article CAS
PubMed Google Scholar * Gao, S. P. _et al_. JAK2 inhibition sensitizes resistant EGFR-mutant lung adenocarcinoma to tyrosine kinase inhibitors. _Sci Signal_ 9, ra33,
doi:10.1126/scisignal.aac8460 (2016). Article PubMed PubMed Central Google Scholar * Doan, P. L. _et al_. Epidermal growth factor regulates hematopoietic regeneration after radiation
injury. _Nat Med_ 19, 295–304, doi:10.1038/nm.3070 (2013). Article CAS PubMed PubMed Central Google Scholar * Odenike, O. Beyond JAK inhibitor therapy in myelofibrosis. _Hematology Am
Soc Hematol Educ Program_ 2013, 545–552, doi:10.1182/asheducation-2013.1.545 (2013). PubMed Google Scholar * Zhao, Y. & Adjei, A. A. The clinical development of MEK inhibitors. _Nat
Rev Clin Oncol_ 11, 385–400, doi:10.1038/nrclinonc.2014.83 (2014). Article CAS PubMed Google Scholar * Blake, T. J., Jenkins, B. J., D’Andrea, R. J. & Gonda, T. J. Functional
cross-talk between cytokine receptors revealed by activating mutations in the extracellular domain of the beta-subunit of the GM-CSF receptor. _J Leukoc Biol_ 72, 1246–1255 (2002). CAS
PubMed Google Scholar * Kitamura, T. _et al_. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. _J Cell
Physiol_ 140, 323–334, doi:10.1002/jcp.1041400219 (1989). Article CAS PubMed Google Scholar * Broxmeyer, H. E. _et al_. Hematopoietic colony formation from human growth factor-dependent
TF1 cells and human cord blood myeloid progenitor cells depends on SHP2 phosphatase function. _Stem Cells Dev_ 22, 998–1006, doi:10.1089/scd.2012.0478 (2013). Article CAS PubMed Google
Scholar * Perugini, M. _et al_. Repression of Gadd45alpha by activated FLT3 and GM-CSF receptor mutants contributes to growth, survival and blocked differentiation. _Leukemia_ 23, 729–738,
doi:10.1038/leu.2008.349 (2009). Article CAS PubMed Google Scholar * Lee, J. C. _et al_. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in
the extracellular domain. _PLoS medicine_ 3, e485, doi:10.1371/journal.pmed.0030485 (2006). Article PubMed PubMed Central Google Scholar * Parker, W. T. _et al_. Sensitive detection of
BCR-ABL1 mutations in patients with chronic myeloid leukemia after imatinib resistance is predictive of outcome during subsequent therapy. _J Clin Oncol_ 29, 4250–4259,
doi:10.1200/JCO.2011.35.0934 (2011). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank all patients who gave samples for this study, and the South
Australian Cancer Research Biobank (SACRB) for their assistance with sample collection and storage. This research was funded by the National Health & Medical Research Council of
Australia (Project Grant #1063443). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Centre for Cancer Biology, SA Pathology and University of South Australia, Adelaide, South Australia,
Australia D. A. Casolari, T. Nguyen, D. G. Iarossi, C. N. Hahn, S. C. Bray, W. T. Parker, J. Feng, K. Z. Y. Maung, A. Wee, L. Vidovic, S. Branford, I. D. Lewis, H. S. Scott & R. J.
D’Andrea * Department of Haematology, SA Pathology and Royal Adelaide Hospital, Adelaide, South Australia, Australia D. A. Casolari, T. Nguyen, C. M. Butcher, D. G. Iarossi, S. C. Bray, K.
Z. Y. Maung, A. Wee, L. Vidovic, I. D. Lewis, D. M. Ross & R. J. D’Andrea * Department of Genetics and Molecular Pathology, SA Pathology, Adelaide, South Australia, Australia C. N. Hahn,
W. T. Parker, J. Feng, S. Branford & H. S. Scott * Basil Hetzel Institute for Translational Health Research and Department of Haematology and Oncology, The Queen Elizabeth Hospital,
Woodville, South Australia, Australia C. M. Butcher, S. C. Bray, P. Neufing, K. Z. Y. Maung, A. Wee & P. G. Bardy * School of Medicine, University of Adelaide, Adelaide, South Australia,
Australia S. C. Bray, K. Z. Y. Maung, A. Wee & D. M. Ross * Cancer Theme, SAHMRI, Adelaide, South Australia, Australia C. H. Kok & D. M. Ross * Queensland Institute of Medical
Research, Brisbane, Queensland, Australia S. W. Lane * Department of Haematology, Flinders University and Medical Centre, Adelaide, South Australia, Australia D. M. Ross * School of Pharmacy
and Medical Sciences, University of South Australia, Adelaide, South Australia, Australia R. J. D’Andrea Authors * D. A. Casolari View author publications You can also search for this
author inPubMed Google Scholar * T. Nguyen View author publications You can also search for this author inPubMed Google Scholar * C. M. Butcher View author publications You can also search
for this author inPubMed Google Scholar * D. G. Iarossi View author publications You can also search for this author inPubMed Google Scholar * C. N. Hahn View author publications You can
also search for this author inPubMed Google Scholar * S. C. Bray View author publications You can also search for this author inPubMed Google Scholar * P. Neufing View author publications
You can also search for this author inPubMed Google Scholar * W. T. Parker View author publications You can also search for this author inPubMed Google Scholar * J. Feng View author
publications You can also search for this author inPubMed Google Scholar * K. Z. Y. Maung View author publications You can also search for this author inPubMed Google Scholar * A. Wee View
author publications You can also search for this author inPubMed Google Scholar * L. Vidovic View author publications You can also search for this author inPubMed Google Scholar * C. H. Kok
View author publications You can also search for this author inPubMed Google Scholar * P. G. Bardy View author publications You can also search for this author inPubMed Google Scholar * S.
Branford View author publications You can also search for this author inPubMed Google Scholar * I. D. Lewis View author publications You can also search for this author inPubMed Google
Scholar * S. W. Lane View author publications You can also search for this author inPubMed Google Scholar * H. S. Scott View author publications You can also search for this author inPubMed
Google Scholar * D. M. Ross View author publications You can also search for this author inPubMed Google Scholar * R. J. D’Andrea View author publications You can also search for this author
inPubMed Google Scholar CONTRIBUTIONS D.M.R. and R.D.A. supervised research; D.A.C., C.M.B., D.M.R. and R.D.A. designed experiments and wrote the manuscript; D.A.C., T.N., C.M.B., D.G.I.,
S.C.B., W.T.P., P.N., A.W. and L.V. performed research; D.A.C., T.N., C.M.B., D.G.I., W.T.P., C.N.H., J.F., K.Z.Y.M. and C.H.K. analysed data; C.M.B., D.G.I., S.C.B., C.N.H., S.B., I.D.L.,
H.S.S. and S.W.L. contributed to preparation of the manuscript; C.M.B., P.G.B., I.D.L. and D.M.R. organised collection of patient samples. CORRESPONDING AUTHOR Correspondence to R. J.
D’Andrea. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare that they have no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE: Springer Nature remains neutral
with regard to jurisdictional claims in published maps and institutional affiliations. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY INFORMATION SUPPLEMENTARY TABLE 3 RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Casolari, D.A.,
Nguyen, T., Butcher, C.M. _et al._ A novel, somatic, transforming mutation in the extracellular domain of Epidermal Growth Factor Receptor identified in myeloproliferative neoplasm. _Sci
Rep_ 7, 2467 (2017). https://doi.org/10.1038/s41598-017-02655-7 Download citation * Received: 08 September 2016 * Accepted: 18 April 2017 * Published: 26 May 2017 * DOI:
https://doi.org/10.1038/s41598-017-02655-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative







