- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Hemorrhagic shock is a leading cause of death in people under the age of 45 and accounts for almost half of trauma-related deaths. In order to develop a treatment strategy based on
potentiating mitochondrial function, we investigated the effect of the orphan drug dichloroacetate (DCA) on survival in an animal model of hemorrhagic shock in the absence of fluid
resuscitation. Hemorrhagic shock was induced in rats by withdrawing 60% of the blood volume and maintaining a hypotensive state. The studies demonstrated prolonged survival of rats subjected
to hemorrhagic injury (HI) when treated with DCA. In separate experiments, using a fluid resuscitation model we studied mitochondrial functional alterations and changes in metabolic
networks connected to mitochondria following HI and treatment with DCA. DCA treatment restored cardiac mitochondrial membrane potential and tissue ATP in the rats following HI. Treatment
with DCA resulted in normalization of several metabolic and molecular parameters including plasma lactate and p-AMPK/AMPK, as well as Ach-mediated vascular relaxation. In conclusion we
demonstrate that DCA can be successfully used in the treatment of hemorrhagic shock in the absence of fluid resuscitation; therefore DCA may be a good candidate in prolonged field care
following severe blood loss. SIMILAR CONTENT BEING VIEWED BY OTHERS DEXMEDETOMIDINE IMPROVES ACUTE LUNG INJURY BY ACTIVATING AUTOPHAGY IN A RAT HEMORRHAGIC SHOCK AND RESUSCITATION MODEL
Article Open access 16 March 2023 ELAMIPRETIDE MITIGATES ISCHEMIA-REPERFUSION INJURY IN A SWINE MODEL OF HEMORRHAGIC SHOCK Article Open access 18 March 2023 LEVOSIMENDAN INCREASES BRAIN
TISSUE OXYGEN LEVELS AFTER CARDIOPULMONARY RESUSCITATION INDEPENDENT OF CARDIAC FUNCTION AND CEREBRAL PERFUSION Article Open access 09 July 2021 INTRODUCTION Trauma is the leading cause of
death for individuals under the age of 45 years old1, 2. Hemorrhage is the most common cause of preventable death in this group3, 4. Hypotensive volume replacement permits maintenance of
limited tissue reperfusion and continued metabolic activities to maintain cell viability following hemorrhagic shock. However, there is a lack of consensus on the use of any specific
resuscitation strategy or adjuncts to fluid resuscitation. Though optimal resuscitation strategy remains controversial, hemorrhage control and resuscitation are high priorities in trauma
care in civilian as well as combat situations5,6,7. When administration of fluid resuscitation is logistically difficult, particularly in combat situations, there is a need for agents that
prolong survival in the absence of fluid resuscitation in prolonged field care to improve survival. Severe haemorrhage and shock lead to whole body tissue hypoxia and nutrient deprivation.
The severe blood loss leads to prolonged hypotension and impaired coronary flow. This causes decreased cardiac perfusion, myocardial hypoxia and cardiac dysfunction, and exemplify the role
of the heart as an important target organ. Hemorrhagic shock is known to cause decreased cardiac output, stroke volume, cardiac contractility, and impaired mitochondrial
bioenergetics8,9,10,11. Hypoxia and reperfusion injury result in dysregulation of molecular pathways and functions including reduced mitochondrial ATP production10, 12,13,14,15. These
evidences suggest that mitochondria play a critical role in maintaining cellular homeostasis following hemorrhagic shock. Hemorrhagic injury (HI) in animal models have demonstrated decreased
activities of electron transport chain complexes, increased release of cytochrome c from the mitochondria, and decreased ATP production16,17,18,19. Our laboratory and others have
demonstrated salutary effect of resveratrol, a mitochondria potentiating agent, following HI in animal models18, 20, 21. Treatment with resveratrol and a synthetic sirtuin 1 (SIRT1)
activator, SRT1720, prolonged life following HI in our animal model, further indicating the possibility of a critical role for mitochondria in outcome following HI22. SIRT1 is a deacetylase
that regulates the activity of a number of transcription factors, and modulate mitochondrial biogenesis and function13, 23. We also found increased expression of pyruvate dehydrogenase
kinase (Pdk) in the heart of rats subjected to HI and restoration of Pdk expression when the animals were treated with resveratrol following HI24. Pdk inhibits pyruvate dehydrogenase (Pdh),
a key enzyme that couples glycolytic pathway to the tricarboxylic acid (TCA) cycle by catalysing the decarboxylation of pyruvate to acetyl-CoA25. In order to further identify a direct role
for mitochondria in HI-mediated organ function and survival, and to test whether inhibition of Pdk can change outcome following HI, we induced HI in rats and treated them with
dichloroacetate (DCA) and monitored survival in the absence of fluid resuscitation. DCA is an inhibitor of Pdk and enhances the activity of Pdh, resulting in increased turnover of pyruvate
to acetyl CoA thereby augmenting oxidative phosphorylation. It has been shown that DCA improves cardiac output and left ventricular function in myocardial ischemia26, 27, and prevented the
transition from cardiac hypertrophy to heart failure in experimental animal models28. Furthermore, clinical studies with this small molecule resulted in reduced lactate levels in patients
with congenital lactic acidosis and sepsis29, 30. The influence of DCA on cellular energetics following hemorrhagic shock in the absence of fluid resuscitation has been unclear. We used our
HI models involving no fluid resuscitation as well as the one with fluid resuscitation, to further characterize the influence of DCA on mitochondrial function and survival following HI.
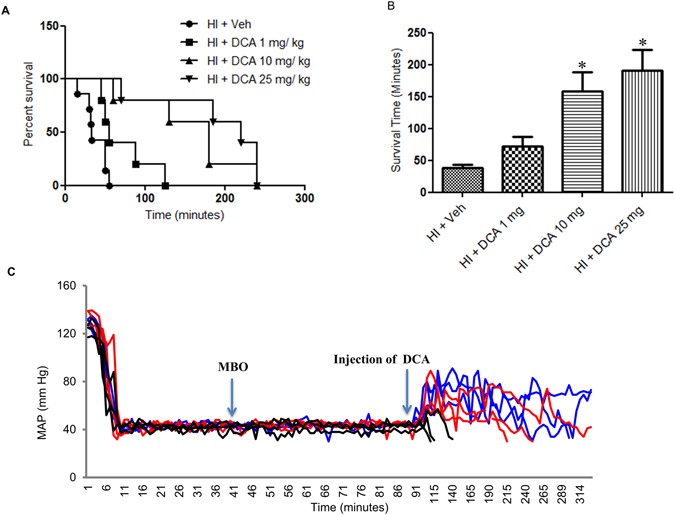
RESULTS DCA IMPROVES SURVIVAL IN THE ABSENCE OF FLUID RESUSCITATION DCA is known to potentiate mitochondrial function. Therefore we first tested whether treatment with DCA can prolong
survival following HI. In the experimental model of HI we subjected rats to soft tissue trauma and severe hemorrhage, but were not resuscitated with fluid following hypovolemic shock period.
Immediately following the shock period, an intravenous dose of DCA was given in 500–600 μl Ringers lactate. The control animals received only 500–600 μl Ringers lactate. We tested three
different doses of DCA, 1 mg, 10 mg and 25 mg per Kg body weight and found that the latter two doses significantly improved survival following HI (Fig. 1A,B and C). However, though 25 mg/Kg
dose had a slight survival advantage, it was not significant from that achieved by 10 mg/Kg dose (Fig. 1A). The mean survival times for each these doses, including vehicle were 38 min, 73
min, 160 min and 200 min for 0, 1, 10 and 25 mg/Kg doses respectively (Fig. 1B). None of the animals that did not DCA (vehicle only) survived more than 55 minutes and the survival period
ranged from15–55 minutes. It is important to note that unlike the control animals, none of the animals that received 10 or 25 mg/Kg dose of DCA died within the first 60 minutes. From Fig.
1C, it is also evident that DCA administration resulted in elevated mean arterial pressure (MAP). EFFECT OF DCA ON HEMODYNAMICS AFTER FLUID RESUSCITATION In the next experiment, the effect
of DCA on hemodynamics and molecular parameters following HI was tested using the fluid resuscitation model. HI was induced and fluid resuscitation was carried out with Ringers Lactate, two
times the volume of shed blood, for one hour. DCA or vehicle was administered 10 minutes after the start of fluid resuscitation. MAP and heart rate were observed for 3 hours and the animals
were then sacrificed. Blood gas values were measured before hemorrhage and after the end of the study (Table 1). As shown in Fig. 2A, following fluid resuscitation MAP increased
significantly in all three groups with significant increase of MAP in the DCA-treated groups (10 mg and 25 mg) compared to untreated groups. Nevertheless, none of the resuscitation groups
fully achieved restoration of MAP equal to the MAP prior to haemorrhage. There was also an increase in the heart rate in all the three groups of animals following fluid resuscitation; the
difference was significant in both vehicle group and the group of rats that received 10 mg/Kg DCA (Fig. 2B) when compared to respective groups at the start of fluid resuscitation. A
significant and comparable blood loss was evident in all the three groups (Fig. 2C). As it is apparent, the decrease in total haemoglobin was consistent with the severe blood loss. The
difference in plasma lactate was very pronounced at 3 hours following fluid resuscitation (Fig. 3A) in the vehicle treated rats subjected to HI. The lactate levels were almost normalized in
the rats treated with DCA. The lactate/pyruvate ratio also improved significantly following DCA treatment (Fig. 3A–C). This was followed by an increased activity of myocardial PDH (Fig. 3D).
DCA TARGETS MITOCHONDRIA DCA is well described as an inhibitor of the mitochondrial enzyme pyruvate dehydrogenase kinase (Pdk)31, 32. In order to test the effect of DCA treatment on Pdk, we
performed Western blot on the heart tissue isolated from rats treated with vehicle or DCA following HI. As shown in Fig. 4A, we observed a significant increase in Pdk expression in the
heart following HI. As Pdk inhibits the enzyme pyruvate dehydrogenase (PDH) that catalyses the conversion of pyruvate to acetyl CoA, the increased Pdk expression following HI was concomitant
with a decreased level of ATP in the heart (Fig. 4B). The protein expression levels of Pdk were restored to normal levels in rats that received DCA at the dose of 10 and 25 mg/Kg. We
observed a corresponding increase in total ATP in the heart tissue demonstrating increased mitochondrial respiratory output. As severe haemorrhage leads to hypoxia, the elevation of Pdk
levels and diminished ATP levels indicated impaired mitochondrial function. In order to determine whether a mitochondrial glycolytic shift takes place following HI, and if a reversal follows
DCA treatment, we tested the activity of the glycolytic enzyme hexokinase and observed significant increase (Fig. 4C). However, we did not see a decline in the hexokinase activity following
DCA treatment indicating a maintenance of heightened enzyme activity even after mitochondrial ATP output increased. Hexokinase may be too far upstream to identify a DCA-mediated effect. It
may also be an adaptive mechanism to increase pyruvate generation to support TCA cycle. MOLECULAR MEDIATORS IN DCA MEDIATED SALUTARY EFFECT In order to identify the molecular mediators
involved in the DCA-induced salutary effect following HI we performed a series of experiments to test changes in the levels of key proteins related to mitochondrial function. Based upon our
previous studies that showed significant impairment of cardiac function following HI, we focussed our molecular studies on heart tissue. One of the critical proteins involved in the
post-translational activation of transcription factors related to mitochondrial function is SIRT1. The amount of this deacetylase enzyme was significantly decreased in the heart of rats
following HI and fluid resuscitation (Fig. 5A). In the animals that received DCA at 10 mg/Kg and 25 mg/Kg dose, the protein level was similar to that was observed in sham operated animals
(Fig. 5A). Consistent with the change in expression level of SIRT1 following HI and DCA treatments, we also observed a significant decrease in the activity of SIRT1 in rats that underwent HI
and restoration of the activity in DCA treated rats (Fig. 5B). Both 10 mg/Kg and 25 mg/Kg DCA doses maintained SIRT1 activity similar to that in the sham. Among the major targets of SIRT1
is Pgc-1α, a mitochondrial biogenesis factor. The transcriptional cofactor Pgc-1α was significantly reduced in the heart of rats subjected to HI (Fig. 5C). Though the protein expression was
not significantly increased with the 10 mg dose of DCA, there was a significant restoration of expression in rats that received 25 mg/kg dose. The two proteins that are important in the
activation of Pgc-1α are SIRT1 and AMPK. AMPK and p-AMPK levels were determined by Western blot and we observed a significant decrease in the phosphorylated fraction of AMPK following HI
(Fig. 5D). However the levels were significantly increased in DCA treated rats when compared to rats that were given only vehicle following HI. Acetyl Co-A carboxylase (ACC) is a direct
target of activated AMPK. Therefore, to further confirm the activation of AMPK, we tested whether ACC phosphorylation is altered when rats were subjected to HI and fluid resuscitation with
or without DCA treatment. We found the pattern of phosphorylation to be similar to that of AMPK, with significant decrease in HI group and normalization of phosphorylation levels in DCA
treated groups, both 10 and 25 mg/kg doses (Fig. 5D) establishing activation of AMPK with DCA treatment. We finally tested one of the markers of endoplasmic reticulum (ER) stress, the ER
chaperone Bip, and found its expression significantly elevated following HI, and normalized when treated with DCA (Fig. 5E). MITOCHONDRIAL MEMBRANE POTENTIAL FOLLOWING HI AND DCA TREATMENT
In order to test whether there is change in mitochondrial membrane potential following HI, we performed the JC1 dye uptake assay using mitochondria isolated from the heart33. There was a
significant decrease in the ratio of aggregate/monomer of the dye as measured by fluorescence intensity ratio at 590/485 in the mitochondria isolated from the heart of rats subjected to HI
and fluid resuscitation (Fig. 6). In cells with decreased mitochondrial membrane potential, the aggregates of dye decreases and emit green fluorescence. The fluorescence intensity ratio was
not significantly different in the mitochondria isolated from rats treated with DCA compared to that from the sham rats. VASCULAR FUNCTION IN HI AND FOLLOWING DCA TREATMENT To understand the
mechanism of salutary effect of DCA we tested the vascular function in aortic rings isolated from the rats subjected to HI, HI with DCA treatment, and sham operation. In this experiment we
specifically determined changes in vascular relaxation following HI and DCA treatment. We compared the vascular relaxation response to acetylcholine (Ach), and sodium nitroprusside (SNP),
and contractile responses to phenylephrine (PE), using isolated aortic rings from sham operated rats and vehicle-treated or DCA-treated rats following HI. When the aortas were treated with
increasing concentrations of Ach or SNP, progressive relaxation was observed with Ach or SNP in all the three groups (Fig. 7A and B). Similarly progressive contraction was seen when treated
with PE (Fig. 7C). The aortic rings from the sham and DCA treated animals demonstrated comparable vascular endothelial-dependent relaxation to Ach as demonstrated by similar robust
responses, as opposed to impaired responses observed with animals subjected to HI and treated with vehicle34 (Fig. 7A). The relaxation responses to the NO donor sodium nitroprusside (SNP),
an endothelium-independent relaxation agent, were not different among the groups. Contractile responses to PE also were not different among the groups (Fig. 7B and C). DISCUSSION Hemorrhagic
shock and fluid resuscitation cause global ischemia-reoxygenation injury and may lead to organ dysfunction and death. The advances in intensive care and patient management have improved
short-term survival following severe injury35, 36 however, the overall hospital mortality for trauma patients admitted to the intensive care unit remains high15, 37. In combat and civilian
situations where care is delayed or fluid resuscitation is logistically difficult, methods to prolong life in the absence of fluid resuscitation are necessitated. Therefore, far-forward
management of severe hemorrhage in combat remains a challenge. Though the precise mechanisms of tissue injury and organ dysfunction following hemorrhagic shock are unclear, recent studies
support the hypothesis that cellular energetics play a critical role in outcome following hemorrhagic shock17, 38,39,40. Therefore, achieving a protracted period of cellular homeostasis
following hemorrhagic shock by treatment with mitochondria potentiating agents is clinically important. Several studies have demonstrated a decline in mitochondrial function in HI41, 42. Our
recent studies have shown that agents such as resveratrol and SRT1720 that have profound effects on mitochondria can rescue rats following HI in the absence of fluid resuscitation22.
Resveratrol was previously shown to improve heart, kidney and liver function after HI in the rodents by several laboratories including ours20, 43, 44. Though improved mitochondrial function
has been observed, there was a lack of direct evidence establishing the role of mitochondria in HI or recovery. The whole body hypoxia and nutrient deprivation following HI and reoxygenation
injury following fluid resuscitation further complicates the molecular signature of HI. A mitochondrial-glycolytic shift due to hypoxic conditions prevailing after severe hemorrhage has
been previously proposed24. Based upon the above premise we performed a systematic investigation of the effect of DCA on survival and mitochondrial function following HI. As shown in Fig. 1,
DCA significantly improved survival in the absence of fluid resuscitation. We tested three different doses of DCA and two higher doses, 10 mg/Kg and 25 mg/Kg, were most effective in
improving survival. This experiment demonstrate that in the absence of fluid resuscitation, an appropriate dose of DCA can prolong life for 2–3 hours following severe hemorrhage and shock in
the rats, though some of the rats survived for the entire duration of the observation (4 hours). The mechanism of action of DCA is well studied and it is attributed to inhibition of Pdk31,
45, 46. During hypoxic conditions, the mitochondrial gatekeeper Pdh is inhibited by Pdk preventing conversion of pyruvate to acetyl CoA. Under hypoxic conditions cells limit oxidative
phosphorylation due to decreased availability of electron acceptors thereby decreasing mitochondrial oxidation. Inhibition of pyruvate decarboxylation to form acetyl CoA is a critical step
in this process. The scenario in cellular response to injury is very similar to the Warburg effect which postulated glucose metabolism yielding lactate in cancer cells despite the
availability of oxygen, which is distinct from that of normal cells47. Nevertheless a profound inhibition of Pdh by Pdk is observed in glycolytic activity within cancer cells and inhibition
of Pdk by DCA has been found to improve mitochondrial oxidative phosphorylation48. DCA can also significantly induce apoptosis of epithelial ovarian cancer cells49. Our experiments
demonstrated an increased expression of Pdk in rats subjected to HI and restoration to normal levels following DCA administration. Warburg suggested a mitochondrial- glycolytic shift that
results in production of ATPs by glycolysis50. A very similar situation may be envisaged in HI with a declining mitochondrial function and an active glycolytic process as evidenced by
increased hexokinase activity and decreased total ATP. Though we expected a normalized hexokinase activity in DCA treated group, we did not observe a decrease in its activity from that of
vehicle treated rats subjected to HI. This demonstrates that the heightened glycolytic activity continues even after DCA treatment. The JC-1 uptake assay demonstrated decreased membrane
potential in the animals treated only with vehicle. The membrane potential in DCA treated animals, as measured by JC-1 aggregation, was similar to that in sham animals. The experiment
demonstrates an overall decline in membrane potential following HI and provides further evidence for the deleterious effect of HI on mitochondria and the salutary actions of DCA in
potentiating mitochondrial function. These experiments demonstrate that by directly intervening on mitochondria, the outcome following HI can be significantly improved. The decompensated
hemorrhagic shock model we used is well-validated, and a depressed cardiac function and myocardial impairment have been well documented in this model16, 20. When DCA is administered
intravenously following HI, it is unclear whether the effect of DCA is predominantly in any particular tissue including the heart. Nevertheless the experiments demonstrate a salutary effect
in the heart at the cellular level following DCA treatment after HI. The experiment to test vascular function demonstrated impaired endothelial-dependent vasorelaxation in response to Ach in
rats following HI, when compared to DCA-treated rats or sham operated rats. We did not observe any significant differences among the groups in response to phenylephrine or sodium
nitroprusside indicating that vascular dysfunction following HI is endothelium-dependent. The involvement of nitric oxide in HI-induced vascular dysfunction and the effect of DCA needs to be
further examined, nevertheless the observation is consistent with previous findings of depressed endothelial function following HI51, 52. We observed a significant decrease in the activity
of SIRT1 following HI and the levels were normalized with DCA treatment. The effect of DCA treatment on SIRT1 is likely secondary due to improved mitochondrial function, though SIRT1 is
known to have profound influence on mitochondrial function. SIRT1 deacetylates Pgc-1α which is a mitochondrial biogenesis factor, and a cofactor for Ppar-α. However the necessary and
sufficient conditions for the activation of Pgc-1α are its deacetylation and phosphorylation. Whereas deacetylation is carried out by SIRT1, AMPK plays a major role in the phosphorylation of
Pgc-1α. Our experiments demonstrated increased ratio of p-AMPK/AMPK in the heart of rats treated with DCA and subjected to HI. The functional relevance of the activated AMPK is further
confirmed by increased phosphorylation of acetyl CoA carboxylase (ACC). ACC is a direct phosphorylation target of AMPK53. The results are consistent with a previous report that showed
increased activation of AMPK when treated with DCA54. The AMPK activation was reported to be due to increased OXPHOS activity. When the animals are treated with DCA, following HI, there is a
switch to pyruvate from lactate and the increased OXPHOS activity is expected to consume NADH thereby increasing the NAD+/NADH ratio resulting in increased SIRT1 activity. Nevertheless,
there is a decrease in SIRT1 activity following HI that is restored with DCA treatment. An inter-relationship between sirtuin activity and AMPK has been proposed elsewhere55 suggesting that
AMPK activation may also be regulated by sirtuin activity. It is known that activation of SIRT1 leads to deacetylation and activation of the AMPK kinase LKB1, resulting in AMPK
activation56,57,58,59. Furthermore potentiating SIRT1 activity by resveratrol was also followed by increased ATP and AMPK activation60. DCA has been used to treat metabolic disorders such as
lactic acidosis and diabetes mellitus due to its ability to reduce circulating glucose and lactate61. DCA treatment was shown to maintain normal circulating lactate levels in patients with
congenital lactic acidosis30. DCA has also been tested in various cancers and other disorders characterized by hypoxia or mitochondrial dysfunction, but with side effects such as peripheral
neuropathy due to chronic use30, 62, 63. The proposed use of DCA as described in this study does not require chronic use. Therefore side-effects or efficacy issues due to chronic use become
less relevant in a single use study for acute conditions such as trauma. There have also been previous attempts testing the effect of DCA in hemorrhagic shock using experimental animal
models, but with varying results64, 65. However, these studies utilized a very high concentration of DCA (150 mg/Kg) and a constant pressure model with unknown or varying shed blood volumes.
The dose response study in our model shows that 10–25 mg/Kg body weight elicits a maximum survival response. Furthermore, the data presented in this study demonstrate that the survival
benefit due to DCA treatment is better than that due to resveratrol treatment reported in a previous study22. The previous study using resveratrol at the maximum employed dose of 10 mg/Kg
demonstrated an average survival period of ~110 minutes with at least one of the six animals having survived the full duration of the study (4 hrs)22. However in the current study, the
maximum mean survival time following DCA treatment was almost doubled (~200 minutes) and most of the animals survived longer, demonstrating that DCA has a more potent effect. A combinatorial
therapy with DCA and resveratrol may also be pursued. Our results show that the concerted mechanisms initiated following potentiation of mitochondrial function triggers a network of
cellular responses to allow a protracted period of metabolic homeostasis. This is further substantiated by the observation of a decrease in the endoplasmic reticulum (ER) chaperone protein
Bip with HI. HI is characterized by whole body hypoxia and nutrient deprivation. One of the first responders to hypoxia and nutrient deprivation is the ER which signals and readies the cells
by an unfolded protein response or ER stress66, 67. These results suggest that cells regain homeostatic balance after HI when treated with DCA; this could be a reason for increased survival
observed in the rats even in the absence of fluid resuscitation. Collectively, the interrogation of metabolic checkpoints following HI demonstrates a major shift in metabolic coordinates
with DCA treatment resulting in improved mitochondrial function and faster restoration of homeostatic balance. We therefore conclude that agents that potentiate mitochondrial function will
be useful in the treatment of hemorrhagic shock. METHODS ANIMALS Male Sprague Dawley rats 295 ± 10 g were obtained from Charles River Laboratory (Wilmington, MA, USA). All animal experiments
performed in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Augusta University. All methods were performed in accordance with the relevant guidelines
and regulations. All animals were housed in Augusta University animal facility during the experiments as per the approved protocol. HEMORRHAGE INJURY PROCEDURE Rats were subjected to sham
or hemorrhagic injury procedure as described before22. Briefly, following the hemorrhage and shock periods rats were subjected to one of the following two procedures: Procedure 1 (called
non-resuscitation model) involved no fluid resuscitation, but the survival was monitored for a total of 4 hours. Procedure 2 (called resuscitation model) involved fluid resuscitation with 2X
Ringer’s lactate and euthanasia after three hours. Hemorrhage and Shock Method: The animals, fasted overnight, were anesthetized with 2.5% isoflurane (Henry Schein, Dublin, OH, USA) and a
midline laparotomy (5 cm) was performed, the incision was closed aseptically in two layers with sutures (Ethilon 5/0, Ethicon, AD Surgical, Sunnyvale, CA, USA). Two femoral arteries and one
femoral vein were cannulated (PE-50 tubing), one artery was connected to a blood pressure analyzer (Digi-Med; Micro-Med Inc., Louisville, KY, USA) to monitor mean arterial pressure (MAP);
bleeding was performed through the other artery. The resuscitation fluid and agents were administered through the femoral vein. Surgical sites were bathed with bupivacaine. Sham animals were
not subjected to bleeding or fluid resuscitation, but they were subjected to laparotomy and groin incisions. Upon awakening, the animals in the HI groups were bled rapidly to a MAP of 40 ±
5 mmHg. The bleeding was continued for 45 min, maintaining the low MAP, until 60% of circulatory blood volume was withdrawn. Following the maximum bleed out (MBO) the animals were maintained
in the state of shock by maintaining MAP at 40 ± 5 for another 45 min. Following this, either of the two procedures mentioned below was performed depending on the experimental goal. Blood
gas values were measured using Opti-Medical (Roswell, GA) OPTI CCA-TS2 analyzer. NON-RESUSCITATION MODEL The animals subjected to this procedure did not receive resuscitation fluid following
the above shock period. The experimental animals were randomly divided into four groups. One of the three doses of dichloroacetate (DCA, Sigma Aldrich, St. Lois, MO) was administered
intravenously at the end of the shock period to the respective groups. In the last group only vehicle was administered. 500–600 μl of Ringer’s Lactate (RL) was used as vehicle. The animals
were continuously observed for 4 hrs and as death cannot be an endpoint as per the IACUC policy, animals were euthanized when their MAP dipped below 30 mmHg. RESUSCITATION MODEL The animals
that underwent this procedure were subjected to fluid resuscitation by administering Ringers lactate, two times the volume of shed blood. Fluid resuscitation was performed for 1 hour from
the end of shock period. DCA or vehicle was administered 10 min following the start of fluid resuscitation. After fluid resuscitation, the animals were observed for three hours following
which they were euthanized, plasma collected and tissues were harvested for molecular studies. LACTATE ASSAY Heparinized blood samples were obtained prior to euthanasia. The plasma was
separated by centrifugation (2,000 g, 10 min) and stored at −80 °C until assayed for lactate levels. An aliquot of plasma was used to determine protein concentration (Bio-Rad DC Protein
Assay). Lactate levels were measured using a Lactate Assay Kit (Sigma St. Louis, MO 63103 USA) and expressed in relation to the total protein to account for hemodilution. PYRUVATE ASSAY
Pyruvate levels were measured using a fluorometric Pyruvate Assay Kit (Cayman Chemical Company, Ann Arbor, Michigan) according to the manufacturer’s directions and normalized to plasma total
protein (Bio-Rad DC Protein Assay). PDH ASSAY Heart tissues were prepared and PDH enzyme activity levels were measured at 450 nm using a colorimetric PDH Enzyme Activity Kit (Abcam,
Cambridge, MA) according to the manufacturer’s directions. Briefly, The PDH enzyme is immunocaptured within the wells of the microplate included with the kit. Activity is determined by
following the reduction of NAD+ to NADH, coupled to the reduction of a reporter dye to yield a coloured reaction product with an increase in absorbance at 450 nm at room temperature. SIRT1
ACTIVITY ASSAY The enzymatic activity of Sirt1 in the heart tissue was assayed by a fluorimetric assay using the SensoLyte Green Sirt1 assay kit (AnaSpec, Fremont, CA, USA) according to the
manufacturer’s directions. Sirtuin containing tissue protein samples were tested with the acetylated p53 peptide substrate provided with the kit. Deacetylation of substrate sensitizes it to
the color developer releasing the green fluorophore. The enzyme activity was assessed from the fluorescence signal generated in proportion to the amount of deacetylation of the lysine. ATP
MEASUREMENT A bioluminescence assay (ATP determination kit; Invitrogen) kit was used for the quantitative determination of ATP. The assay employed recombinant firefly luciferase and its
substrate D-luciferin. Briefly, reaction solution containing luciferase and luciferin was plated and background luminescence measured. ATP standard solution or sample containing ATP was
added to respective wells and luminescence was measured. ATP concentration was deduced from the standard curve and normalized to total protein concentration. HEXOKINASE ACTIVITY Hexokinase
activity was measured using the hexokinase colorimetric assay kit (Sigma-Aldrich. St. Louis, MO) according to the manufacturer’s instructions. Absorbance was measured at 563 nm using a
SpectraMax M5 plate reader (Molecular Devices). One unit of hexokinase is the amount of enzyme that will generate 1.0 mM of NADH per min at pH 8.0 and room temperature. The results were
normalized to the amount of total protein compared to the sham. WESTERN BLOT ANALYSIS The heart tissues were homogenized in Pierce RIPA lysis buffer (Thermo Scientific, Chicago, IL)
containing 25 mmol/L Tris-HCl pH 7.6, 150 mmol/L NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO, USA) in a D1000 handheld
homogenizer (Benchmark Scientific, Sayreville, NJ, USA). Heart tissue lysates were centrifuged at 14,000 g for 10 min and the supernatant saved for protein estimation and analysis. Protein
aliquots were combined with 4X Laemmli buffer (Bio-Rad, Hercules, CA, USA) and resolved on a 10% SDS polyacrylamide gel, transferred to PVDF membrane, blocked using 5% (w/v) nonfat dried
milk or 5% BSA in Tris-buffered saline containing 25 mmol/L Tris-HCl (pH 7. 4), 0.13 mol/L NaCl, 0.0027 mol/L KCl and 0.1% Tween 20 for 1 h at room temperature (RT) and then incubated with
respective antibodies overnight at 4 °C or for 1 h at RT. The membranes were probed with antibodies to Sirt-1, Pgc-1α, Bip, Pdk, P-AMPK, AMPK, P-ACC, and ACC. The membranes were subsequently
washed and incubated with horseradish peroxidase conjugated secondary antibody for 1 h at RT and developed using enhanced western lightning plus-ECL (PerkinElmer). Protein bands developed
on X-ray films were scanned from the original film and the Western Blot bands are presented in the manuscript without alteration (Supplementary Figures 3 and 4). The bands were quantified
using the ImageJ software (Wayne Rasband, NIH, Rockville, MD, USA). JC-1 ASSAY FOR MITOCHONDRIAL MEMBRANE POTENTIAL The experiment was performed using Isolated Mitochondria Staining kit from
Sigma Chemical Co., St. Louis, MO. Briefly, 5 µg sample of mitochondria isolated from the heart (Biovision, CA) was stained with a 0.2 µg/mL JC-1 solution. For a control, valinomycin, an
ionophore, was added to an identical 5 µg sample of isolated mitochondria to a final concentration of 0.5 μg/ml. This sample was placed on ice for 10 minutes to allow complete dissipation of
the membrane potential, and then stained with a 0.2 µg/mL JC-1 solution and assayed in parallel. Samples were read over a period of 30 minutes using a BIOTEK (Synergy HT) fluorescence plate
reader (excitation wavelength = 485 nm, emission wavelength = 590 nm). Results are expressed as a percentage of the valinomycin baseline control fluorescence. VASCULAR FUNCTION ASSESSMENT
Aorta was rapidly excised and placed in chilled Krebs solution of the following composition (mM): NaCl, 118; NaHCO3, 25; glucose, 5.6; KCl, 4.7; KH2PO4, 1.2; MgSO4 7H2O, 1.17 and CaCl2 2H2O,
2.5. After periadventitial adhering fat was removed, endothelial intact aorta were cleaned and cut into 2 mm rings. The aortic rings were mounted under resting tension of 10 mN in myograph
organ bath chambers (Danish Myo Technology A/S) filled with Krebs solution at 37 °C (pH 7.4) and continuously bubbled with a mixture of 95% O2 and 5% CO2. Isometric force was recorded using
a PowerLab/8SP data acquisition system (Software Chart, version 5, AD Instrument, Colorado Springs, CO, USA). The tissues were allowed to equilibrate for 1 h before starting the experiments.
After equilibration, aortic rings were contracted with high KCl solution (80 mM) to verify the viability of the preparations. After washing out KCl, cumulative concentration-response curves
to acetylcholine (ACh; 0.001–10 µM), an endothelium-dependent vasodilator or sodium nitroprusside (SNP; 0.0001–10 µM), a NO donor were generated with aortic rings. The rings were contracted
with phenylephrine (PE, an α1-adrenergic receptor agonist). Cumulative concentration-response curve to PE were also performed in aortic rings (0.001–10 µM) from Sham, HI and HI+ DCA treated
rats. STATISTICS Survival analysis was performed by Kaplan-Meier method and significance between survival curves was determined using GraphPad software (La Jolla, CA). Multigroup
comparisons were carried out and significance determined by one-way ANOVA followed by Tukey’s test using GraphPad software (Graphpad Prism, WA). Two-group comparisons for significance were
done by unpaired t test using GraphPad software. A p value of less than 0.05 is considered significant. REFERENCES * Camazine, M. N. _et al_. Massive transfusion policies at trauma centers
participating in the American College of Surgeons Trauma Quality Improvement Program. _J Trauma Acute Care Surg_ 78, S48–53, doi:10.1097/TA.0000000000000641 (2015). Article PubMed Google
Scholar * Minino, A. M., Anderson, R. N., Fingerhut, L. A., Boudreault, M. A. & Warner, M. Deaths:injuries, 2002. 1–124 (Centers for Disease Control and Prevention, National Center for
Health Statistics, National Vital Statistics System 2006). * Curry, N. _et al_. The acute management of trauma hemorrhage: a systematic review of randomized controlled trials. _Crit Care_
15, R92, doi:10.1186/cc10096 (2011). Article PubMed PubMed Central Google Scholar * Perel, P. _et al_. Predicting early death in patients with traumatic bleeding: development and
validation of prognostic model. _BMJ_ 345, e5166, doi:10.1136/bmj.e5166 (2012). Article PubMed PubMed Central Google Scholar * Shrestha, B. _et al_. Damage-control resuscitation
increases successful nonoperative management rates and survival after severe blunt liver injury. _J Trauma Acute Care Surg_ 78, 336–341, doi:10.1097/TA.0000000000000514 (2015). Article
PubMed Google Scholar * Holcomb, J. B. _et al_. Challenges to effective research in acute trauma resuscitation: consent and endpoints. _Shock_ 35, 107–113, doi:10.1097/SHK.0b013e3181f7fd01
(2011). Article PubMed Google Scholar * Spinella, P. C. & Holcomb, J. B. Resuscitation and transfusion principles for traumatic hemorrhagic shock. _Blood Rev_ 23, 231–240,
doi:10.1016/j.blre.2009.07.003 (2009). Article PubMed PubMed Central Google Scholar * Kozlov, A. V. _et al_. Mitochondrial dysfunction and biogenesis: do ICU patients die from
mitochondrial failure? _Ann Intensive Care_ 1, 41, doi:10.1186/2110-5820-1-41 (2011). Article PubMed PubMed Central Google Scholar * Szalay, L. _et al_. Estradiol improves cardiac and
hepatic function after trauma-hemorrhage: role of enhanced heat shock protein expression. _Am J Physiol Regul Integr Comp Physiol_ 290, R812–818, doi:10.1152/ajpregu.00658.2005 (2006).
Article CAS PubMed Google Scholar * Lu, A. _et al_. Mitochondria play an important role in 17beta-estradiol attenuation of H(2)O(2)-induced rat endothelial cell apoptosis. _Am J Physiol
Endocrinol Metab_ 292, E585–593, doi:10.1152/ajpendo.00413.2006 (2007). Article CAS PubMed Google Scholar * Raju, R., Bland, K. I. & Chaudry, I. H. Estrogen: a novel therapeutic
adjunct for the treatment of trauma-hemorrhage-induced immunological alterations. _Mol Med_ 14, 213–221, doi:10.2119/2008-00001.Raju (2008). CAS PubMed PubMed Central Google Scholar *
Kalogeris, T., Baines, C. P., Krenz, M. & Korthuis, R. J. Cell biology of ischemia/reperfusion injury. _Int Rev Cell Mol Biol_ 298, 229–317, doi:10.1016/B978-0-12-394309-5.00006-7
(2012). Article CAS PubMed PubMed Central Google Scholar * Ham, P. B. 3rd & Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. _Prog Neurobiol_.
doi:10.1016/j.pneurobio.2016.06.006 (2016). PubMed Google Scholar * Poulose, N. & Raju, R. Sirtuin regulation in aging and injury. _Biochim Biophys Acta_ 1852, 2442–2455,
doi:10.1016/j.bbadis.2015.08.017 (2015). Article CAS PubMed PubMed Central Google Scholar * Thiessen, S. E., Van den Berghe, G. & Vanhorebeek, I. Mitochondrial and endoplasmic
reticulum dysfunction and related defense mechanisms in critical illness-induced multiple organ failure. _Biochim Biophys Acta_, doi:10.1016/j.bbadis.2017.02.015 (2017). * Hsieh, Y. C. _et
al_. Upregulation of mitochondrial respiratory complex IV by estrogen receptor-beta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following
trauma-hemorrhage. _J Mol Cell Cardiol_ 41, 511–521, doi:10.1016/j.yjmcc.2006.06.001 (2006). Article CAS PubMed Google Scholar * Poulose, N. & Raju, R. Aging and injury: alterations
in cellular energetics and organ function. _Aging Dis_ 5, 101–108, doi:10.14336/AD.2014.0500101 (2014). PubMed PubMed Central Google Scholar * Wang, H. _et al_. Resveratrol Rescues Kidney
Mitochondrial Function Following Hemorrhagic Shock. _Shock_ 44, 173–180, doi:10.1097/SHK.0000000000000390 (2015). Article CAS PubMed PubMed Central Google Scholar * Wang, H. _et al_.
Resveratrol ameliorates mitochondrial dysfunction but increases the risk of hypoglycemia following hemorrhagic shock. _J Trauma Acute Care Surg_ 77, 926–933, doi:10.1097/TA.0000000000000358
(2014). Article CAS PubMed PubMed Central Google Scholar * Jian, B., Yang, S., Chaudry, I. H. & Raju, R. Resveratrol improves cardiac contractility following trauma-hemorrhage by
modulating Sirt1. _Mol Med_ 18, 209–214, doi:10.2119/molmed.2011.00365 (2012). Article CAS PubMed Google Scholar * Yu, H. P., Yang, S. C., Lau, Y. T. & Hwang, T. L. Role of
Akt-dependent up-regulation of hemeoxygenase-1 in resveratrol-mediated attenuation of hepatic injury after trauma hemorrhage. _Surgery_ 148, 103–109, doi:10.1016/j.surg.2009.12.008 (2010).
Article PubMed Google Scholar * Ayub, A., Poulose, N. & Raju, R. Resveratrol Improves Survival and Prolongs Life Following Hemorrhagic Shock. _Mol Med_ 21, 305–312,
doi:10.2119/molmed.2015.00013 (2015). Article CAS PubMed PubMed Central Google Scholar * Gomes, A. P. _et al_. Declining NAD(+) induces a pseudohypoxic state disrupting
nuclear-mitochondrial communication during aging. _Cell_ 155, 1624–1638, doi:10.1016/j.cell.2013.11.037 (2013). Article CAS PubMed PubMed Central Google Scholar * Jian, B., Yang, S.,
Chaudry, I. H. & Raju, R. Resveratrol restores sirtuin 1 (SIRT1) activity and pyruvate dehydrogenase kinase 1 (PDK1) expression after hemorrhagic injury in a rat model. _Mol Med_ 20,
10–16, doi:10.2119/molmed.2013.00077 (2014). Article PubMed Google Scholar * Harris, R. A., Bowker-Kinley, M. M., Huang, B. & Wu, P. Regulation of the activity of the pyruvate
dehydrogenase complex. _Adv Enzyme Regul_ 42, 249–259 (2002). Article CAS PubMed Google Scholar * Bersin, R. M. & Stacpoole, P. W. Dichloroacetate as metabolic therapy for myocardial
ischemia and failure. _Am Heart J_ 134, 841–855 (1997). Article CAS PubMed Google Scholar * Lewis, J. F., DaCosta, M., Wargowich, T. & Stacpoole, P. Effects of dichloroacetate in
patients with congestive heart failure. _Clin Cardiol_ 21, 888–892 (1998). Article CAS PubMed Google Scholar * Kato, T. _et al_. Analysis of metabolic remodeling in compensated left
ventricular hypertrophy and heart failure. _Circ Heart Fail_ 3, 420–430, doi:10.1161/CIRCHEARTFAILURE.109.888479 (2010). Article PubMed Google Scholar * Abdelmalak, M. _et al_. Long-term
safety of dichloroacetate in congenital lactic acidosis. _Mol Genet Metab_ 109, 139–143, doi:10.1016/j.ymgme.2013.03.019 (2013). Article CAS PubMed PubMed Central Google Scholar *
James, M. O. _et al_. Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. _Pharmacol Ther_ 170, 166–180, doi:10.1016/j.pharmthera.2016.10.018 (2017).
Article CAS PubMed Google Scholar * Kankotia, S. & Stacpoole, P. W. Dichloroacetate and cancer: new home for an orphan drug? _Biochim Biophys Acta_ 1846, 617–629,
doi:10.1016/j.bbcan.2014.08.005 (2014). CAS PubMed Google Scholar * Constantin-Teodosiu, D. Regulation of muscle pyruvate dehydrogenase complex in insulin resistance: effects of exercise
and dichloroacetate. _Diabetes Metab J_ 37, 301–314, doi:10.4093/dmj.2013.37.5.301 (2013). Article PubMed PubMed Central Google Scholar * Zholobak, N. M. _et al_. Effect of tilorone and
its analogues on the change of mitochondrial potential of rat hepatocytes. _Fiziol Zh_ 58, 39–43 (2012). CAS PubMed Google Scholar * Romero, M. J. _et al_. Diabetes-induced coronary
vascular dysfunction involves increased arginase activity. _Circ Res_ 102, 95–102, doi:10.1161/CIRCRESAHA.107.155028 (2008). Article CAS PubMed Google Scholar * Barringer, M. L.,
Thomason, M. H., Kilgo, P. & Spallone, L. Improving outcomes in a regional trauma system: impact of a level III trauma center. _Am J Surg_ 192, 685–689, doi:10.1016/j.amjsurg.2005.11.006
(2006). Article PubMed Google Scholar * Tansley, G., Schuurman, N., Amram, O. & Yanchar, N. Spatial Access to Emergency Services in Low- and Middle-Income Countries: A GIS-Based
Analysis. _PLoS One_ 10, e0141113, doi:10.1371/journal.pone.0141113 (2015). Article PubMed PubMed Central Google Scholar * Ulvik, A., Wentzel-Larsen, T. & Flaatten, H. Trauma
patients in the intensive care unit: short- and long-term survival and predictors of 30-day mortality. _Acta Anaesthesiol Scand_ 51, 171–177, doi:10.1111/j.1399-6576.2006.01207.x (2007).
Article CAS PubMed Google Scholar * Choudhry, M. A. _et al_. Gender differences in acute response to trauma-hemorrhage. _Shock_ 24(Suppl 1), 101–106 (2005). Article PubMed Google
Scholar * Toth, B. _et al_. Sex differences in hepatic heme oxygenase expression and activity following trauma and hemorrhagic shock. _Arch Surg_ 138, 1375–1382,
doi:10.1001/archsurg.138.12.1375 (2003). Article CAS PubMed Google Scholar * Kimmoun, A., Novy, E., Auchet, T., Ducrocq, N. & Levy, B. Hemodynamic consequences of severe lactic
acidosis in shock states: from bench to bedside. _Crit Care_ 19, 175, doi:10.1186/s13054-015-0896-7 (2015). Article PubMed PubMed Central Google Scholar * Gomez, H. _et al_. Inhaled
Carbon Monoxide Protects against the Development of Shock and Mitochondrial Injury following Hemorrhage and Resuscitation. _PLoS One_ 10, e0135032, doi:10.1371/journal.pone.0135032 (2015).
Article PubMed PubMed Central Google Scholar * Rao, G., Xie, J., Hedrick, A. & Awasthi, V. Hemorrhagic shock-induced cerebral bioenergetic imbalance is corrected by pharmacologic
treatment with EF24 in a rat model. _Neuropharmacology_ 99, 318–327, doi:10.1016/j.neuropharm.2015.07.033 (2015). Article CAS PubMed PubMed Central Google Scholar * Powell, R. D. _et
al_. Resveratrol attenuates hypoxic injury in a primary hepatocyte model of hemorrhagic shock and resuscitation. _J Trauma Acute Care Surg_ 76, 409–417, doi:10.1097/TA.0000000000000096
(2014). Article CAS PubMed Google Scholar * Yu, H. P., Hwang, T. L., Hwang, T. L., Yen, C. H. & Lau, Y. T. Resveratrol prevents endothelial dysfunction and aortic superoxide
production after trauma hemorrhage through estrogen receptor-dependent hemeoxygenase-1 pathway. _Crit Care Med_ 38, 1147–1154, doi:10.1097/CCM.0b013e3181cd124e (2010). Article CAS PubMed
Google Scholar * Miquel, E. _et al_. Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis.
_PLoS One_ 7, e34776, doi:10.1371/journal.pone.0034776 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Sutendra, G. _et al_. Mitochondrial activation by inhibition of
PDKII suppresses HIF1a signaling and angiogenesis in cancer. _Oncogene_ 32, 1638–1650, doi:10.1038/onc.2012.198 (2013). Article CAS PubMed Google Scholar * Vander Heiden, M. G., Cantley,
L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. _Science_ 324, 1029–1033, doi:10.1126/science.1160809 (2009). Article ADS
Google Scholar * Bonnet, S. _et al_. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. _Cancer Cell_ 11, 37–51,
doi:10.1016/j.ccr.2006.10.020 (2007). Article CAS PubMed Google Scholar * Saed, G. M., Fletcher, N. M., Jiang, Z. L., Abu-Soud, H. M. & Diamond, M. P. Dichloroacetate induces
apoptosis of epithelial ovarian cancer cells through a mechanism involving modulation of oxidative stress. _Reprod Sci_ 18, 1253–1261, doi:10.1177/1933719111411731 (2011). Article CAS
PubMed Google Scholar * Gogvadze, V., Orrenius, S. & Zhivotovsky, B. Mitochondria in cancer cells: what is so special about them? _Trends Cell Biol_ 18, 165–173,
doi:10.1016/j.tcb.2008.01.006 (2008). Article CAS PubMed Google Scholar * Ba, Z. F. _et al_. Attenuation of vascular endothelial dysfunction by testosterone receptor blockade after
trauma and hemorrhagic shock. _Arch Surg_ 136, 1158–1163 (2001). Article CAS PubMed Google Scholar * Hierholzer, C. & Billiar, T. R. In _Surgical treatment: evidence-based and
problem-oriented_ (eds Holzheimer, R. & John, A. Mannick) xxiii, 843 p. (Zuckschwerdt, 2001). * Jager, S., Handschin, C., St-Pierre, J. & Spiegelman, B. M. AMP-activated protein
kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. _Proc Natl Acad Sci USA_ 104, 12017–12022, doi:10.1073/pnas.0705070104 (2007). Article ADS PubMed PubMed
Central Google Scholar * Allende-Vega, N. _et al_. The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism. _Oncotarget_
6, 19228–19245, doi:10.18632/oncotarget.4653 (2015). Article PubMed PubMed Central Google Scholar * Ruderman, N. B. _et al_. AMPK and SIRT1: a long-standing partnership? _Am J Physiol
Endocrinol Metab_ 298, E751–760, doi:10.1152/ajpendo.00745.2009 (2010). Article CAS PubMed PubMed Central Google Scholar * Hou, X. _et al_. SIRT1 regulates hepatocyte lipid metabolism
through activating AMP-activated protein kinase. _J Biol Chem_ 283, 20015–20026, doi:10.1074/jbc.M802187200 (2008). Article CAS PubMed PubMed Central Google Scholar * Ivanov, V. N. _et
al_. Resveratrol sensitizes melanomas to TRAIL through modulation of antiapoptotic gene expression. _Exp Cell Res_ 314, 1163–1176, doi:10.1016/j.yexcr.2007.12.012 (2008). Article CAS
PubMed Google Scholar * Lan, F., Cacicedo, J. M., Ruderman, N. & Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in
AMP-activated protein kinase activation. _J Biol Chem_ 283, 27628–27635, doi:10.1074/jbc.M805711200 (2008). Article CAS PubMed PubMed Central Google Scholar * Zaha, V. G. & Young,
L. H. AMP-activated protein kinase regulation and biological actions in the heart. _Circ Res_ 111, 800–814, doi:10.1161/CIRCRESAHA.111.255505 (2012). Article CAS PubMed PubMed Central
Google Scholar * Price, N. L. _et al_. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. _Cell Metab_ 15, 675–690,
doi:10.1016/j.cmet.2012.04.003 (2012). Article CAS PubMed PubMed Central Google Scholar * Stacpoole, P. W., Nagaraja, N. V. & Hutson, A. D. Efficacy of dichloroacetate as a
lactate-lowering drug. _J Clin Pharmacol_ 43, 683–691 (2003). Article CAS PubMed Google Scholar * Sun, Y. _et al_. Dichloroacetate treatment improves mitochondrial metabolism and reduces
brain injury in neonatal mice. _Oncotarget_ 7, 31708–31722, doi:10.18632/oncotarget.9150 (2016). PubMed PubMed Central Google Scholar * Zhu, D. _et al_. Crosstalk among proteome,
acetylome and succinylome in colon cancer HCT116 cell treated with sodium dichloroacetate. _Sci Rep_ 6, 37478, doi:10.1038/srep37478 (2016). Article ADS CAS PubMed PubMed Central Google
Scholar * Kline, J. A. _et al_. Activation of pyruvate dehydrogenase improves heart function and metabolism after hemorrhagic shock. _J Mol Cell Cardiol_ 29, 2465–2474,
doi:10.1006/jmcc.1997.0483 (1997). Article CAS PubMed Google Scholar * Barbee, R. W., Kline, J. A. & Watts, J. A. Depletion of lactate by dichloroacetate reduces cardiac efficiency
after hemorrhagic shock. _Shock_ 14, 208–214 (2000). Article CAS PubMed Google Scholar * Boyce, M. & Yuan, J. Cellular response to endoplasmic reticulum stress: a matter of life or
death. _Cell Death Differ_ 13, 363–373, doi:10.1038/sj.cdd.4401817 (2006). Article CAS PubMed Google Scholar * Kincaid, M. M. & Cooper, A. A. ERADicate ER stress or die trying.
_Antioxid Redox Signal_ 9, 2373–2387, doi:10.1089/ars.2007.1817 (2007). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS The study was supported by grants from the
National Institute of General Medical Sciences (R01 GM 101927) to RR and laboratory start up assistance (to R.R.) from Augusta University (formerly Georgia Regents University), Augusta, GA.
AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Laboratory Sciences, Augusta University, Augusta, GA, 30912, United States of America Kumar Subramani, Sumin Lu, Marie Warren,
Xiaogang Chu & Raghavan Raju * Department of Surgery, Augusta University, Augusta, GA, 30912, United States of America Raghavan Raju * Department of Biochemistry and Molecular Biology,
Augusta University, Augusta, GA, 30912, United States of America Raghavan Raju * Department of Pharmacology and Toxicology, Augusta University, Augusta, GA, 30912, United States of America
Haroldo A. Toque & R. William Caldwell * Department of Obstetrics and Gynaecology, Augusta University, Augusta, GA, 30912, United States of America Michael P. Diamond Authors * Kumar
Subramani View author publications You can also search for this author inPubMed Google Scholar * Sumin Lu View author publications You can also search for this author inPubMed Google Scholar
* Marie Warren View author publications You can also search for this author inPubMed Google Scholar * Xiaogang Chu View author publications You can also search for this author inPubMed
Google Scholar * Haroldo A. Toque View author publications You can also search for this author inPubMed Google Scholar * R. William Caldwell View author publications You can also search for
this author inPubMed Google Scholar * Michael P. Diamond View author publications You can also search for this author inPubMed Google Scholar * Raghavan Raju View author publications You can
also search for this author inPubMed Google Scholar CONTRIBUTIONS Conceived the experiments: M.P.D. and R.R. Designed the experiments: K.S. and R.R. Experiments for Figures 1 and 2 and all
animal experiments: KS. Experiments for Figures 3, 4 and 5: K.S., S.L. and X.C. Experiments for Figure 6: M.W. Experiments for Figure 7: H.A.T. Wrote the manuscript: K.S., R.W.C., M.P.D. and
R.R. All authors reviewed the manuscript. CORRESPONDING AUTHOR Correspondence to Raghavan Raju. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare that they have no competing
interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. ELECTRONIC
SUPPLEMENTARY MATERIAL SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Subramani, K., Lu, S., Warren, M. _et al._ Mitochondrial targeting by dichloroacetate improves outcome following hemorrhagic shock. _Sci Rep_
7, 2671 (2017). https://doi.org/10.1038/s41598-017-02495-5 Download citation * Received: 20 September 2016 * Accepted: 12 April 2017 * Published: 01 June 2017 * DOI:
https://doi.org/10.1038/s41598-017-02495-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative










