- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Studies have characterized the immune escape landscape across primary tumors. However, whether late-stage metastatic tumors present differences in genetic immune escape (GIE)
prevalence and dynamics remains unclear. We performed a pan-cancer characterization of GIE prevalence across six immune escape pathways in 6,319 uniformly processed tumor samples. To address
the complexity of the HLA-I locus in the germline and in tumors, we developed LILAC, an open-source integrative framework. One in four tumors harbors GIE alterations, with high mechanistic
and frequency variability across cancer types. GIE prevalence is generally consistent between primary and metastatic tumors. We reveal that GIE alterations are selected for in tumor
evolution and focal loss of heterozygosity of _HLA-I_ tends to eliminate the HLA allele, presenting the largest neoepitope repertoire. Finally, high mutational burden tumors showed a
tendency toward focal loss of heterozygosity of _HLA-I_ as the immune evasion mechanism, whereas, in hypermutated tumors, other immune evasion strategies prevail. SIMILAR CONTENT BEING
VIEWED BY OTHERS IMMUNOGENOMIC PAN-CANCER LANDSCAPE REVEALS IMMUNE ESCAPE MECHANISMS AND IMMUNOEDITING HISTORIES Article Open access 03 August 2021 FREQUENCY-DEPENDENT SELECTION OF
NEOANTIGENS FOSTERS TUMOR IMMUNE ESCAPE AND PREDICTS IMMUNOTHERAPY RESPONSE Article Open access 25 June 2024 PERSISTENT MUTATION BURDEN DRIVES SUSTAINED ANTI-TUMOR IMMUNE RESPONSES Article
Open access 26 January 2023 MAIN Cancer immune escape is the process whereby tumor cells prevent their elimination by the immune system1,2. Tumors acquire this capacity as a response to the
accumulation of tumor-specific alterations, which may be presented—in the form of neoepitopes—by the major histocompatibility complex class I (MHC-I). Escape from immune system recognition
often involves tumor-specific genomic alterations in immune-related pathways, a process named genetic immune escape (GIE). GIE alterations operate through different mechanisms, including
partial or complete abrogation of neoepitope presentation3 or suppression of proapoptotic signals from the surrounding immune cells4. Therefore, identification of GIE events across human
cancers is key to understanding the interplay between cancer cells and the immune system, as well as to enable effective precision medicine based on immunotherapy. Previous studies have
performed cancer type-specific molecular profiling of GIE events and their phenotypic implications in several cancer types, including non-small-cell lung cancer5,6 (NSCLC) and colorectal
carcinoma7, among others8,9. Others have performed an extensive analysis of loss of heterozygosity (LOH) of _HLA-I_ across thousands of tumor samples10. However, a pan-cancer analysis of the
prevalence and impact of diverse GIE events is currently lacking. In addition, the focus of these studies was to portray GIE in early stage primary tumors, whereas the changes induced by
exposure to treatment and by the metastatic bottleneck have not been comprehensively addressed. One of the main challenges to perform such analyses lies in the extraordinary diversity of the
_HLA-I_ locus, with >15,000 different sequences of the _HLA-A_, _HLA-B_ and _HLA-C_ genes reported to date11. This extensive polymorphism hampers the identification of tumor-specific
somatic alterations, prompting the development of tools that specifically identify LOH of _HLA-I_ (ref. 12) or _HLA-I_ somatic mutations13 from whole-exome sequencing (WES) and whole-genome
sequencing (WGS) data. However, none of these tools provides an integrative characterization of the _HLA-I_ tumor status in both the germline and the tumor, which includes _HLA-I_ typing,
allelic imbalance, LOH of _HLA-I_ and somatic mutation annotation. In the present study, we present a pan-cancer landscape of the GIE prevalence in primary (represented by the PCAWG
(pan-cancer analysis of whole genomes) cohort) and unmatched metastatic patients (represented by the Hartwig cohort). Furthermore, to address the complexity of the _HLA-I_ locus, we
developed LILAC, an open-source integrative framework that characterizes the _HLA-I_ locus, including its tumor status from WGS data. We applied LILAC and a universal tumor-processing
pipeline to establish a comprehensive portrait of GIE events and their positive selection landscape across six different pathways associated with an immune evasion phenotype: the _HLA-I_
locus, the antigen presentation machinery, interferon (IFN)-γ signaling pathway, the programmed cell death ligand 1 (PD-L1) immune checkpoint, the costimulatory signaling by the CD58
receptor and epigenetic immune escape driven by _SETDB1_ (Fig. 1a and Supplementary Table 1). We also studied how the tumor mutational burden (TMB) and other genomic and environmental
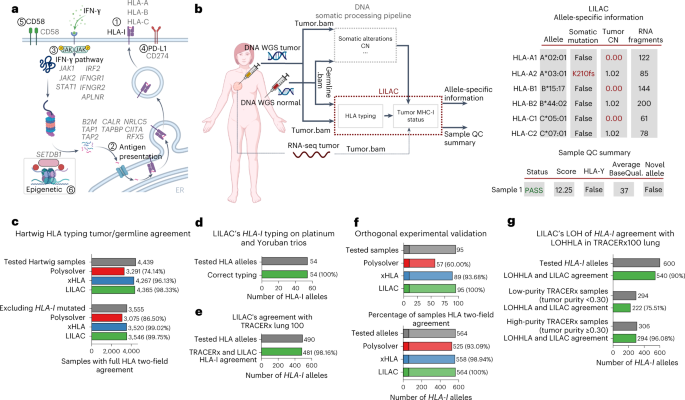
features influence the prevalence of GIE alterations, providing insights into tumorigenesis and its interplay with the immune system. RESULTS INFERENCE OF _HLA-I_ TUMOR STATUS WITH LILAC
Inference of the correct _HLA-I_ tumor status is fundamental to identifying GIE alterations (Fig. 1a), to estimate the neoepitope repertoire and burden and to predict the response to immune
checkpoint inhibitors14,15 (ICIs). We have developed LILAC, a framework that performs _HLA-I_ typing for the germline of each patient, as well as determining the status of each of those
alleles in the tumor using WGS data on tumor-normal paired samples as input. LILAC also allows for detection of novel human leukocyte antigen (HLA) alleles and provides allele-specific and
sample-level, quality control measurements (Fig. 1b and Supplementary Note 1). We first assessed LILAC’s _HLA-I_ typing robustness by independently calculating the germline and tumor _HLA-I_
two-field-calling agreement across 6,279 patients, including 4,439 patients from the Hartwig16 dataset and 1,839 from the PCAWG17 cohort. LILAC showed the highest agreement compared with
two state-of-the-art _HLA_ typing tools, Polysolver13 and xHLA18 (Fig. 1c, Extended Data Fig. 1a and Supplementary Data 1). The Hartwig dataset showed higher normal-tumor agreement for all
tools, possibly due to the higher sequencing coverage and read quality of this dataset. In a three-way comparison, LILAC also displayed the highest overlap with the predictions from the
other tools across both datasets (Extended Data Fig. 1b,c). Moreover, LILAC’s _HLA-I_ typing performance on three family trios with diverse genetic ancestries showed a perfect agreement with
previously reported _HLA-I_ types (Fig. 1d). Next, we demonstrated WES applicability by running LILAC on the TRACERx100 lung cohort, where it showed a 98.16% agreement with the _HLA-I_
types originally reported in the publication12 (Fig. 1e). Finally, we evaluated LILAC _HLA-I_ typing sensitivity in a set of 95 samples with challenging _HLA-I_ types—including 10 from tumor
biopsies—with an independent orthogonal and clinically validated _HLA-I_ typing approach (Supplementary Note 1). LILAC showed a perfect 100% two-field agreement across the 564 alleles,
higher than Polysolver (93.09%) and xHLA (98.94%) agreements (Fig. 1f and Supplementary Data 1). To conclude, LILAC reported nine somatic mutations in seven of the tumor biopsies evaluated.
All of them were perfectly matched by the orthogonal approach (Supplementary Data 1). HLA allele-specific, tumor copy number (CN) determination is key to identify LOH of _HLA-I_ genes in
tumors, a well-established mechanism of immune evasion10,12. LILAC annotates allele-specific ploidy levels of each _HLA-I_ allele based on the purity-corrected local tumor CN estimations and
the number of fragments assigned to each allele (Supplementary Note 1). WGS data provide adequate resolution to annotate purity-adjusted minor and major allele ploidy in the HLA-I locus
(Extended Data Fig. 1d,e). Moreover, we quantified LILAC’s agreement with LOHHLA12 in the TRACERx100 lung WES cohort. LILAC and LOHHLA estimates displayed a global 90% agreement (Fig. 1g and
Supplementary Data 1). Importantly, high tumor purity samples showed considerably better concordance than low-purity samples (96.08% in samples with tumor purity ≥0.3, 75.51% when tumor
purity <0.3), reflecting increased challenges for genome-wide CN loss calling in low-purity WES samples. Finally, the three tumor samples harboring LOH of _HLA-I_, according to our
framework and evaluated by the orthogonal approach, displayed a strong allelic imbalance in the experimental validation (Supplementary Data 1). GIE PREVALENCE ACROSS CANCER TYPES We then
combined LILAC with the Hartwig tumor analytical cancer WGS pipeline16,19 to annotate GIE events across 6 pathways strongly associated with immune escape (Fig. 1a and Supplementary Table 1)
across 6,319 uniformly processed WGS samples20, including 1,880 primary patients from PCAWG and 4,439 patients with metastases from Hartwig (Fig. 2a, Extended Data Fig. 2a, Supplementary
Table 2 and Supplementary Note 1). In total, these patients were classified into 58 cancer types, which included 30 tumor types with sufficiently high representativeness (that is, number of
patients ≥15) in the metastatic cohort, 27 in the primary dataset and 20 cancer types with sufficient representation in both datasets (Fig. 2b, Extended Data Fig. 2b,c and Supplementary
Table 2). GIE prevalence showed high mechanistic and frequency variability across primary and metastatic cancer types (Fig. 2c,d, top panels and Supplementary Data 2). The median proportion
of patients harboring GIE alterations per cancer type was 0.27 for the metastatic cohort and 0.20 for primary tumors, both showing highly dispersed distributions (±0.15 s.d. and ±0.19 s.d.
in metastatic and primary tumors, respectively). In certain cancer types, such as pancreatic neuroendocrine (PANET, metastatic), diffuse large B-cell lymphoma (DLBCL, metastatic) and kidney
chromophobe cancer (KICH, primary), GIE was present in >50% of patient samples (65%, 55% and 74%, respectively) whereas in others, such as lung neuroendocrine (LUNET, metastatic), GIE was
an extremely rare event. Overall, one in four patients (26% in metastatic and 24% in primary) presented GIE alterations based on the six investigated pathways (Fig. 2c,d, bottom panels).
The most frequent GIE alteration was partial loss of the _HLA_-_I_ locus (including both LOH of _HLA-I_ and homozygous deletions of _HLA-I_ genes that were grouped as LOH of _HLA-I_ for
simplicity), which was present in 783 (18%) of metastatic and 319 (17%) of primary cancer patients, followed by IFN-γ inactivation (4% in metastatic and 3% in primary) and alterations in the
antigen presentation pathway (4% in metastatic and 3% in primary). CD58 inactivation was the least frequent immune escape event present in only 16 metastatic and 8 primary patients. The
high GIE rates of KICH and PANET were exclusively due to LOH of _HLA-I_ (Fig. 2e), whereas other cancer types displayed a wider range of GIE mechanisms (Fig. 2f). Of note, we did not observe
a significant mutual exclusivity between LOH of _HLA-I_ and other GIE events in cancer types with sufficient representation of multiple GIE mechanisms (Supplementary Data 2). This suggests
that certain tumors may require complementary GIE alterations, such as concurrent alterations that disrupt _HLA-I_-mediated neoepitope presentation and _CD58_ loss21, to effectively escape
immune surveillance. HIGH AGREEMENT BETWEEN PRIMARY AND METASTATIC GIE RATES We next sought to investigate whether there was a GIE prevalence difference between early stage primary and
late-stage metastatic tumors. Comparison by tumor type across the 20 cancer types with sufficient representation showed a broad agreement between both stages (Fig. 3a). Although nine cancer
types showed a certain degree of metastatic enrichment (log2(odds ratio) (log2(OR)) > 0.5; Fig. 3a,b), only in prostate carcinoma (PRAD) and thyroid cancer (THCA) was this difference
statistically significant (Fisher’s exact test corrected _P_ < 0.01). The significant enrichment in these two cancer types might be connected to the substantial genome transformation at
the metastatic transition20. Breaking down pathway-specific differences revealed that THCA metastatic enrichment is the result of increased LOH of _HLA-I_ incidence, whereas the
discrepancies in PRAD are the result of a widespread enrichment across several pathways (Fig. 3b). In general, LOH of _HLA-I_ showed a nonsignificant trend toward metastatic enrichment
across seven of the nine metastatic-enriched cancer types. None of the cancer types showed a significantly higher GIE incidence in primary tumors. POSITIVE SELECTION OF _HLA-I_ ALTERATIONS
We next examined to what extent somatic alterations in _HLA-I_ genes (that is, _HLA-A_, _HLA-B_ and _HLA-C_) were positively selected during tumorigenesis. First, a pan-cancer-grouped
_HLA-I_ analysis revealed a nonsynonymous:synonymous substitution (dN:dS) ratio >1 for nonsense, splice site and truncating variants in both the metastatic and the primary datasets (Fig.
4a), indicating that these genes are subject to positive selection. Next, pan-cancer and gene-specific dN:dS ratios showed that _HLA-A_ and _HLA-B_, but not _HLC-C_, are positively selected
and are mostly enriched in truncating variants but not in missense mutations (Fig. 4b,c). Finally, gene and cancer type-specific analysis showed that _HLA-A_ and _HLA-B_ were deemed as
drivers across several cancer types, including metastatic colorectal, NSCLC and DLBCL as well as the pan-cancer cohorts (Fig. 4d,e and Supplementary Data 3). Somatic point mutations and
small indels (insertions and deletions) of _HLA-I_ genes were evenly distributed along their sequences (Fig. 4f and Extended Data Fig. 3a). The main exception was the recurrent _HLA-A_
Lys210 frameshift indel (chromosome 6 at position 29911899), which was observed in six mismatch repair-deficient (MMRd) metastatic tumors. This genomic region overlaps with a (C)7
homopolymer repeat, which probably explains its susceptibility for the observed base indel. No enrichment for mutations in amino acids involved in the peptide binding was observed. Such
uniform distribution was in agreement with previous observations22 and with the expected profile in tumor-suppressor genes dominated by inactivating variants23. LOH of _HLA-I_ trims the
repertoire of _HLA-I_-presented epitopes in _HLA-I_ heterozygous individuals. Therefore, to further shed light on the tumorigenic role of LOH of _HLA-I_, we developed a randomization
strategy that pinpoints cancer types where the LOH of _HLA-I_ rates were significantly higher than the expected, given their background LOH rates using three genomic resolutions (that is,
nonfocal LOH including all LOH events spanning >75% of the chromosome arm length, focal LOH for those events <75% of the chromosome arm and highly focal LOH for LOH events <3 Mb).
In spite of the global correlation with background genome-wide LOH rates (Extended Data Fig. 3b), our analyses revealed higher-than-expected rates of LOH of _HLA-I_ across several cancer
types in both the metastatic and the primary datasets (G-test goodness of fit _q_ value <0.1; Fig. 4d,e and Supplementary Data 3). PANET (Fig. 4g) and KICH (Extended Data Fig. 3c) showed
nonfocal LOH of _HLA-I_ enrichment. Others, such as metastatic cervix carcinoma (Fig. 4h), metastatic colorectal cancer (Fig. 4i) or primary DLBCL, showed focal or highly focal LOH of
_HLA-I_ patterns. Furthermore, 33 patients with nonsynonymous mutations of _HLA-I_ genes (20% of the total 159 patients with mutations in _HLA-I_ genes) displayed the concurrent loss of the
alternative allele by LOH, potentially leading to complete inactivation of the _HLA_ gene. Finally, we did not observe any biallelic deletion of the entire _HLA-I_ locus (Supplementary Data
3), suggesting that homozygous deletions within the _HLA-I_ might be constrained by purifying selection, featuring the importance of expressing a minimal amount of _HLA-I_ molecules to avoid
immune-alerter signals24. DIFFERENCES BETWEEN FOCAL AND NONFOCAL LOH OF HLA-I Our results suggest that LOH of _HLA-I_ is a positively selected genomic event in certain tumor types. However,
it remains unclear whether these losses target a specific allele and whether both focal and nonfocal LOH of HLA events display similar selective patterns. To address these questions, we
assessed whether LOH of _HLA-I_ tends to involve the allele(s) with the highest neoepitope ratio (that is, higher number of predicted neoepitopes compared with the alternative allele; Fig.
5a). We observed a positive association between the neoepitope ratio and the frequency of the allele with highest neoepitope repertoire to be lost in both the metastatic and the primary
cohorts (Fig. 5b,c). This trend was significantly different from a neutral scenario where both alleles are equally likely to be lost independently of their neoepitope repertoire
(Kolmogorov–Smirnov test metastatic _P_ = 2.47 × 10−5 and primary _P_ = 2.24 × 10−7). Remarkably, the association between neoepitope ratio and the loss frequency became stronger when
selecting for focal LOH of _HLA-I_ events (Fig. 5d,e; _P_ = 1.71 × 10−9 and _P_ = 1.17 × 10−10 for metastatic and primary, respectively). However, it was indistinguishable from a neutral
scenario for nonfocal LOH of _HLA-I_ (Fig. 5f,g; _P_ = 0.32 and _P_ = 0.99 for metastatic and primary, respectively), showing that nonfocal LOH of _HLA-I_ does not select for the allele with
the highest neoepitope repertoire and that its high recurrency in several cancer types may be associated with other selective forces operating on chromosome 6. Furthermore, the majority of
focal LOH of _HLA-I_ events were CN neutral (81% in metastatic tumors and 70% in primary), which was considerably higher than for nonfocal events (65% in metastatic and 35% in primary),
providing further support for the notion that the loss of neoepitope repertoire, and not gene dosage, is the main driving force behind focal LOH of _HLA-I_. POSITIVE SELECTION OF GIE
ALTERATIONS BEYOND _HLA-I_ Alterations in other pathways beyond the HLA-I locus may also lead to immune escape. Hence, we explored signals of positive selection across 18 genes associated
with 5 immune escape pathways (pathways 2–6 in Fig. 1a). Grouped pan-cancer analysis of the dN:dS ratio in these pathways (covering a total of 16 genes, excluding those with an oncogenic
mechanism based on CN amplification; Methods) revealed a >1 ratio for nonsense, splice site and truncating variants in both the metastatic and the primary datasets (Fig. 6a), which was
indicative of positive selection. Refining the analysis for specific genes and cancer types revealed that two genes from the antigen presentation pathway (that is, _B2M_ and _CALR_)
displayed recurrent patterns of inactivating mutations and focal biallelic deletions across several tumor types, as well as in the pan-cancer cohorts (Fig. 6b–d). Moreover,
higher-than-expected frequencies of focal biallelic deletions for several IFN-γ pathway genes, including _JAK1_, _JAK2_ and _IRF2_, were also observed. _CD58_ also harbored a
higher-than-expected number of nonsynonymous mutations and homozygous deletions in DLBCL and the pan-cancer primary cohort. Finally, the chromatin modifier _SETDB1_ was recurrently focally
amplified in multiple cancer types, including metastatic NSCLC (Fig. 6e) and primary breast cancer. Full results are available in Supplementary Data 3. GIE ASSOCIATION WITH CANCER GENOMIC
FEATURES We next investigated whether, aside from cancer-type intrinsic differences, there were other cancer genomic and environmental features associated with GIE prevalence. Thus, we
performed a cancer type-specific univariate logistic regression of 99 tumor genomic features and 366 driver genes against the presence of GIE events (excluding nonfocal LOH of _HLA-I_)
across 38 cancer types (Supplementary Note 3). Moreover, to control for associations that may be secondary to increased mutation and CN variant (CNV) background rates, we filtered out
significant associations that were found in our GIE simulations (Supplementary Note 3). Overall, 35 genomic features and 5 driver genes showed a statistically significant association with
GIE in at least one cancer type (Fig. 7a and Extended Data Fig. 4a). Even after controlling for background mutation rates, TMB and patient’s neoepitope load were strongly associated with GIE
events in DLBCL, pancreas carcinoma and skin melanoma (_q_ value < 0.05, log2(OR) > 0.0 and simulated GIE prevalence ≤2%; Fig. 7a and Extended Data Fig. 4a–c). It is interesting that
clonal TMB and clonal neoepitope load showed a strong positive association with GIE, whereas subclonal TMB and neoepitope load showed a modest correlation (Fig. 7a–c), highlighting the
relevance of mutation cellularity in triggering immune responses. Finally, fusion-derived neoepitopes were significantly associated with GIE in DLBCL and NSCLC (Fig. 7a), which emphasizes
the importance of considering noncanonical sources of neoepitopes beyond small nonsynonymous variants in coding regions. Exposure to certain endogenous and exogenous mutational processes
have been correlated with increased immunogenicity25 and response to ICIs26,27. After controlling for molecular age and excluding non-GIE exclusive associations (that is, associations also
observed in the GIE simulations) several mutational processes showed significant association with GIE incidence (Fig. 7a and Extended Data Fig. 4a). First, MMRd mutational processes were
broadly associated with increased GIE incidence. Similarly, exposure to the APOBEC family of cytidine deaminases was strongly associated with GIE in multiple cancer types, including breast
carcinomas (Fig. 7d and Extended Data Fig. 4d). Last, the mutation burden associated with several exogenous mutational processes, such as ultraviolet light in skin melanoma (Fig. 7e and
Extended Data Fig. 4e) and platinum treatment in NSCLC (Fig. 7f and Extended Data Fig. 4f), was also significantly linked to an increased incidence of GIE events in these cancer types. We
also identified other tumor genomic features that were correlated with GIE. For instance, in colorectal cancers, which also include some patients with anal cancer, human papillomavirus DNA
integration was positively associated with GIE incidence (Fig. 7a). Moreover, high-immune infiltration, as determined by several RNA-sequencing-based deconvolution measurements
(Supplementary Note 3), was significantly linked with higher GIE incidence in this cancer type (Fig. 7g and Extended Data Fig. 4g), which is in agreement with previous reports7. Certain
driver alterations, beyond the GIE pathways considered in the present study, also showed a strong association with GIE events. Specifically, _CASP8_, _KMT2D_, _RPL22_ and _TGFBR2_
alterations tended to co-occur with GIE in patients with colorectal cancer. Of note, _CASP8_ (ref. 13) and _TGFBR2_ (ref. 28) alterations have previously been linked to immune surveillance
escape. Finally, other factors, such as the _HLA-I_ supertype, the germline _HLA-I_ divergence, patient chronological age or exposure to previous treatments, including immunotherapy, failed
to attain significant association with GIE (or the association was also observed in the simulated GIE). All the screened molecular features alongside their cancer type-specific significance
coefficients are available in Supplementary Data 4. THE SELECTED IMMUNE EVASION MECHANISMS DEPENDS ON TMB An increase in mutational load leads to the generation of neoepitopes susceptible to
recognition as neoantigens by the adaptive immune system. Therefore, we investigated the relationship between the frequency of GIE alterations (excluding nonfocal LOH of _HLA-I_) and the
TMB across 20 evenly distributed TMB buckets (Methods). We first observed that GIE frequency steadily increased with the TMB (Fig. 8a; observed GIE) and that this trend was not fully
explained by an increased background mutation and CNV rate (Fig. 8a; simulated GIE). More specifically, as the TMB increases, the observed GIE frequency deviates from the expected frequency
given by the GIE simulations. This is particularly noticeable for (ultra)hypermutated tumors, which showed a GIE incidence two- to threefold higher than the simulations. This trend was still
consistent after controlling for the cancer type (Extended Data Fig. 5a) and mutation clonality (Extended Data Fig. 5b). Using the burden of predicted neoepitopes based on the germline
_HLA-I_ profile as baseline also revealed an almost uniformly increasing distribution across the neoepitope buckets, which becomes sharper and higher than expected after the 17th bucket
(Fig. 8b). It is interesting that, in the bucket grouping samples with ~10–13 mutations per Mb, which is the minimal threshold regularly used as a response to ICIs, we observed an average
GIE frequency of 0.30 ± 0.03 s.d. Similarly, in the group of samples between 26 and 36 mutations per Mb, mostly including hypermutated tumors, the average frequency was 0.42 ± 0.06 s.d.,
whereas beyond ~95 mutations per Mb (considered to be ultra-hypermutated tumors29) we identified GIE alterations in >70% of samples (0.72 ± 0.06 s.d.). Our results thus showed that an
important fraction of patients eligible for ICIs harbored tumor alterations that may hinder recognition and/or elimination by the immune system. We then analyzed the relationship between the
TMB and the presence of specific GIE alterations across the six immune escape pathways included in the present study. Overall, the observed frequency distributions across these pathways
were remarkably different (Fig. 8c and Extended Data Fig. 5c). In fact, different types of _HLA-I_ alterations showed a distinctive frequency distribution along the TMB buckets. Nonfocal LOH
of _HLA-I_ was primarily present in low-TMB tumors, whereas focal LOH of _HLA-I_ showed a clear enrichment for mid and high TMB tumors, peaking around ~10–20 mutations per Mb (average
frequency of 0.22 ± 0.04 s.d.) and displaying an inverted U-shaped distribution. Finally, mutations in _HLA-I_ genes were more frequent in hypermutated tumors (that is, from ~26 mutations
per Mb to 36 mutations per Mb). Similarly, alterations in the antigen presentation machinery and the IFN-γ pathway were predominantly found in hypermutated tumors (Extended Data Fig. 5c).
The remaining pathways did not show any clear TMB preference, probably due the lower prevalence of these alterations in our dataset. Finally, using the number of predicted neoepitopes as
baseline revealed consistent distributions (Fig. 8d and Extended Data Fig. 5d). DISCUSSION In the present study, we have characterized the prevalence and impact of GIE alterations involved
in six major pathways across thousands of uniformly processed primary and metastatic tumors from fifty-eight cancer types. Moreover, we addressed the complexity of identifying tumor-specific
_HLA-I_ alterations by developing LILAC. Our results revealed that, on average, one in four patients bears a GIE event, primarily as a result of LOH of _HLA-I_. However, GIE incidence and
the targeted pathways showed high diversity across cancer types. Importantly, the fact that we did not observe mutual exclusivity between GIE alterations targeting different pathways
suggests that multiple GIE alterations may concur to effectively avoid immune surveillance. Remarkably, our analyses also showed that the frequency of GIE alterations in metastatic patients
are comparable to their primary counterparts across most cancer types. This result is also supported by independent studies6,30, denoting that early stages of tumorigenesis have already
acquired the capacity to escape from immune system recognition. Immune escape alterations were often positively selected during tumor evolution. Specifically, loss-of-function mutations in
_HLA-A_ and _HLA-B_, as well as multiple genes from other immune escape pathways, displayed higher-than-expected frequencies across several cancer types. Nevertheless, _HLA-C_ did not show a
significant enrichment in inactivating variants which may imply that its expression is needed to avoid natural killer-mediated immunity31 and that the neoepitope repertoire of this gene is
generally lower compared with _HLA-A_ and _HLA-B_. Finally, we also observed higher-than-expected LOH of _HLA-I_ rates across multiple cancer types. Related to this, focal and nonfocal LOH
of _HLA-I_ undergo divergent mechanisms of selection. Focal LOH of _HLA-I_ was primarily a CN-neutral event that tended to target the HLA allele with the largest neoepitope repertoire,
indicating an active role in immune evasion. On the contrary, we did not observe such allelic preference for nonfocal LOH of _HLA-I_, suggesting that alternative selective forces, such as
_DAXX_ haploinsufficiency32, are operating in these large-scale chromosome 6 events. Multiple tumor intrinsic and extrinsic features displayed a significant association with increased GIE
incidence. However, in our cohort, a patient’s exposure to previous cancer therapies, including immunotherapies, did not attain a significant association with GIE frequency, indicating that
the efficacy of GIE alterations may be compromised when dealing with the strong immune pressure released by ICIs. The tumor mutation and neoepitope burden influenced both the GIE frequency
and the targeted GIE pathway. Although focal LOH of _HLA-I_ was the most frequent mechanism in mid and high TMB tumors, the loss of certain _HLA-I_ alleles was apparently not sufficient to
cope with the neoepitope load of (ultra)hypermutated tumors, where a nontargeted GIE mechanism, such as antigen presentation abrogation, is probably needed. However, we cannot rule out the
fact that such differences may also be partially shaped by mutation and CNV rate differences across cancer types. It is important to mention that the GIE escalation as the TMB increases was
not entirely attributed to the underlying increase in background mutation rate, particularly in hypermutated tumors. Although the modeling of background GIE rates could be sensitive to the
selected randomization strategy, our results are supported by independent studies based on orthogonal analytical approaches30, evidencing the robustness of our conclusions. The present study
considered a collection of highly confident GIE alterations across six well-characterized, immune-related pathways. However, in our dataset, three of four patients did not harbor GIE
events, highlighting the need to characterize other mechanisms of immune evasion. These may involve not only alternative molecular pathways such as the _HLA-II_ (ref. 33), but also other
types of alterations such as germline variants34 and epigenetic modifications5,35. Finally, tumor extrinsic factors such as clonal hematopoiesis, tumor-associated microbiome or the tissue
architecture may also play an important role in tumor immune evasion. We expect that the combination of cancer genomics with high-resolution characterization of the tumor microenvironment
will aid in further understanding of the interplay between tumor evolution and the immune system. METHODS DATA COLLECTION AND PROCESSING The Hartwig Medical Foundation sequences and
characterizes the genomic landscape for a large number of patients with metastases. A detailed description of the consortium and the whole patient cohort has been given in detail in
Priestley et al.16. In the present study, the Hartwig cohort included 4,784 metastatic tumor samples from 4,468 patients. The Hartwig patient samples have been processed using the Hartwig
analytical pipeline5 (https://github.com/hartwigmedical/pipeline5) implemented in Platinum (v.1.0) (https://github.com/hartwigmedical/platinum). Briefly, Platinum is an open-source pipeline
designed for analyzing WGS tumor data. It enables a comprehensive characterization of tumor WGS samples (for example, somatic point mutations and indels, structural variants, CN changes) in
one single run. Hartwig samples that failed to provide a successful pipeline output, potential nontumor samples, with purity <0.2, with TMB < 50 SNVs/indels, lacking sufficient
informed consent for the present study or without enough read coverage to perform two-field HLA typing (Supplementary Note 1) were discarded. Similarly, for patients with multiple biopsies,
we selected the tumor sample with the most recent biopsy date and, if this information did not exist, we selected the sample with the highest tumor purity. However, some Hartwig patients had
biopsies from different primary tumor locations. In these cases, we kept at least one sample from each primary tumor location and, when there were multiple samples from the same primary
tumor location, we applied the aforementioned biopsy date and tumor purity-filtering criteria. A total number of 4,439 Hartwig samples were whitelisted and used in the present study
(Extended Data Fig. 2a and Supplementary Table 2). Preprocessed RNA-sequencing (RNA-seq) data by ISOFOX (https://github.com/hartwigmedical/hmftools/tree/master/isofox) were available for
1,864 Hartwig samples and were consequently used in the immune infiltration deconvolution analysis. Patient clinical data were obtained from the Hartwig database. Cancer-type labels were
harmonized to maximize the number of samples that had tumor types comparable with the PCAWG dataset (Supplementary Table 2). The PCAWG cohort consisted of 2,835 patient tumors and access for
raw sequencing data for the PCAWG-US was approved by the National Institutes of Health (NIH) for the dataset General Research Use in The Cancer Genome Atlas (TCGA) and downloaded via the
dbGAP download portal. Raw sequencing access to the non-US PCAWG samples was granted via the Data Access Compliance Office (DACO). A detailed description of the consortium and the whole
patient cohort has been given in Campbell et al.17. The samples were fully processed using the same cancer analytical pipeline applied to the Hartwig cohort (BWA38 v.0.7.17, GATK39 v.3.8.0,
SAGE16 v.2.2, GRIDSS40 v.2.9.3, PURPLE16 v.2.53 and LINX41 v.1.17). This enabled a harmonized analysis and eliminated the potential biases introduced by applying different methodological
approaches. Samples that failed to provide a successful pipeline output, with a tumor purity <0.2, potential nontumor samples, blacklisted by the PCAWG original publication17 or without
enough read coverage to perform two-field HLA typing were discarded. Similarly, for patients with multiple samples, we selected the first according to the aliquot ID alphabetical order. A
total number of 1,880 were whitelisted and used in the present study (Extended Data Fig. 2a and Supplementary Table 2). For more details about the re-processing of the PCAWG dataset and the
technical validation see Martínez-Jiménez et al.20. Preprocessed gene level expression data were downloaded for 1,118 samples from the International Cancer Genome Consortium (ICGC) portal
(https://dcc.icgc.org/releases/PCAWG/transcriptome/gene_expression/tophat_star_fpkm_uq.v2_aliquot_gl.tsv.gz). ENSEMBL identifiers were mapped to HUGO symbols. Of these samples, 930 belonged
to biopsies selected for the present study and were therefore used for the RNA analysis in PCAWG samples. The most recent clinical data were downloaded from the PCAWG release page
(https://dcc.icgc.org/releases/PCAWG) on August 2021. Cancer-type labels were harmonized to maximize the number of samples that had tumor types comparable with the Hartwig dataset
(Supplementary Table 2). LILAC All information relative to LILAC’s algorithm, implementation and validation is described in Supplementary Note 1. DEFINITIONS OF GIE ALTERATIONS We searched
in the literature for somatic genomic alterations that are robustly and recurrently associated with immune evasion. We stratified the reported alterations into six major pathways (Fig. 1a
and Supplementary Table 1): * (1) The _HLA-I_: somatic alterations in the _HLA-A_, _HLA-B_ and _HLA-C_ genes have been extensively reported as a mechanism for immune evasion across several
cancer types9,10,12,22. We considered LOH of _HLA-I_, homozygous deletions and somatic nonsynonymous mutations on these genes as immune evasion alterations. We defined LOH for _HLA-A_,
_HLA-B_ and _HLA-C_ as those cases with a minor allele ploidy <0.3 and a major allele ploidy >0.7 according to LILAC annotation. We also relied on LILAC mapping of somatic mutations
into _HLA-A_, _HLA-B_ and _HLA-C_ alleles to report samples with nonsynonymous alterations. Finally, we also used LILAC allele-specific tumor CN estimations to annotate samples with
homozygous deletions of _HLA-A_, _HLA-B_ and _HLA-C_ genes. A gene was homozygous deleted in a sample if the estimated minimum tumor CN of the gene was <0.5. * (2) The antigen
presentation pathway: several studies have reported the immunomodulatory effect of somatic inactivation of genes involved in the antigen presentation machinery (see Supplementary Table 1 for
gene-specific references). The most recurrent alteration is _B2M_ inactivation, but there are other genes involved in antigen presentation and antigen presentation activation, the
inactivation of which has been linked to increased immune evasion, including _CALR_, _TAP1_, _TAP2_, _TAPBP_, _NLRC5_, _CIITA_ and _RFX5_. We defined inactivation events as monoallelic and
biallelic clonal loss-of-function mutations (frameshift variant, stop gained, stop lost, splice acceptor variant, splice donor variant, splice region variant and start lost), biallelic
clonal nonsynonymous mutations not included in the former group (for example, missense mutations) and homozygous deletions. A gene was homozygous deleted in a sample if the estimated minimum
tumor CN of the gene was <0.5. * (3) The IFN-γ pathway: IFN-γ is a cytokine with known proapoptotic and immune booster capacities. Hence, it has been reported that tumors frequently
leverage somatic alterations targeting IFN-γ receptors and downstream effectors to evade immune system surveillance (see Supplementary Table 1 for gene-specific references). More
specifically, we considered that inactivation events (see above for specifics of which type of alterations are included) in _JAK1_, _JAK2_, _IRF2_, _IFNGR1_, _IFNGR2_, _APLNR_ and _STAT1_
have been probed to have the ability to provide an immune evasion phenotype. * (4) The PD-L1 receptor: the PD-L1 receptor, encoded by the _CD274_ gene, plays a major role in suppressing the
adaptive immune system. It has been reported how overexpression of PD-L1 in tumor cells leads to impaired recruitment of immune effectors42. We therefore considered _CD274_ CN amplification
as a genetic mechanism of immune evasion. We defined a _CD274_ CN amplification event as samples with _CD274_ minimum tumor CN >3× the average sample ploidy. * (5) The CD58 receptor: the
CD58 receptor, encoded by _CD58_, plays an essential role in T-cell recognition and stimulation. It has been extensively reported that _CD58_ alterations in B-cell lymphomas lead to immune
evasion21. Moreover, a recent study identified _CD58_ loss as one of the major effectors of impaired T-cell recognition43. Consequently, we considered inactivation events (see above) in
_CD58_ as alterations able to provide an immune escape phenotype. * (6) Epigenetic driven immune escape: it has been recently reported how _SETDB1_ amplification leads to epigenetic
silencing of tumor intrinsic immunogenicity44. _SETDB1_ amplification was recurrently found across several cancer types and was therefore considered in the present study as a mechanism of
immune evasion. We defined a _SETDB1_ CN amplification event as samples with SETDB1 minimum tumor CN >3× the mean sample ploidy. A summary table with all 21 considered genes, their
associated pathway, references and their type of somatic alterations is presented in Supplementary Table 1. GIE MUTUAL EXCLUSIVITY To assess whether LOH of HLA-I events were mutually
exclusive with other GIE events, we performed two statistical tests. First, we performed a left-sided Fisher’s exact test comparing two groups of annotations (LOH of _HLA-I_ and other GIE
events) in a cancer type-specific manner. Second, for each cancer type, we compared the number of samples bearing both LOH of _HLA-I_ and other GIE events with the expected given by 10,000
randomization, using the observed alteration frequency of both groups in the specific cancer type (LOH of _HLA-I_ and other GIE alterations). The significance was computed using an empirical
one-sided _P_ value (that is, number of randomizations with co-occurring events lower than the real observed value divided by the total number of randomizations). PRIMARY AND METASTATIC GIE
PREVALENCE The prevalence of a pathway alteration for a particular cohort was calculated as the number of samples with at least one alteration in the pathway divided by the total number of
cohort samples. The presence of a genetic immune alteration in a given sample was annotated if there was at least one pathway with an alteration in that sample. For the primary versus
metastatic comparison, we performed a tumor type-specific Fisher’s exact test comparing pathway-specific and global escaped status prevalence across the two cohorts. _P_ values were adjusted
with a multiple-testing correction using the Benjamini–Hochberg procedure (_α_ = 0.05). POSITIVE SELECTION: SOMATIC POINT MUTATIONS AND INDELS Positive selection analysis based on somatic
point mutation and small indels was performed using dNdScv and the hg19 reference genome. The analysis was performed in a cohort-specific, cancer-type and pan-cancer manner across the two
datasets. The analysis was restricted to datasets with sufficient representativeness (that is, number of samples ≥15). Global grouped dN:dS ratios of the _HLA-I_ (_HLA-A_, _HLA-B_ and
_HLA-C_) and the 16 non-_HLA-I_ genes potentially targeted by mutations (that is, excluding _SETDB1_ and _CD274_ because their immune escape phenotype is associated with CN gains;
Supplementary Table 1) were calculated in a pan-cancer manner using the gene_list attribute of the dndscv function. We used the geneci() function of dNdScv to estimate the pan-cancer and
gene-specific dN:dS ratios, which include confidence intervals (CIs), of the _HLA-I_ genes. POSITIVE SELECTION: CNAS We devised a statistical test to assess positive selection in LOH,
homozygous deletion (HD) and CN amplification (AMP) events. LOH was defined as those genomic regions where the minor allele ploidy of this gene was <0.3 and the major allele ploidy
>0.7. HD was defined as those regions with estimated minimum CN < 0.5. Similarly, AMP events were defined as those genomic regions with the minimum tumor CN >3× the mean sample
ploidy. For a particular type of genomic event overlapping with a gene, this test compares the number of observed samples bearing the alteration with the expected number after whole-genome
randomization. More specifically, these are the steps followed: * (1) Let us first denote _E_ as the type of query alteration (that is, LOH, HD or AMP), _S_ as a group of samples (usually
samples from the same cancer type and same dataset) and _G_s as the genomic scale (that is, nonfocal for segment lengths >75% chromosome arm, focal for segments <75% of the chromosome
arm and highly focal for segments <3 Mb). * (2) For every sample _S__i_ in {_S_1,_S_2,…_S__T_} we first gather the number and length of observed (_O__i_) segments targeted by _E_ within
that _G_s. Only _E_ events overlapping with autosomes are considered in the present study. Samples that do not harbor any event of type _E_ within that _G_s are ignored. * (3) Next, for
every sample _S__i_ we performed 10 independent randomizations (_R__i_1, _R__i_2, … _R__i_10) of the _O__i_ events, by randomly shuffling these events _E_ along the autosomes. For this, we
used the shuffle function from pybedtools45 with the following parameters (genome=‘hg19’, noOverlapping=True, excl=‘sexual_chomosomes’, allowBeyondChromEnd=False). In certain samples, with
an extremely high segment load (_O__i_ > 10,000) or with mean ploidy of ~1 (that is, monoploid genome), the noOverlapping flag was set to False because the randomization would not
converge. * (4) We then binned the autosomes into 28,824 bins of 100 kb and counted for each bin _k__j_ {_k_1, … _k_28,842} the total number of observed events _O__Tj_ as the sum of observed
events O1k, … OTK overlapping with that bin across all _S_ samples. * (5) Similarly, for each _R__i_th (_R_1, … _R_10) randomization and each bin _k__j_{_k_1,_… k_28,842}, we counted the
total number of simulated events as the sum of events—in that _i_th randomization and overlapping with that bin across all samples in _S_. * (6) We then performed a bin-specific comparison
of the _O__Tk_ with the average number of simulated events _R__TK_ across the ten simulations and performed a statistical test of significance using a G-test goodness of fit. As chromosome
starting bins were highly depleted in the simulated group (_R__TK_), we also computed the global simulated mean across all bins _k__j_{_k_1, … _k_28,842}, and used this as the expected
number of events for the statistical significance assessment. * (7) The _P_ values were adjusted (that is, converted to _q_ values) with a multiple-testing correction using the
Benjamini–Hochberg procedure (_α_ = 0.05). * (8) For each gene, overlapping with one or with multiple _k__j_ bins, we used the minimal adjusted _P_-value significance of the bin(s)
overlapping with the genomic location of the specific gene-coding sequence. Therefore, by definition, two genes sharing the same bins would have a similar _q_ value. We used ENSEMBL v.88 to
perform the annotation of gene exonic regions to hg19 genomic coordinates. We observed that LINE insertions near the HLA-I locus (LINE activation site at chr6:29,920,000) in some esophageal
cancer samples had an incorrect CN estimation due to multiple insertions originating from almost the same site in the same sample. Consequently, these samples were not considered in the
_HLA-I_ homozygous deletion analysis. DISTRIBUTION OF MUTATIONS IN _HLA-I_ GENES LILAC mapped the _HLA-A_, _HLA-B_ and _HLA-C_ somatic mutations detected by SAGE into the inferred _HLA-I_
alleles (see LILAC section). LILAC provides the consequence type and coding sequence position of _HLA-I_ alterations, which was used to display the distribution of mutations across the
_HLA-_I coding sequence. The 34 amino acids involved in peptide presentation were gathered from our neoepitope prioritization pipeline (see below). Pfam _HLA-A_, _HLA-B_ and _HLA-C_ domains
were manually downloaded from the Pfam36 website. TUMOR-SPECIFIC NEOEPITOPES The methodology for the identification and prioritization of neoepitopes is extensively described in
Supplementary Note 2. CALCULATION AND RANDOMIZATION OF NEOEPITOPE RATIO We wanted to evaluate whether LOH of _HLA-I_ tends to select the _HLA-I_ allele with highest neoepitope repertoire.
Let us first introduce the neoepitope allele ratio (nr). Given an _HLA-I_ gene, _G_, we defined nr as _G_A1/2 = _G_A1/(_G_A1 + _G_A2), where _G_A1/2 is the number of predicted neoepitopes of
allele 1 and allele 2, respectively. For each patient tumor sample, the assignment of allele number (that is, allele 1 or allele 2) was randomly performed. Then, we followed the next steps:
* (1) For each patient sample with LOH of _HLA-I_ we calculated the nr across the _HLA-I_ genes targeted by the LOH. Homozygous _HLA-I_ cases were not considered, because their nr is by
definition 0.5. * (2) We then grouped the nr into eight buckets: (0.0–0.35), (0.35, 0.4), (0.4, 0.45), (0.45, 0.5), (0.5, 0.55), (0.55, 0.6), (0.6, 0.65) and (0.65, 1.0). Consequently, each
bucket included _n_ allele pairs with an nr within the bound limits. * (3) Next, we performed 100 bootstraps by randomly subsampling 75% of the total number of available allele pairs in the
bucket. * (4) For each bootstrap iteration _i_th (_i_ \(\in\) 1, … 100) and each bucket we estimated the frequency of allele 1 loss (_F_A1_loss) as the number of cases with allele 1 loss
compared with the total number of cases in that bucket. Similarly, we computed the expected frequency (_F_A1exp) by randomly assigning LOH events to the allele 1 (background probably of
0.5). * (5) We then computed the bucket-specific average and s.d. of _F_A1loss and _F_A1exp values across the 100 bootstraps. * (6) Finally, we performed a Kolmogorov–Smirnov test to compare
the observed distribution with the expected given random distribution of the LOH events. This test was applied to LOH of _HLA-I_, focal LOH of _HLA-I_ and nonfocal LOH of _HLA-I_ events
across the metastatic (Hartwig) and primary (PCAWG) datasets. GIE AND TUMOR GENOMIC FEATURES Check Supplementary Note 3 for a full description of the methods for this section. GIE AND TMB
ASSOCIATION We aggregated the two datasets, metastatic and primary, to increase the robustness of this analysis. We then defined 20 evenly arranged buckets (10 for the cancer type-specific
analyses) of the log10(TMB) scale, starting from the 1st percentile and ending in the 99th percentile values. Next, each sample with a log10(TMB) = _S_tmb, was allocated to the _i_th (_i_
\(\in\) 1, … 20) bucket such as log10(TMB)i−1 < _S_tmb ≤ log10(TMB)i. Samples with an _S_tmb greater than the last bucket threshold (that is, log10(TMB)20) were allocated into the last
bucket. The number of mutations in each bucket was displayed as the number of mutations per megabase by dividing the total number of mutations by 3,000 (that is, approximated number of human
genome megabases). Finally, the GIE frequency (GIEfreq) of the _i_th bucket was defined as the number of GIE samples in the _i_th bucket divided by the total number of available samples in
that bucket. To enable calculation of the uniformity in GIE frequency among samples in the same TMB bucket, we performed _n_ (where _n_ = 1,000) bootstraps of the 50% of samples allocated to
each bucket. We then calculated the average and s.d. of the GIEfreq across the bootstraps. A similar approach was conducted to analyze the relationship between the predicted neoepitope load
and GIE frequency. The number of neoantigens of each bucket was estimated as 1% (ref. 46) and 5% (ref. 47) of the total predicted neoepitopes assigned to that bucket threshold. For the
simulated GIE control, we estimated the average and s.d. across the 100 simulated GIE iterations for each TMB bucket. STATISTICS AND REPRODUCIBILITY Sample sizes were determined by the
availability of samples with sufficient quality from the two datasets included in the present study (PCAWG and Hartwig). Sample-exclusion criteria are thoroughly described in Methods,
Supplementary Note 1 and the original publication describing the harmonized cohort20. The statistical tests and randomization strategies used in each specific analysis are described in
Methods and the figure legends. Unless otherwise specified, the scipy48 (v.1.5.3) library from python v.3.6.9 was used to carry out the statistical tests. All the code and data to reproduce
the analyses presented in the present article have been deposited in public repositories as described in Data availability and Code availability. REPORTING SUMMARY Further information on
research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The Hartwig dataset used in the present study is freely available for
academic use from the Hartwig Medical Foundation through standardized procedures and request forms that can be found at https://www.hartwigmedicalfoundation.nl/en/applying-for-data. This
includes raw sequencing data (.bam files and unmapped reads for hg19 reference genome) as well as the processed data through the latest version of the Hartwig tumor-processing pipeline. The
re-processed PCAWG data using the Hartwig Medical Foundation pipeline (for hg19 reference genome) have also been made available for academic purposes. The ICGC part of the PCAWG dataset17
can be accessed now through the ICGC platform (https://dcc.icgc.org/releases/PCAWG/Hartwig), following their standard access control mechanisms originally put in place. Similarly, users with
authorized access can download the re-processed TCGA portion of the PCAWG dataset at https://icgc.bionimbus.org/files/5310a3ac-0344-458a-88ce-d55445540120. We refer to the accompanying
publication20, including the description of the entire primary and metastatic cohort, for further information about the technical aspects of the re-processing of the PCAWG dataset. Raw
sequencing data of the high-resolution HLA typing performed by GenDx can also be downloaded via European Genome-phenome Archive (http://www.ebi.ac.uk/ega) under accession no.
EGAD00001008643. _HLA-I_ typing, sample-specific GIE events and processed data are now shared as Supplementary Data and Supplementary Tables. CODE AVAILABILITY The Hartwig analytical
processing pipeline is available at https://github.com/hartwigmedical/pipeline5 and implemented in Platinum (https://github.com/hartwigmedical/platinum). LILAC’s source code is available at
https://github.com/hartwigmedical/hmftools/tree/master/lilac. The source code of the neoepitope prioritization pipeline is available at
https://github.com/hartwigmedical/hmftools/tree/master/neo. The source code to reproduce the figures and analysis of the manuscript is available at
https://github.com/UMCUGenetics/Genetic-Immune-Escape. REFERENCES * Vinay, D. S. et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. _Semin. Cancer Biol._ 35,
S185–S198 (2015). PubMed Google Scholar * Jhunjhunwala, S., Hammer, C. & Delamarre, L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. _Nat.
Rev. Cancer_ 21, 298–312 (2021). CAS PubMed Google Scholar * Dhatchinamoorthy, K., Colbert, J. D. & Rock, K. L. Cancer immune evasion through loss of MHC class I antigen presentation.
_Front. Immunol._ 12, 636568 (2021). CAS PubMed PubMed Central Google Scholar * Castro, F., Cardoso, A. P., Gonçalves, R. M., Serre, K. & Oliveira, M. J. Interferon-gamma at the
crossroads of tumor immune surveillance or evasion. _Front. Immunol._ 9, 847 (2018). PubMed PubMed Central Google Scholar * Rosenthal, R. et al. Neoantigen-directed immune escape in lung
cancer evolution. _Nature_ 567, 479–485 (2019). CAS PubMed PubMed Central Google Scholar * Pennycuick, A. et al. Immune surveillance in clinical regression of preinvasive squamous cell
lung cancer. _Cancer Discov._ 10, 1489–1499 (2020). CAS PubMed PubMed Central Google Scholar * Grasso, C. S. et al. Genetic mechanisms of immune evasion in colorectal cancer. _Cancer
Discov._ 8, 730–749 (2018). CAS PubMed PubMed Central Google Scholar * Restifo, N. P. et al. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving
immunotherapy. _J. Natl Cancer Inst._ 88, 100–108 (1996). CAS PubMed Google Scholar * Fangazio, M. et al. Genetic mechanisms of HLA-I loss and immune escape in diffuse large B cell
lymphoma. _Proc. Natl Acad. Sci. USA_ 118, e2104504118 (2021). CAS PubMed PubMed Central Google Scholar * Montesion, M. et al. Somatic HLA class I loss Is a widespread mechanism of
immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. _Cancer Discov._ 11, 282–292 (2021). CAS PubMed Google Scholar * Robinson,
J. et al. IPD-IMGT/HLA database. _Nucleic Acids Res._ 48, D948–D955 (2020). CAS PubMed Google Scholar * McGranahan, N. et al. Allele-specific HLA loss and immune escape in lung cancer
evolution. _Cell_ 171, 1259–1271.e11 (2017). CAS PubMed PubMed Central Google Scholar * Shukla, S. A. et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA
genes. _Nat. Biotechnol._ 33, 1152–1158 (2015). CAS PubMed PubMed Central Google Scholar * Chowell, D. et al. Patient HLA class I genotype influences cancer response to checkpoint
blockade immunotherapy. _Science_ 359, 582–587 (2018). CAS PubMed Google Scholar * Naranbhai, V. et al. HLA-A*03 and response to immune checkpoint blockade in cancer: an epidemiological
biomarker study. _Lancet Oncol._ 23, 172–184 (2022). CAS PubMed Google Scholar * Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. _Nature_ 575, 210–216
(2019). CAS PubMed PubMed Central Google Scholar * Campbell, P. J. et al. Pan-cancer analysis of whole genomes. _Nature_ 578, 82–93 (2020). Google Scholar * Xie, C. et al. Fast and
accurate HLA typing from short-read next-generation sequence data with xHLA. _Proc. Natl Acad. Sci. USA_ 114, 8059–8064 (2017). CAS PubMed PubMed Central Google Scholar * Roepman, P. et
al. Clinical validation of whole genome sequencing for cancer diagnostics. _J. Mol. Diagn._ 23, 816–833 (2021). CAS PubMed Google Scholar * Martínez-Jiménez, F. et al. Pan-cancer whole
genome comparison of primary and metastatic solid tumors. _Nature_ https://doi.org/10.1038/s41586-023-06054-z (2023). * Challa-Malladi, M. et al. Combined genetic inactivation of
β2-microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. _Cancer Cell_ 20, 728–740 (2011). CAS PubMed PubMed Central Google Scholar *
Castro, A. et al. Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes. _BMC Med. Genomics_ 12, 107 (2019). PubMed PubMed Central Google
Scholar * Martínez-Jiménez, F. et al. A compendium of mutational cancer driver genes. _Nat. Rev. Cancer_ 20, 555–572 (2020). PubMed Google Scholar * Waldhauer, I. & Steinle, A. NK
cells and cancer immunosurveillance. _Oncogene_ 27, 5932–5943 (2008). CAS PubMed Google Scholar * Boichard, A. et al. APOBEC-related mutagenesis and neo-peptide hydrophobicity:
implications for response to immunotherapy. _OncoImmunology_ 8, 1550341 (2019). PubMed Google Scholar * Rousseau, B. et al. The spectrum of benefit from checkpoint blockade in hypermutated
tumors. _N. Engl. J. Med._ 384, 1168–1170 (2021). PubMed PubMed Central Google Scholar * Litchfield, K. et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to
checkpoint inhibition. _Cell_ 184, 596–614.e14 (2021). CAS PubMed PubMed Central Google Scholar * Dhainaut, M. et al. Spatial CRISPR genomics identifies regulators of the tumor
microenvironment. _Cell_ 185, 1223–1239.e20 (2022). CAS PubMed PubMed Central Google Scholar * Campbell, B. B. et al. Comprehensive analysis of hypermutation in human cancer. _Cell_ 171,
1042–1056.e10 (2017). CAS PubMed PubMed Central Google Scholar * Lakatos, E. et al. Evolutionary dynamics of neoantigens in growing tumors. _Nat. Genet._ 52, 1057–1066 (2020). CAS
PubMed PubMed Central Google Scholar * Blais, M.-E., Dong, T. & Rowland-Jones, S. HLA-C as a mediator of natural killer and T-cell activation: spectator or key player? _Immunology_
133, 1–7 (2011). CAS PubMed PubMed Central Google Scholar * Jiao, Y. et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. _Science_
331, 1199–1203 (2011). CAS PubMed PubMed Central Google Scholar * Nijland, M. et al. HLA dependent immune escape mechanisms in B-cell lymphomas: implications for immune checkpoint
inhibitor therapy? _Oncoimmunology_ 6, e1295202 (2017). PubMed PubMed Central Google Scholar * Sayaman, R. W. et al. Germline genetic contribution to the immune landscape of cancer.
_Immunity_ 54, 367–386.e8 (2021). CAS PubMed PubMed Central Google Scholar * Berglund, A. et al. Methylation of immune synapse genes modulates tumor immunogenicity. _J. Clin. Invest._
130, 974–980 (2020). CAS PubMed PubMed Central Google Scholar * Mistry, J. et al. Pfam: The protein families database in 2021. _Nucleic Acids Res._ 49, D412–D419 (2021). CAS PubMed
Google Scholar * Davoli, T., Uno, H., Wooten, E. C. & Elledge, S. J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. _Science_
355, eaaf8399 (2017). PubMed PubMed Central Google Scholar * Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. _Bioinformatics_ 25, 1754–1760
(2009). CAS PubMed PubMed Central Google Scholar * McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. _Genome Res._
20, 1297–1303 (2010). CAS PubMed PubMed Central Google Scholar * Cameron, D. L. et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph
assembly. _Genome Res._ 27, 2050–2060 (2017). CAS PubMed PubMed Central Google Scholar * Shale, C. et al. Unscrambling cancer genomes via integrated analysis of structural variation and
copy number. _Cell Genomics_ 2, 100112 (2022). CAS PubMed PubMed Central Google Scholar * Hamanishi, J. et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes
are prognostic factors of human ovarian cancer. _Proc. Natl Acad. Sci. USA_ 104, 3360–3365 (2007). CAS PubMed PubMed Central Google Scholar * Frangieh, C. J. et al. Multimodal pooled
Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. _Nat. Genet._ 53, 332–341 (2021). CAS PubMed PubMed Central Google Scholar * Griffin, G. K. et al.
Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. _Nature_ 595, 309–314 (2021). CAS PubMed PubMed Central Google Scholar * Dale, R. K., Pedersen, B. S. &
Quinlan, A. R. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. _Bioinforma_ 27, 3423–3424 (2011). CAS Google Scholar * Gartner, J. J. et al. A
machine learning model for ranking candidate HLA class I neoantigens based on known neoepitopes from multiple human tumor types. _Nat. Cancer_ 2, 563–574 (2021). CAS PubMed PubMed Central
Google Scholar * Wells, D. K. et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. _Cell_ 183, 818–834.e13 (2020).
CAS PubMed PubMed Central Google Scholar * Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. _Nat. Methods_ 17, 261–272 (2020). CAS PubMed
PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This publication and the underlying study have been made possible partly on the basis of the data that the Hartwig
Medical Foundation and the Center of Personalised Cancer Treatment have made available to the study. We thank A. Van Hoeck and A. G. Perez for critical reading of the manuscript and their
valuable input. We also thank A. Van Hoeck for assistance in the dataset collection and L. Nyguen for help in the mutational signature extraction. We thank H. W. Lee for technical assistance
in the TRACERx dataset processing. The present study makes use of data generated by the TRAcking Non-small Cell Lung Cancer Evolution Through Therapy (TRACERx) Consortium and provided by
the UCL Cancer Institute and the Francis Crick Institute. The TRACERx study is sponsored by University College London (UCL), funded by Cancer Research UK and coordinated through the Cancer
Research UK and UCL Cancer Trials Centre. We also thank N. McGranahan, O. Pich and C. Puttick for sharing the original _HLA-I_ typing and LOHHLA calls used in their original publication.
Finally, we thank W. Mulder, M. Penning and H. Merkens for discussions on LILAC’s orthogonal validation. AUTHOR INFORMATION Author notes * Francisco Martínez-Jiménez Present address: Vall
d’Hebron Institute of Oncology, Barcelona, Spain * These authors contributed equally: Francisco Martínez-Jiménez, Peter Priestley. AUTHORS AND AFFILIATIONS * Center for Molecular Medicine
and Oncode Institute, University Medical Center Utrecht, Utrecht, the Netherlands Francisco Martínez-Jiménez & Edwin Cuppen * Hartwig Medical Foundation, Amsterdam, the Netherlands
Francisco Martínez-Jiménez & Edwin Cuppen * Hartwig Medical Foundation Australia, Sydney, New South Wales, Australia Peter Priestley, Charles Shale & Jonathan Baber * GenDx, Utrecht,
the Netherlands Erik Rozemuller Authors * Francisco Martínez-Jiménez View author publications You can also search for this author inPubMed Google Scholar * Peter Priestley View author
publications You can also search for this author inPubMed Google Scholar * Charles Shale View author publications You can also search for this author inPubMed Google Scholar * Jonathan Baber
View author publications You can also search for this author inPubMed Google Scholar * Erik Rozemuller View author publications You can also search for this author inPubMed Google Scholar *
Edwin Cuppen View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS F.M.J., E.C. and P.P. conceived the study. F.M.J., P.P., J.B. and C.S.
provided the methodology. P.P., J.B. and C.S. provided the software. F.M.J., P.P. and E.R. validated the study. F.M.J. and P.P. performed the formal analysis. F.M.J. and P.P. carried out the
investigations. E.C. and P.P. provided the resources. F.M.J., P.P., C.S. and E.R. curated the data. F.M.J. wrote the original draft of the MS. E.C. and P.P. reviewed and edited the MS.
F.M.J. visualized the study. F.M.J. and E.C. supervised the study. F.M.J. and E.C. administered the project. E.C. acquired funding. CORRESPONDING AUTHORS Correspondence to Francisco
Martínez-Jiménez or Edwin Cuppen. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Genetics_ thanks Jimmy Van
den Eynden, Teresa Davoliand and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA EXTENDED DATA FIG. 1 LILAC’S VALIDATION.
A) _HLA-I_ typing tumor and germline agreement in PCAWG. Venn diagrams representing the overlap of germline samples with perfect 2-field _HLA-I_ allotype match in B) Hartwig and C) PCAWG.
D) Left, copy number of minor and major alleles of _HLA-A_, _HLA-B_ and _HLA-C_ in Hartwig dataset. Right, proportion of samples with integer minor and major copy number (copy number
<=0.3 or >=0.7) of _HLA-I_ genes (orange) compared to the average of 1,000 randomly selected genes (gray) in the Hartwig cohort (N=4,439). E) analogous for PCAWG samples (N=1,880).
Box-plots: center line, median; box limits, first and third quartiles; whiskers, lowest/highest data points at first quartile minus/plus 1.5× IQR. N, number of samples. EXTENDED DATA FIG. 2
GIE IN PRIMARY AND METASTATIC TUMORS. A) Workflow of the sample processing pipeline used in this study in Hartwig (left) and PCAWG (right). Each rectangle represents a processing step. The
resulting number of selected samples for this study are displayed at the bottom. Sample-exclusion criteria for LILAC is described in Supp. Note 1. B) number of metastatic (Hartwig) samples
across cancer types that lack sufficient representation in the primary (PCAWG) dataset. C) Analogous representation for primary (PCAWG) samples. Vertical dashed lines (N>= 15 samples)
represent the threshold of samples to consider a cohort as sufficiently populated. EXTENDED DATA FIG. 3 POSITIVE SELECTION OF _HLA-I_ GENES. A) Needle plots representing the pan-cancer
distribution of somatic mutations along the _HLA-A_, _HLA-B_ and _HLA-C_ protein sequences in the primary (PCAWG) dataset. Mutations are colored according to the consequence type. Rectangles
represent the Pfam domains. B) Correlation of LOH of _HLA-I_ rates (y-axis) with the mean genome-wide LOH rates (x-axis) across the metastatic (left) and primary (right) datasets. Each dot
represents a cancer type. Horizontal lines represent genome-wide LOH standard deviation across samples from each cancer type. The regression and 95% confidence intervals of the linear
regression are represented as a solid line and the adjacent shaded area, respectively. Confidence intervals are calculated using 1,000 bootstraps. P-values are obtained from a two-sided test
of no-correlation. C) Distribution of non-focal LOH events along the autosomes in the primary kidney chromophobe cancer cohort. X-ticks represent the chromosomal starting position. Dashed
horizontal lines represent the expected mean after randomization. Vertical dashed lines highlight the _HLA-I_ genomic location. CDS pos, coding sequence position. Std., standard deviation.
EXTENDED DATA FIG. 4 GIE ASSOCIATION WITH CANCER GENOMIC FEATURES. A) Heatmap displaying the association of genomic features with GIE frequency across cancer types. Significant associations
that can not be explained by higher background mutation rate are highlighted by a red border line. Significant associations found in >2% of the GIE simulations are highlighted by a black
border line. B) Comparison of the TMB between samples bearing GIE alterations and non-GIE samples in diffuse large B-cell lymphoma (DLBCL). Left boxplot, using observed GIE events. Right
using simulated GIE events. C) Similar comparison for neoepitope burden in skin melanoma. D) Comparison of the APOBEC mutational exposure between samples bearing simulated GIE alterations
and wild-type (no simulated GIE) in breast cancer in one randomly selected simulation. E) Analogous for ultraviolet light (UV) associated double base substitutions (DBSs) in skin melanoma
and F) for platinum treatment associated DBSs in non-small cell lung cancer (NSCLC). G) Comparison of immune infiltration estimates from Davoli et al.37 between samples bearing simulated GIE
alterations and wild-type (no simulated GIE) in colorectal cancer. Boxplots: center line, median; First section out from the centerline contains 50% of the data. The next sections contain
half the remaining data until we are at the outlier level. Each level out is shaded lighter. N, number of samples. P-values of the boxplots are calculated using a two-sided Mann–Whitney U
test. One of the 100 simulations was randomly selected for all the simulated GIE boxplots. SBS, single base substitution. EXTENDED DATA FIG. 5 IMMUNE EVASION MECHANISMS AND TMB. A) Analogous
to Fig. 7a, but restricted to (from left to right) non-small cell lung cancer, breast cancer and colorectal cancer patients. Top panels represent the total number of bucket-assigned (white
bars with a black contouring line) and GIE-positive (pink bars with pink contouring lines) samples across the ten evenly arranged TMB buckets. bottom panel, representation of observed (pink)
and simulated (gray) GIE frequency across these buckets. For the observed GIE values, average (represented as pink dots) and standard deviation (std, vertical error bars and shaded pink
area) values are computed using 1,000 bootstraps from the total number of cancer type samples classified into each bucket (from the top panel). For the simulated GIE values, average (gray
triangle) and standard deviation (std, vertical bars and shaded gray area) values are computed from 100 GIE simulations using the total number of cancer type samples assigned into each
bucket. B) Analogous to panel a) but using the entire cohort (N=6,319) and twenty evenly arranged buckets as well as clonal TMB (left) and subclonal TMB (right) as baseline. C) Equivalent to
panel a) but using the entire cohort (N=6,319) and twenty evenly arranged buckets and grouping GIE events according to the assigned GIE pathway. Label number represents the assigned immune
escape pathway (from Fig. 1a) D) Similar to panel c) but using the number of predicted neoepitopes as baseline for the buckets. Bottom labels, number of estimated neoantigens as a relative
percentage (1% and 5%) of the number of predicted neoepitopes in the bucket. N, number of samples. Muts/Mb, mutations per megabase. ICI, immune checkpoint inhibitor. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION Supplementary Tables 1 and 2 and Data 1–4 captions, and Supplementary Notes 1–3. REPORTING SUMMARY PEER REVIEW FILE SUPPLEMENTARY TABLES 1 AND 2
Supplementary Table 1 Pathways and genes involved in genetic immune escape. Table 2 Dataset metadata and cancer-type representation. SUPPLEMENTARY DATA 1 _HLA-I_ typing for Hartwig and PCAWG
patients and validation of LILAC. SUPPLEMENTARY DATA 2 Sample-specific GIE annotation and cohort-wise GIE frequency. SUPPLEMENTARY DATA 3 Positive selection in _HLA-I_ and non-_HLA-I_
genes. SUPPLEMENTARY DATA 4 Tumor genomics and clinical features and GIE association. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Martínez-Jiménez, F., Priestley, P., Shale, C. _et al._ Genetic immune escape
landscape in primary and metastatic cancer. _Nat Genet_ 55, 820–831 (2023). https://doi.org/10.1038/s41588-023-01367-1 Download citation * Received: 28 June 2022 * Accepted: 10 March 2023 *
Published: 10 May 2023 * Issue Date: May 2023 * DOI: https://doi.org/10.1038/s41588-023-01367-1 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative




.jpg?rect=0%2C0%2C1536%2C806)


