- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Download PDF Article Open access Published: 07 August 2024 A virally encoded tRNA neutralizes the PARIS antiviral defence system Nathaniel Burman ORCID: orcid.org/0000-0002-3732-31591 na1,
Svetlana Belukhina2 na1, Florence Depardieu3 na1, Royce A. Wilkinson ORCID: orcid.org/0000-0001-8831-20811, Mikhail Skutel ORCID: orcid.org/0009-0002-1351-32382, Andrew Santiago-Frangos
ORCID: orcid.org/0000-0001-9615-065X1, Ava B. Graham1, Alexei Livenskyi4,5, Anna Chechenina2, Natalia Morozova6, Trevor Zahl1, William S. Henriques1, Murat Buyukyoruk1, Christophe Rouillon
ORCID: orcid.org/0000-0003-4038-315X3, Baptiste Saudemont3, Lena Shyrokova ORCID: orcid.org/0000-0002-0361-63597, Tatsuaki Kurata7, Vasili Hauryliuk ORCID:
orcid.org/0000-0003-2389-50577,8,9, Konstantin Severinov4,10, Justine Groseille11,12,13, Agnès Thierry11, Romain Koszul ORCID: orcid.org/0000-0002-3086-117311, Florian Tesson ORCID:
orcid.org/0000-0003-4038-115414, Aude Bernheim ORCID: orcid.org/0000-0003-0212-777X14, David Bikard ORCID: orcid.org/0000-0002-5729-12113, Blake Wiedenheft ORCID:
orcid.org/0000-0001-9297-53041 & …Artem Isaev ORCID: orcid.org/0000-0002-9593-27452 Show authors Nature volume 634, pages 424–431 (2024)Cite this article
24k Accesses
143 Altmetric
Metrics details
Subjects BiochemistryStructural biology An Author Correction to this article was published on 04 December 2024
This article has been updated
AbstractViruses compete with each other for limited cellular resources, and some deliver defence mechanisms that protect the host from competing genetic parasites1. The phage antirestriction induced
system (PARIS) is a defence system, often encoded in viral genomes, that is composed of a 55 kDa ABC ATPase (AriA) and a 35 kDa TOPRIM nuclease (AriB)2. However, the mechanism by which AriA
and AriB function in phage defence is unknown. Here we show that AriA and AriB assemble into a 425 kDa supramolecular immune complex. We use cryo-electron microscopy to determine the
structure of this complex, thereby explaining how six molecules of AriA assemble into a propeller-shaped scaffold that coordinates three subunits of AriB. ATP-dependent detection of foreign
proteins triggers the release of AriB, which assembles into a homodimeric nuclease that blocks infection by cleaving host lysine transfer RNA. Phage T5 subverts PARIS immunity through
expression of a lysine transfer RNA variant that is not cleaved by PARIS, thereby restoring viral infection. Collectively, these data explain how AriA functions as an ATP-dependent sensor
that detects viral proteins and activates the AriB toxin. PARIS is one of an emerging set of immune systems that form macromolecular complexes for the recognition of foreign proteins, rather
than foreign nucleic acids3.
Similar content being viewed by others Anti-viral defence by an mRNA ADP-ribosyltransferase that blocks translation Article Open access 23 October 2024Architecture and activation mechanism of the bacterial PARIS defence system Article 07 August 2024 Evasion of antiviral bacterial immunity by phage tRNAs Article Open access 11 November 2024
Main
Antiviral defence systems in bacteria and archaea are extraordinarily diverse and many are mechanistically similar to immune responses in eukaryotic cells4. The recent expansion of bacterial
and archaeal immune systems stems from the appreciation that they tend to colocalize in the genome5 and are often carried by mobile genetic elements, including prophages and satellite
viruses6. The phage antirestriction induced system (PARIS) is one of several defence systems recently identified within a hotspot of genetic diversity carried by P4 phage satellites2. The
PARIS system of Escherichia coli strain B185 protects against phage T7 infection through a mechanism triggered by sensing the T7 overcoming classical restriction (Ocr) protein. Ocr is a DNA
mimic that inhibits type I restriction-modification and BREX defence systems. Thus PARIS is an ‘anti-antirestriction’ system that senses a viral immune suppressor.
The PARIS defence system consists of two proteins, AriA and AriB (Fig. 1a,b). AriA contains a conserved ABC ATPase domain whereas AriB is a domain of unknown function (DUF4435), which
includes a TOPRIM nuclease7,8. AriA and AriB are usually separate proteins, although they are occasionally fused into a single polypeptide. Several defence systems share ABC ATPase and
TOPRIM domains, including the protein-overcoming lysogenization defect (OLD) produced by the P2 prophage, which blocks infection by phage lambda9. OLD does not directly sense a phage
protein, but rather inactivation of the RecBCD exonuclease10. Activation of OLD results in translation inhibition9, but the underlying mechanism has not been determined.
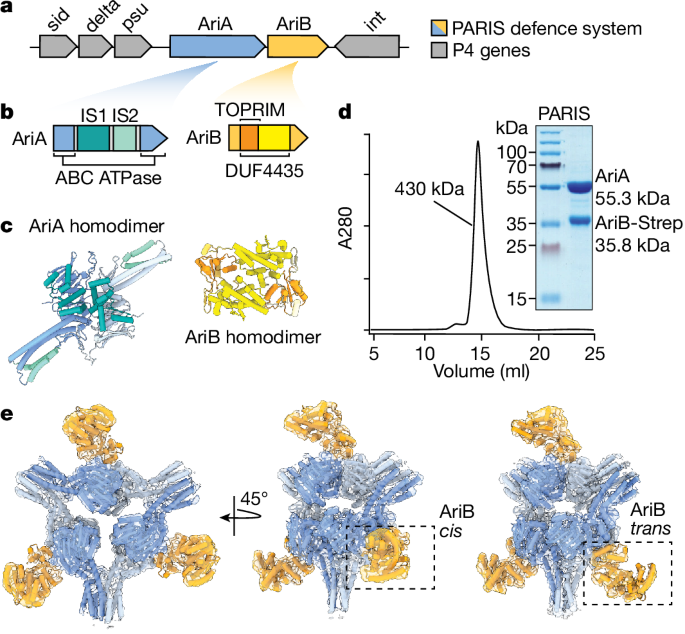
Fig. 1: PARIS isa two-component system that assembles into a propeller-shaped supramolecular complex.
a, Schematic of the integrated P4 satellite in the E. coli genome. Grey arrows labelled sid (size determination), delta (δ transcriptional activator), psu (polarity suppression) and int
(integrase) are P4 genes flanking AriA and AriB genes. b, PARIS genes coloured according to domain organization. AriA is an ABC ATPase whereas AriB is a DUF4435 that includes a TOPRIM
nuclease. IS1 is composed of three helices flanked by the ABC ATPase domain, whereas IS2 adds two additional helices to the coiled-coil resulting in a helical bundle. c, AlphaFold2-predicted
structures of AriA and AriB as homodimers coloured according to the structural features shown in b. d, Size exclusion chromatography showing that AriA and AriB copurify as a complex of
roughly 430 kDa. The size exclusion chromatography profile is representative of three independent biological experiments. e, Experimentally determined density maps of the PARIS complex
showing a homohexameric assembly of AriA with D3 symmetry. The AriA hexamer is decorated by AriB subunits that attach to AriA homodimers in one of two potential orientations: a ‘cis’
arrangement where the three AriB subunits are facing the same direction, or a ‘trans’ arrangement where one AriB subunit is rotated by 180° relative to the other two.
Full size imageAssociation between the ABC ATPase and TOPRIM domains is an emerging theme in diverse recently discovered immune systems11. Examples include the Gabija system12,13, effector toxin proteins
of retron immunity14 and bona fide toxin–antitoxin modules15. This implies a broad adaptability of the conserved ABC ATPase and TOPRIM nuclease architecture to a variety of antigenic
stimuli, resulting in diversification of the immune modules probably acting through abortive mechanisms. We currently have little understanding of the molecular mechanisms underpinning how
these systems sense and respond to viral infections.
Here we use cryo-electron microscopy (cryo-EM) to show that the E. coli B185 PARIS system components assemble into a 425 kDa, propeller-shaped complex with six molecules of AriA and three
subunits of AriB. We show that AriA senses the T7 Ocr antirestriction protein leading to the release of the AriB nuclease effector. Release of AriB from the AriA scaffold is necessary for
potentiation of AriB, which forms a nuclease-active dimer that degrades host lysine transfer RNA (tRNALys), thereby blocking translation and leading to growth arrest or cell death. We find
that phage T5 carries its own tRNALys isoacceptor that rescues the phage from PARIS-mediated defence. Finally, we perform a phylogenetic analysis of PARIS systems that provides insights into
their evolutionary history and shows how they relate to other defence systems that use ABC ATPases.
PARIS forms a propeller-shaped complexThe PARIS defence system consists of two proteins, AriA and AriB, which protect cells from phages through a previously undetermined mechanism2. Here, we focused on the system from the P4
satellite integrated into the E. coli (strain B185) genome (Fig. 1a,b). Based on published experimental structures of related proteins8,13 and AlphaFold2 (ref. 16) predictions (Fig. 1c and
Extended Data Fig. 1a–d), we anticipated that AriA/AriB would assemble into a multisubunit complex. To determine how AriA and AriB assemble, we overexpressed AriA with a C-terminally tagged
AriB (AriB-Strep), pulled down on AriB, and purified the complex using size exclusion chromatography (Fig. 1d). The AriA and AriB proteins coelute in a single peak with an estimated
molecular weight of 430 kDa. Next, we used cryo-EM to determine the structures of the PARIS complex (Fig. 1e). The structure explains how six molecules of AriA combine to form a D3 symmetric
scaffold decorated by three subunits of AriB. The helical domains, which typically provide a platform for DNA binding in related ABC ATPases17, instead function as additional dimerization
interfaces that enable a trimeric assembly of AriA homodimers (Fig. 1c and Extended Data Fig. 2). Dimerization of the helical domains from adjacent AriA homodimers form three blades of the
propeller (Fig. 1e and Extended Data Fig. 2). Additional density is evident between each of the AriA blades, where AriB attaches to AriA near the ATP-binding sites. However, density in this
region of the reconstruction shows conformational heterogeneity, consistent with AriB attaching to either, but not both, AriA molecules at the dimer interface (Extended Data Fig. 1e,f and
Supplementary Video 1). Using multiple rounds of focused three-dimensional classification, we resolved two distinct isomers of the complex (Fig. 1e and Extended Data Fig. 3d). In one
reconstruction, AriB attaches to three symmetrically equivalent AriA molecules (3.7 Å resolution, cis configuration) and, in the other, one of the three AriB molecules binds the opposing
AriA, which flips AriB 180° relative to the other two AriB subunits (3.9 Å resolution, trans configuration; Extended Data Fig. 3g,h and Supplementary Video 1).
AriA sequesters AriBAlthough the homohexameric assembly of AriA was apparent in the early stages of reconstruction, conformational heterogeneity limited resolution (Extended Data Fig. 3 and Supplementary Video
1). To improve resolution, we used local refinement to determine a 3.2 Å nominal resolution reconstruction of one asymmetric unit of the PARIS complex (AriA2–AriB1), into which we built an
atomic model (PDB: 8UX9; Fig. 2 and Extended Data Table 1). Each asymmetric unit of the PARIS complex is composed of two AriA molecules that form a head-to-tail homodimer18,19,20, and one
AriB molecule that binds directly above the AriA dimer interface.
Fig. 2: Interaction between AriA ATPase and AriB is essential for PARIS-mediated defence.a, Each asymmetric unit of the PARIS complex contains an AriA homodimer with two NBDs. A 90° rotation of the asymmetric unit, showing 1–53 N-terminal residues (yellow) of AriB positioned
over the ATPase active sites of AriA. b, AriB attaches to AriA through electrostatic interactions near helix α2 of AriB. TOPRIM active site residues predicted to participate in metal binding
(grey spheres) and catalysis are shown as sticks. c, Close-up of NBD1 showing ATPγS bound in an open conformation. d, Close-up of NBD2 showing the bound ligand trapped in a closed
precleavage state. e, Plaque assays were performed in duplicate with two independent clones of each mutant, using a serial dilution of phage T7 on E. coli MG1655 carrying the WT or mutated
PARIS system.
Full size imageAriB is a metal-dependent TOPRIM nuclease and the predicted effector of the PARIS defence system2,7,8. The structure shows a series of spatially conserved, negatively charged residues (E26,
D30, D88, E90 and E122) that are characteristic of TOPRIM domains that bind two metal ions7. The E26A mutation inactivates PARIS-mediated defence2 (Fig. 2b). Glutamic acid 26 is just
upstream of an N-terminal alpha helix (α2) on AriB, which includes two arginine residues (R31 and R28) that form salt bridges with negatively charged residues on AriA, which is positioned
directly above the ATP-binding sites (Fig. 2b, Extended Data Fig. 2 and Supplementary Video 1). Mutations that interfere with the AriA–AriB interface (AriB R28E, AriB R31E and AriB R31E/R28E
or AriA D401N/E404Q) limit phage protection by PARIS (Fig. 2e), which suggests that the association between AriA and AriB is required to provide protection.
The AriA ABC ATPase domain resembles that of Rad50 (root mean squared deviation 1.1 Å across 86 C-alpha atom pairs), a universally conserved protein involved in double-stranded DNA break
repair21,22,23. Structural comparison of AriA with Rad50 confirms that AriA contains the sequence motifs required for ATPase activity, along with two unique insertion sequences, insertion
sequences 1 and 2 (IS1 and IS2, respectively; Fig. 1b and Extended Data Fig. 2). Insertion sequence 2 contains a series of aromatic residues located on one face of helix α8 (F230, F234, F238
and Y304) that coordinate interactions between AriA homodimers, and explains how AriA homodimers assemble into a trimer of dimers (Extended Data Fig. 2), a rare assembly state for this
family of proteins21. Like other ABC ATPases, the head-to-tail assembly of the AriA homodimer results in two symmetrically positioned nucleotide-binding domains (NBDs) at the dimer interface
(Fig. 2a). In the presence of a non-hydrolysable ATP analogue (that is, ATPγS), we observe density for the ligand in both NBDs but in different coordination states (Fig. 2c,d). Previous
studies of ABC ATPases showed that ATP hydrolysis occurs at the two NBDs in an alternating fashion21,24, which is consistent with the observed ‘open’ and ‘closed’ conformations of NBDs (Fig.
2c,d and Supplementary Video 1).
AriA binds to Ocr and releases AriBThe T7 phage antirestriction protein Ocr was previously identified as a trigger of the PARIS system, and its expression in the presence of PARIS leads to growth arrest or cell death2. To
determine whether AriA–AriB senses Ocr through direct interaction, we coexpressed AriA with a non-toxic active site mutant of AriB (E26A) and Ocr-Strep. AriA, but not AriB, copurified with
Ocr-Strep and the complex migrated according to the predicted mass of the AriA hexamer (Fig. 3a and Extended Data Fig. 5a,b). This is consistent with a direct interaction between AriA and
Ocr and release of AriB. A pulldown with AriB-Strep confirmed that AriB dissociates from the complex in the presence of Ocr (Fig. 3b).
Fig. 3: AriA interacts with Ocr in anATPase-dependent manner, leading to the release of AriB from the PARIS complex.
a, Ocr-Strep pulls down AriA, but not AriB. A mutation in the ATPase active site of AriA (K39A) blocks binding to Ocr-Strep. b, Ocr releases AriB (E26A) from WT AriA (lane 3), but Ocr does
not trigger AriB release from an ATP-binding-deficient variant of AriA (K39A) (lane 5). c, A 4.4-Å-resolution reconstruction of AriA that was purified using Ocr-Strep as a bait. The
structure shows three radial pores symmetrically positioned around a central pore. The pores contain disordered loops with several positively charged residues. Ocr (PDB: 1S7Z) is shown as an
electrostatic surface. d, Mutations in the central pore (R116A or R119A) reduce the efficiency of phage protection. e, Ocr-Strep binds WT AriA and ejects AriB (E26A), and the charge swap
mutations in the central pore of AriA (R116E/R119E) limit interaction with Ocr-Strep. f, AriB-Strep (E26A) pulls down WT AriA but not AriA (R116E/R119E). g, ATP and ADP were separated using
thin-layer chromatography (Extended Data Fig. 7), and the rates of ATP hydrolysis for PARIS, PARIS mixed with tenfold excess Ocr and PARIS with an ATPase mutation in AriA (E393A) were
measured. Experiments were performed in triplicate, and error bars show ±s.e.m. Two-sided statistical analysis performed using a post hoc Dunnett’s test, ****P < 0.0001. h, Size exclusion
chromatography of AriB-Strep (around 36 kDa) following Ocr-mediated release from AriA. The column was calibrated using molecular weight standards (grey lines). AriB elutes in a single peak
with an estimated molecular weight of 81 kDa (Extended Data Fig. 5a). i, Glutaraldehyde (GA) cross-linking assay with activated AriB (E26A). j, Mutations predicted to block AriB dimerization
(E285R and F137A) prevent PARIS-mediated defence (Extended Data Fig. 1g). Plaque assays were performed in duplicate with two independent clones of each mutant.
Full size imageTo determine how Ocr triggers the release of AriB, we used the purified AriA–Ocr-Strep complex for structure determination. The 4.4-Å-resolution structure shows a D3 symmetric, homohexamer
of AriA with no AriB (Fig. 3c and Extended Data Fig. 6). At this resolution, the structure of AriA purified using Ocr-Strep is indistinguishable from that of AriA purified using AriB-Strep.
We anticipated that the structure would include Ocr, because Ocr-Strep was used to pull down the AriA hexamer and the complex remained stable during gel filtration. However, multiple
data-processing strategies failed to resolve Ocr, suggesting that Ocr has several binding sites on AriA and/or that the interaction is conformationally flexible.
Because Ocr is a negatively charged DNA mimic25,26, we hypothesized that the binding site on AriA would be positively charged. An electrostatic surface of the AriA hexamer shows a central
pore, flanked by a series of three positively charged radial pores (Fig. 3c). These radial pores contain 2 flexible loops (one from each of the adjacent AriA homodimers) with 14 unmodelled
residues (161–174, SSDVGYERRVIRSS), with the central pore containing 6 loops (one from each AriA) incorporating 12 unmodelled residues (113–124, ESERHLRERDVK). To determine the role of these
loops in phage defence, we tested the effects of alanine substitutions and charge swap mutations on positively charged residues. Arginine-to-alanine substitutions (R116A and R119A) in the
central pore resulted in partial loss of phage protection (Fig. 3d). Attempts to generate other mutations in the central (R116E, R119E) or radial pores (R168E, R168A, R172E or R172A) of AriA
consistently failed when we used a vector that also included wild-type (WT) AriB. We hypothesized that these mutations in AriA mimic trigger binding, which releases AriB and results in
toxicity. To test this hypothesis, we expressed the central pore mutant (R116E/R119E) with the catalytically inactive AriB (E26A), which is non-toxic. Pulldown experiments performed with
Ocr-Strep (Fig. 3e) or AriB-Strep (Fig. 3f) demonstrated that AriA (R116E/R119E) has limited interactions with both Ocr and AriB. Collectively, these results suggest that either the central
or radial pores, or both, are involved in interaction with the trigger.
AriB alone does not provide phage defence and is only mildly toxic when overexpressed2 (Extended Data Fig. 4). However, activated AriB (that is, AriB released from PARIS) is significantly
more toxic. Moreover, mutations in AriA that disrupt interaction with AriB limit AriB toxicity and abolish phage defence. Collectively, these data suggest that the toxicity of AriB depends
on its initial association with AriA.
The ATPase activity of AriA is also required for phage defence2, which suggests that the ATPase of AriA is necessary for loading of AriB onto AriA, or release of activated AriB. To
differentiate between loading or release of AriB, we performed pulldown assays using AriB-Strep and WT or an ATP-binding-defective mutant of AriA (K39A in Walker A). AriB-Strep associates
with mutant and WT AriA at similar efficiency (Fig. 3b), demonstrating that the ATPase mutant of AriA loads, but does not release, AriB in the presence of Ocr. We reasoned that the lack of
AriB release in the presence of AriA K39A is due to a defect in trigger recognition. To confirm the role of ATP in AriA-mediated trigger recognition, we conducted pulldowns using Ocr-Strep
and WT or K39A AriA. The AriA K39A mutant failed to copurify with Ocr, indicating that trigger recognition is ATP dependent (Fig. 3a). To determine whether or how the trigger impacts the
ATPase activity of AriA, we measured the rate of PARIS-mediated ATP hydrolysis in the presence or absence of Ocr and compared these results with an ATPase-defective AriA mutant (E393A in
Walker B; Fig. 3g). The results demonstrated that Ocr significantly (P = 0.0001) reduced ATP turnover by AriA (Fig. 3g and Extended Data Fig. 7).
TOPRIM-containing proteins are known to form homodimers27,28, but a predicted homodimer of AriB generated using AlphaFold2 clashed with AriA (Extended Data Fig. 1f). Thus, we hypothesized
that AriB forms an active homodimer following trigger-mediated release from AriA. The toxicity of PARIS following activation makes purification of active AriB challenging. To overcome this
problem, we mixed lysates from cells expressing AriA–AriB-Strep with lysates from cells expressing Ocr, and then recovered activated AriB using affinity chromatography (Extended Data Fig.
5c). Size exclusion chromatography indicated that activated AriB forms a homodimer (Fig. 3h and Extended Data Fig. 5a), which was further confirmed by glutaraldehyde cross-linking
experiments (Fig. 3i). Mutations in the AriB dimerization interface (E285R and F137A), as predicted from the AlphaFold2 model (Fig. 1c and Extended Data Fig. 1g), resulted in the loss of
phage defence (Fig. 3j), highlighting that AriB dimerization is essential for PARIS activity. Collectively, these data show that the Ocr trigger interacts with AriA in an ATP-dependent
manner, leading to the release and dimerization of AriB.
Activated PARIS inhibits translationWe next investigated the consequences of PARIS activation by Ocr. The expression of Ocr from a plasmid in E. coli is sufficient to cause cell death in the presence of PARIS (Fig. 4a).
Monitoring of PARIS+ cells under the microscope using live/dead staining with propidium iodide showed the accumulation of cells with permeabilized membrane within 20 min of Ocr induction
(Fig. 4b and Supplementary Video 2). The TOPRIM domain, probably responsible for AriB toxicity, is frequently associated with DNase or RNase activities29, but RNA or DNA preparations from
AriB-activated cells did not show evidence of indiscriminate nuclease activity (Extended Data Fig. 8a–c). However, DAPI staining showed that PARIS activation results in nucleoid compaction
(Fig. 4b), reminiscent of the translation inhibition phenotype30. To understand the nature of this compaction, we performed chromosome conformation capture (Hi-C) experiments. The results
showed a loss of chromosomal structures similar to that observed following treatment with chloramphenicol, a known inhibitor of translation (Extended Data Fig. 9).
Fig. 4: Activation ofPARIS leads to cell death and inhibition of translation.
a, Expression of Ocr from the pBAD vector in PARIS+ culture resulted in cell toxicity, whereas the empty vector (EV) had no impact. b, Ocr expression in PARIS+ cells induced cell death
(propidium iodide staining, red) and DNA compactization (DAPI staining, blue). Arrows indicate zoomed-in images of nucleoid structures from PARIS+/Ocr (top) and PARIS−/Ocr (bottom). Cells
were imaged 1 h following induction of Ocr expression. c, Metabolic labelling experiments show a drop in 3H-methionine incorporation following induction of Ocr, indicative of translation
inhibition following PARIS activation. d, In vitro translation in the presence of activated AriB (WT) or active site mutant (E26A). Translation of firefly luciferase mRNA was monitored by
luminescence. In the presence of activated AriB, luciferase signal was reduced markedly, further indicating that PARIS functions through a mechanism of translational inhibition. c,d, Mean
and s.d. were calculated for each time point for three independent experiments. Scale bars, 10 µm. a.u., Arbitrary units; 3H-t, 3H-thymidine.
Source Data
Full size imageTo better understand the nature of PARIS-induced translational inhibition, we conducted metabolite-labelling experiments and in vitro translation assays. Production of Ocr in PARIS+ cells
resulted in (1) rapid inhibition of 3H-methionine (3H-met) incorporation, consistent with translation inhibition (Fig. 4c) and (2) a moderate increase in 3H-uridine (3H-u) uptake. This
increase is a known consequence of translation inhibition that can be explained by enhanced transcription of ribosomal RNA genes15. The importance of activated AriB in translational arrest
was further corroborated using a luciferase reporter in an in vitro translation assay. The presence of activated AriB in the reaction mixture interferes with the production of luciferase,
whereas the active site mutant (AriB E26A) has no impact (Fig. 4d). Collectively, these results demonstrate that PARIS activation results in translational inhibition.
Triggers and asuppressor of PARIS
Comparative analysis of related phages has provided considerable insight into both defence and antidefence strategies31. While testing the activity of PARIS against phages of the T5 family,
we noticed distinct plaquing phenotypes among T5 variants (Fig. 5a). PARIS does not protect cells from T5WT phage infection, but efficiently blocks infection by a variant of T5 from the
Moscow phage collection (T5Mos). Consistent with translation arrest following PARIS activation, infection of PARIS+ cells with T5Mos at high multiplicity of infection (MOI) resulted in rapid
growth inhibition, which occurred more rapidly than the time required for lysis of PARIS− cells (Extended Data Fig. 10a). Aside from a large deletion (Δ30661–38625), the T5Mos genome is
nearly identical to the T5 archetype (Fig. 5b). We hypothesized that both phages contain a trigger that activates PARIS, but that the T5 archetype also contains a suppressor of PARIS that
has been lost in T5Mos. To determine how T5Mos triggers PARIS, we isolated and sequenced T5Mos mutants that escape PARIS immunity. All PARIS-escape mutants carried mutations in two genes
encoding highly acidic proteins of unknown function: open reading frame (ORF)094 and ORF103 (ref. 32) (Fig. 5a–d). Expression of these proteins from a plasmid resulted in activation of PARIS
toxicity (Fig. 5e), confirming that T5 carries two novel PARIS triggers. Mutated variants encoded by escape mutants did not trigger PARIS. We named these proteins PARIS triggers 1 and 2
(Ptr1, ORF094 and Ptr2, ORF103). These results show that structurally diverse, negatively charged viral proteins can activate the PARIS system. The presence of genes ptr1 and ptr2 in the
genome of T5wt, which is not affected by PARIS, further indicates that this phage contains a suppressor of PARIS immunity.
Fig. 5: PARIS cleaves E. coli tRNALys and T5 encodes anon-cleavable tRNALys.
a, Plaque assays conducted using cells with (+) or without (−) PARIS against mutants T5WT, T5Mos and T5Mos (T5Mut1–Mut4). b, Graphical map of the WT T5 genome. ORFs are represented by grey
arrows and tRNAs by red ovals. T5Mos is missing an 8 kb fragment (red), with T5123 having a smaller 3 kb deletion (blue). c, T5Mos phages acquire mutations in ORF094 (Ptr1) and ORF103 (Ptr2)
(black arrows) to escape PARIS-mediated immunity (mut1–mut4). d, AlphaFold2-predicted structures for Ptr1 and Ptr2 compared with T7 Ocr. Protein surfaces coloured by electrostatic
potential. e, Expression of WT triggers, but not mutant, cause toxicity in PARIS+ cells but not in PARIS− cells. f, Overexpression of T5 tRNALys in PARIS+ cells rescues infection by phage
T5123, which lacks tRNALys. g, RNA blot performed on total RNA extracted from E. coli MG1655 carrying the PARIS system or a GFP control, and Ocr under the control of a PPhlF promoter. RNA
was extracted either 15 or 30 min (15′ and 30′, respectively) following induction. Identification of 5S rRNA (green) and E. coli tRNALys (red) was made using distinct probes. h, Northern
blot performed on total RNA extracted from E. coli carrying PARIS or GFP, Ocr and T5 tRNALys under the control of an araBAD promoter. RNA was extracted following 30 min of induction. The
tRNALys (red) and T5 tRNALys (yellow) probes show that the latter was not degraded during PARIS-mediated defence. i, Small RNA extracted from E. coli showing a cleavage product when
incubated with AriB in a metal-dependent manner. j, Primer extension assay. AriB cleaves E. coli tRNALys between positions 40 and 41. k, Schematic representation of the AriB cleavage site on
E. coli tRNALys. Scale bars, 2 kb. aa, Amino acids; l-ara, l-arabinose; pI, isoelectric point.
Full size imageViral tRNA is resistant to AriB cleavageThe T5 genome encodes 23 tRNAs, 16 of which have been lost in T5Mos. Given the inhibitory effect of activated AriB on translation, we hypothesized that phage-encoded tRNAs are responsible
for overcoming PARIS toxicity. To narrow the search for a PARIS suppressor, we tested a collection of T5 deletion mutants for their sensitivity to PARIS33. Among the variants tested, T5123
carries the smallest deletion in the tRNA genes region (Δ29191–32442) and is still sensitive to PARIS (Extended Data Fig. 10b). The DNA fragment missing from T5123 was divided into three
segments, which were separately cloned on an expression plasmid and introduced into PARIS+ cells. A fragment encoding tRNAPro, tRNAfMet, tRNALys and tRNAVal partially rescued the ability of
phage T5123 to infect PARIS+ cells, whereas other fragments had no effect (Extended Data Fig. 10c). Next, we cloned each of the tRNAs individually and tested their ability to rescue T5123
infection. Overexpression of the T5 tRNALys completely restored T5123 infectivity (Fig. 5f), demonstrating that this phage-encoded tRNA neutralizes PARIS-induced toxicity and suggesting that
the host tRNALys is targeted by AriB.
To confirm that the E. coli tRNALys is indeed degraded following PARIS activation, we performed RNA blot with total RNA extracted from PARIS+ cells 15 or 30 min following induction of the
Ocr trigger. tRNALys was degraded, whereas SYBR Gold staining did not show degradation of other RNAs (Fig. 5g). We note that we cannot exclude the possibility that other E. coli tRNAs might
be specifically degraded by AriB, and that T5123 complements their loss through some of the other tRNAs that it carries. More work is necessary to fully characterize the activity of AriB.
To determine how T5 tRNALys suppresses PARIS immunity, we analysed RNA extracted from PARIS-activated cells expressing the phage tRNA from a plasmid. Although E. coli tRNALys was still
degraded, T5 tRNALys remained intact (Fig. 5h and Extended Data Fig. 11a,b). The phage-encoded variant of tRNALys contains mutations in the anticodon stem-loop (Extended Data Fig. 10d),
which could be responsible for the lack of AriB recognition. To test this hypothesis, we introduced mutations from T5 tRNA into the E. coli tRNALys counterpart. The resulting chimeric tRNA
rescued infection by phage T5123 in the presence of PARIS, confirming that AriB recognizes the tRNA anticodon stem-loop (Extended Data Fig. 10e). To determine the precise cleavage site of
AriB, we incubated tRNAs extracted from E. coli with activated AriB, which resulted in cleavage products only in the presence of Mg2+ and with the catalytically active AriB (Fig. 5i). A
primer extension assay performed using a probe complementary to the 3′ end of the host tRNALys showed that the AriB cleavage site lies between residues 40 and 41 of E. coli tRNALys (Fig.
5j,k). This cleavage site was corroborated by Sanger sequencing of the cleaved 3′ fragment (Extended Data Fig. 11c). Collectively, these results indicate that phage T5 encodes a
non-cleavable variant of tRNA that carries a mutation in the AriB cleavage site, compensating for the loss of host tRNA depleted following PARIS activation.
Evolution of the PARIS defencesystem
To inform our understanding of the diversity of PARIS systems and their relationship with other ABC ATPase-powered defence systems, we conducted phylogenetic analyses. First, we generated
trees for AriA and AriB showing a diverse set of systems that can be grouped into 11 distinct clades, including 2 in which ariA and ariB merged into a single ariAB gene (Fig. 6a,b). ariA and
ariB genes from the same system consistently fall in matching clades, showing that they coevolve and are not frequently swapped between systems. A rooted version of the AriB tree shows that
AriAB emerged once from clade 9 of two gene systems in a single event and was then split into two different clades (Extended Data Fig. 11d,e). Interestingly, sequence alignments of AriA
show that residues associated with the central and radial pores of the complex are poorly conserved (Extended Data Fig. 11d,e). This suggests that the signal transduction and effector parts
of PARIS immunity are shared across homologues, whereas the central and radial pores have evolved to recognize different viral triggers.
Fig. 6: Phylogenies of PARIS- and related ABCATPase-powered defence systems.
a,b, The phylogenetic trees of AriA (a) and AriB (b). The two trees share similar branching patterns. Different colours represent 11 subclades of PARIS, including 2 in which ariA and ariB
merged into a single ariAB gene (merged 1 and 2). c, Phylogenetic tree of AAA15/21 ATPase-containing defence proteins. Pfam accessions AAA15 (PF13175), AAA21 (PF13304), TOPRIM-Old (PF20469),
DUF4435 (PF14491), RloB (PF13707), DUF4276 (PF14103), UvrD_N (PF00580), UvrD_C (PF13361), DUF262 (PF03235), DUF3996 (PF13161), HNH endonuclease (PF14279, PF10107) and RVT1 (PF00078) were
used for domain annotations. Tree scale bars, 1.
Source Data
Full size imagePARIS was recently assigned to a family of OLD-like defence systems that share an ABC ATPase and a TOPRIM nuclease11. This analysis identified four classes, in which the single-gene systems
such as P2 Old are categorized as class 1, Gabija as class 2, reverse transcriptase-containing systems as class 3 and PARIS systems as class 4. We built a tree of known antiphage defence
systems that carry an ABC ATPase of the AAA15/21 family (Fig. 6c). Our analysis adds AbiL and MADS3–4 systems to the list of systems sharing an ABC ATPase and TOPRIM nuclease; it also shows
how this ABC ATPase can be found in association with a variety of other effector domains, demonstrating the evolutionary success of this domain as the probable sensor of viral infection in
diverse immune systems.
DiscussionOur results demonstrate how AriA forms a propeller-shaped hexamer capable of binding three AriB monomers. AriA is an ATP-dependent sensor that detects viral immune suppressors (for example,
Ocr), which trigger the release of AriB. Released AriB assembles into a homodimer that cleaves specific host tRNAs, resulting in rapid interruption of translation followed by loss of
membrane integrity. As opposed to prototypical toxin–antitoxin systems, AriB has modest toxicity when expressed alone. This suggests that AriA activates AriB through a structural
modification that enables dimerization or an unidentified post-translational modification. More work will be necessary to determine the mechanism of AriB release and activation.
Interestingly, we found that the PARIS system from E. coli B185 is triggered not only by the phage T7 Ocr protein but also by the Ptr1 and Ptr2 proteins of phage T5. These proteins are small
and negatively charged but lack obvious sequence or structural similarities. Determination of how PARIS detects such diverse proteins, and whether different systems have distinct trigger
sensitivities, presents an intriguing avenue for future research.
Our work adds to a growing list of defence systems that cause translational arrest following infection. The PrrC and Retron I A (that is, PtuAB proteins) systems induce tRNA degradation
following detection of foreign proteins, whereas Cas13a degrades tRNA following detection of foreign RNA34,35,36. However, translational arrest is not limited to tRNA degradation, Mogila et
al.37 recently identified a CRISPR–Cas system that cleaves mRNA associated with the ribosome, which arrests translation, and the protease late inhibition of T4 (Lit) degrades the translation
elongation factor EF-Tu following detection of the major capsid protein of phage T4 (ref. 38).
We show that some phages circumvent PARIS-induced toxicity by expression of non-cleavable tRNA variants. Many tailed phages, such as those from the families Demerecviridae (T5-like phages),
Straboviridae (T4-like phages) or Ackermannviridae, encode large arrays of tRNAs previously thought to be associated with translation optimization due to codon bias in the host and viral
genome39. However, estimation of the abundance of host and phage tRNAs during infection, compared with the corresponding codon frequencies in produced mRNAs, does not directly support this
hypothesis39. A recent computational analysis suggests that phages may have evolved these variants in response to bacterial tRNA-targeting toxins, as evidenced by distinct anticodon loop
structures aimed at evasion of host nucleases40. This hypothesis is further supported by a parallel work demonstrating that phage T5 tRNATyr provides protection against the PtuAB toxin of
the Eco7 (Ec78) retron36.
Interestingly, this strategy of encoding tRNA clusters is also observed in some eukaryotic viruses, such as those from the families Herpesviridae, Mimiviridae and Phycodnaviridae41, possibly
serving a similar function in evasion of host immunity. Strikingly, an anticodon nuclease was also recently found to protect humans against Pox viruses, demonstrating that the inhibition of
translation through the inactivation of specific tRNAs is a conserved antiviral strategy used across domains of life42.
MethodsBacterial strains, phages and plasmidsThe phages, bacterial strains and plasmids used in the study are listed in Supplementary Table 1 and the primers are listed in Supplementary Table 2. Infection with T5 phage was carried out
in Luria-Bertani (LB) medium supplemented with 1 mM CaCl2. Unless otherwise indicated, 0.2% l-ara, 0.1 mM isopropyl-ß-d-thiogalactopyranoside (IPTG) and 0.2 µg ml−1 anhydrotetracycline (aTc)
were used for induction.
For structural studies, the coding sequences for AriA and AriB from E. coli B185 (ref. 2) were cloned under the control of a T7 promoter into a pRSF-Duet vector with a C-terminal strep-tag
II on AriB. For coexpression of PARIS with the T7 Ocr trigger, the TOPRIM nuclease of AriB was inactivated (E26A) and strep-tag II was removed by site-directed mutagenesis. The coding
sequence for T7 Ocr protein was cloned into a pET-Duet vector with a C-terminal strep-tag II. For in vitro binding assays, the coding sequence for T7 Ocr protein was cloned into a pET-Duet
vector with an N-terminal His6-TwinStrep-SUMO tag. All in vivo experiments were performed with pFR85, encoding PARIS from E. coli B185 with its native promoter and an inducible Ptet promoter
upstream. For in vivo protein pulldowns, C-terminal AriB strep-tag II was introduced to pFR85. Mutants of PARIS were constructed from pFR85 using either the Gibson method43 or Q5
site-directed mutagenesis kit (NEB). PARIS triggers both Ptr1 (T5 ORF094) and Ptr2 (T5 ORF103) from the T5 phage, and T5 genome fragments, T5 tRNALys and E. coli tRNALys were cloned on the
pBAD18 vector under the control of the araBAD promoter using the Gibson method. Twenty base pairs upstream and downstream of tRNA, genes were preserved during cloning. T5 nucleotide
positions are provided according to genome assembly AY543070.1. All constructions were verified using Sanger sequencing.
Expression and purification of the PARIS complexAriA and AriB-Strep were coexpressed from a pRSF-Duet plasmid in BL21-AI cells (Invitrogen). Cultures grown at 37 °C to 0.4–0.5 OD600 were induced with 0.1 mM IPTG and 0.1% l-ara. Following
induction, cultures were incubated overnight at 16 °C. Cells were pelleted (3,000g, 10 min at 4 °C), resuspended in lysis buffer (25 mM Tris pH 8.5, 150 mM NaCl and 1 mM EDTA) and lysed by
sonication. Cellular debris was removed by centrifugation (10,000g, 25 min at 4 °C) and the AriAB complex was purified by affinity chromatography on 5 ml of streptactin resin (IBA) and
eluted with 2.5 mM desthiobiotin (IBA) in lysis buffer. The eluted protein was concentrated (100K MWCO PES Spin-X UF concentrator, Corning) and further purified by size exclusion
chromatography (Sup200 column, Cytiva) in lysis buffer with 2% glycerol. Fractions of interest were combined and concentrated to 5 µM (100K MWCO PES concentrator, Pierce) and used
immediately for vitrification on cryo-EM grids.
In vivo protein pulldownFor determination of protein-of-interest interacting partners in vivo, either AriB-Strep expressed from pFR85 or Ocr-Strep expressed from pBAD was used as bait, and pulldowns were performed
in E. coli BW25113. Three litres of cell culture were grown in LB medium at 37 °C in Thomson flasks with aerated lids (1.5 litres per flask). Expression of proteins was induced with either
0.2 µg ml−1 aTc or 0.2% l-ara at an approximate OD600 of 0.1, and cells were harvested by centrifugation after reaching an approximate OD600 of 0.8–1.0. Cells were processed as described
above, and Strep-tagged proteins purified on two stacked 5 ml StrepTrap HP (Cytiva) columns. The identity of protein bands following SDS–polyacrylamide gel electrophoresis (PAGE) was
determined by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Roughly one-third of the Coomassie-stained band was cleaved from the gel, and samples were
prepared with Trypsin Gold (Promega) in accordance with the manufacturer’s instructions. Mass spectra were obtained using the rapifleX system (Bruker).
For the production of activated AriB, AriB-Strep from pFR85 and non-tagged Ocr from pBAD were expressed in separate cultures and cells grown as previously described. Cell cultures were
mixed, and cell walls disrupted by sonication on ice (60% power, 10 s pulse, 20 s pause, 30–60 min) on a Qsonica sonicator with 6 mm sonotrode, followed by StrepTrap HP purification of
activated AriB-Strep. The molecular weight of protein complexes was determined following size exclusion chromatography performed on a Superdex 200 Increase 10/300 column (GE Healthcare)
calibrated with the High Molecular Weight calibration kit (GE Healthcare).
Glutaraldehyde cross-linking assayTo determine the oligomeric state of activated AriB in solution, we performed a glutaraldehyde cross-linking assay. Concentrated AriB aliquots were mixed with glutaraldehyde (no.
253857.1611, PanReac) to a final concentration of 0.025, 0.125 or 0.25% v/v and incubated for 30 min at room temperature. The reaction was quenched by the addition of one sample volume of 1
M Tris-HCl at pH 8.0 and a half-sample volume of 5× concentrated SDS sample buffer. Samples were heated for 5 min at 60 °C and separated by SDS–PAGE.
In vitro protein pulldownThe T7 Ocr protein with an N-terminal His6-TwinStrep-SUMO was expressed from a pET-Duet plasmid in BL21(DE3) cells. Cultures grown at 37 °C to 0.4–0.5 OD600 were induced with 0.5 mM IPTG.
Following induction, cultures were incubated overnight at 16 °C. Cells were pelleted (3,000g, 10 min at 4 °C), resuspended in lysis buffer (25 mM Tris pH 8.5, 150 mM NaCl and 1 mM EDTA) and
lysed by sonication. Cellular debris was removed by centrifugation (10,000g, 25 min at 4 °C) and HSS-Ocr was purified by affinity chromatography on 5 ml of streptactin resin (IBA) and eluted
with 2.5 mM desthiobiotin (IBA) in lysis buffer. The eluted protein was incubated with SUMO protease at 4 °C overnight before washing over 5 ml of Ni-NTA resin (25 mM Tris pH 7.5, 300 mM
NaCl, 1 mM TCEP and 20 mM Imidazole) to remove the SUMO-cleaved tag. Flow-through protein was concentrated (10K MWCO PES Spin-X UF concentrator, Corning) and further purified by size
exclusion chromatography (Sup6 10 300 column, Cytiva) in lysis buffer with 2% glycerol. Fractions of interest were combined and concentrated (10K MWCO PES Spin-X UF concentrator, Corning).
Cleaved Ocr (3.8 μM) was incubated with WT PARIS (11.3 μM) at 37 °C for 15 min before analysis via size exclusion chromatography (Sup6 10 300 column, Cytiva) in lysis buffer with 2%
glycerol. Fractions of interest were combined, concentrated (10K MWCO PES Spin-X UF concentrator, Corning) and analysed via SDS–PAGE (15% acrylamide, 150 V for about 1 h).
Cryo-EM samplepreparation and data collection
All samples were subjected to vitrification on 1.2/1.3 Carbon Quantifoil grids, which were glow-discharged using an easiGlow (Pelco) glow-discharge station (glow 45 s, hold 15 s). Following
the application of 3 µl of 5 µM PARIS + 1 mM ATPγS, grids were subjected to double-sided blotting using a Vitrobot Mk IV, with a force of 5 for 4 s at 4 °C and 100% relative humidity.
Following plunge-freezing in liquid ethane, grids were clipped and stored in liquid nitrogen.
For PARIS obtained from AriB pulldowns, cryo-EM grids were screened and all data collected using a Talos Arctica microscope (ThermoFisher) operating at 200 kV and equipped with a K3 direct
electron detector (Gatan). SerialEM44 software v.3.8.6 was used to automate data collection. For final data collection, a total of 7,340 movies were collected in super-resolution mode with a
pixel size of 0.552 Å and a total dose of 56.42 eÅ−2 distributed over 50 subframes.
For determination of the structure of AriA, samples were prepared for cryo-EM analysis as described above. Following screening of grids, 10,340 movies were collected using a Talos Arctica
microscope (ThermoFisher) operating at 200 kV and equipped with a K3 direct electron detector (Gatan). SerialEM software v.3.8.6 was used to automate data collection. Videos were recorded in
super-resolution mode with a pixel size of 0.552 Å and a total dose of 56.31 eÅ−2 distributed over 50 subframes.
Cryo-EM data processing of inactive PARISAll data were processed in cryoSPARC v.4.41 (ref. 45). Following patch motion correction and contrast transfer function (CTF) estimation, micrographs with CTF fits below 8 Å were discarded,
yielding 5,998 movies for particle picking. Following processing of a subset of 200 micrographs, the AriA6–AriB3 assembly of the PARIS complex was apparent. Using this low-resolution initial
volume, AlphaFold2-predicted structures of AriA and AriB were fit into the map and used to generate a 20 Å low-pass-filtered map using the Molmap command in ChimeraX v.1.7 (ref. 46). This
volume was imported to cryoSPARC and used to generate templates for particle picking. A total of 4,078,384 particles were extracted and, following initial two-dimensional classification,
1,643,515 were retained for further processing. These particles were sorted using a three-class ab initio reconstruction that showed two junk classes and a volume corresponding to the
assembled PARIS complex containing 940,782 particles. After a second round of two-dimensional classification, 934,763 particles remained and were subjected to a further round of ab initio
reconstruction and heterogeneous refinement. This produced a consensus volume containing 532,010 particles with conformational heterogeneity for two AriB subunits, with one asymmetric unit
of the complex being well aligned. Masked local refinement was used to determine a 3.2-Å-resolution map of one asymmetric unit of the complex. The consensus refinement with 532,010 particles
was also subjected to multiple rounds of masked three-dimensional classification to isolate particles in the cis and trans arrangements. The fully assembled PARIS complex demonstrates
flexibility based on three-dimensional variability analysis, which limited the attainable resolution of the fully assembled complex. The density maps for both local refinement and the full
complex are available at EMDB-42719, 43103 and 43104, and raw micrographs are available at EMPIAR-11832.
Cryo-EM data processing of Ocr pulldownAll data were processed in cryoSPARC v.4.41. Of the 10,340 movies collected, 9,399 were selected for further processing based on the CTF fit criteria described above. Using a blob picker
with a particle diameter of 200 Å, particles were extracted and subjected to two-dimensional classification. In the two-dimensional classes there were two distinct macromolecular complexes
present, one of which was interpreted to be the AriA scaffold of the PARIS complex, with the other interpreted as corresponding to RNA polymerase.
From 7 selected classes corresponding to the AriA hexamer, 667,479 particles were selected for downstream refinement. From a two-class ab initio reconstruction of these particles, 392,989
were retained. Iterative sorting by multiclass ab initio reconstruction and heterogenous refinements yielded a volume containing 62,732 particles. AriA subunits can be fit into this density,
but poor resolution rendered this structure indistinguishable from the AriA scaffold identified in the intact PARIS complex. No density was observed for Ocr, which suggests that multiple
binding sites may be present on the PARIS complex, or that Ocr is attached to AriA in a flexible state. The density map is available at EMDB-43105 and raw micrographs at
EMPIAR-11833.
Model building and refinementTo determine the structure of the asymmetric unit of the PARIS complex, density half-maps were obtained from cryoSPARC local refinements before being provided as inputs to DeepEMhancer
v.0.14 (ref. 47) for map sharpening. AlphaFold2-predicted structures of AriA and AriB were then docked into the map density, and map-to-model fit was improved using the ISOLDE v.1.7.1 (ref.
48) molecular dynamics simulation environment in ChimeraX v.1.7. Following initial equilibration, PDB files were saved and used as inputs for real-space refinement using PHENIX v.1.20.1
(ref. 49). Following initial refinement, problematic areas were manually inspected and modified using COOT v.0.9.8.1 (ref. 50), and side-chains were removed from regions with resolution
below 4 Å. Following addressing of clashes and non-rotameric side-chain orientations, iterative refinements were carried out in Phenix until Clashscores, Ramachandran and side-chain outliers
failed to improve. The final model and corresponding map have been deposited (PDB ID: 8UX9, EMDB-42719).
To generate the biological assemblies for the PARIS complex atomic model, half-maps from C3 symmetric and non-uniform refinements of particles corresponding to the cis and trans orientations
were obtained from cryoSPARC and sharpened in DeepEMhancer v.0.14. Following fitting of the three copies of the asymmetric unit into the cis and trans density maps using the ChimeraX v.1.7
fitmap command, the find NCS tool in Phenix was then used to calculate the appropriate symmetry operators for the assembled forms of the complex. These symmetry operators were applied to the
asymmetric unit to generate biological assemblies 1 and 2 using the Apply NCS tool in Phenix. Maps for the cis and trans orientations of the PARIS complex are available at EMDB-43104 and
EMDB-43105, respectively.
ATPase assaysATPase activity was measured using 6 µM Ocr, 100 nM PARIS and 128 µM ATP supplemented with 10 nM [α-32 P]-ATP (PerkinElmer). The reaction buffer included 20 mM Tris-HCl pH 8.0, 150 mM sodium
chloride, 1 mM DTT and 5 mM magnesium chloride. Reactions were incubated at 37 °C, and 10 µl aliquots were quenched with phenol at 1, 2, 4, 8, 16 and 32 min. For trigger-only reactions, 100
nM Ocr was mixed with 128 µM ATP supplemented with 10 nM [α- 32 P]-ATP (PerkinElmer) in the reaction buffer. Reactions were incubated for 32 min at 37 °C and quenched with phenol. The ATP
and ADP makers were created using T4 polynucleotide kinase (NEB). T4 PNK was mixed with 128 µM ATP, supplemented with 10 nM [α-32 P]-ATP and 1 mM DNA oligo, in T4 polynucleotide kinase
buffer (NEB); this mixture was then incubated at 37 °C for 1 h. All reaction products were phenol-chloroform extracted and resolved on silica TLC plates (Millipore). Reaction products were
spotted 2.5 cm above the bottom of the TLC plate. Plates were placed in a TLC developing chamber filled to around 1.5 cm with developing solvent (0.2 M ammonium bicarbonate pH 9.3, 70%
ethanol and 30% water) and covered with aluminium foil for 4 h at room temperature. Plates were then exposed to a phosphor screen, imaged using a Typhoon phosphor imager and quantified with
the ImageQuant software package (Cytiva).
EOP assayThe activity of PARIS mutants was measured in comparison with the WT system by performing efficiency of plaquing (EOP) assays with phage T7. E. coli K-12 MG1655 carrying each of the AriA or
AriB mutants, the control plasmid pFR66 (sfGFP) or the WT PARIS system (pFR85) was grown overnight in LB + kanamycin (50 µg ml−1). Bacterial lawns were prepared by mixing 100 µl of a
stationary culture with 5 ml of LB + 0.5% agar, and the mixture was poured onto a Petri dish containing LB + kanamycin (50 µg ml−1). Tenfold serial dilutions of high-titre stock of T7 phage
were spotted on each plate and incubated for 5 h at 37 °C. EOPs with T5 and T5-like phages were performed with E. coli BW25113 as described above, except that plates were incubated at 37 °C
overnight and the top agar was supplemented with 1 mM CaCl2. Induction of tRNA genes or phage T5 fragments was achieved by supplementing the top agar with the indicated amount of l-ara.
Plaque assays were performed in at least two independent replicates.
Liquid culture phage infectionTo monitor the dynamics of T5Mos phage infection of PARIS+ (pFR85) and PARIS− (pFR66) cultures in liquid, we used an EnSpire Multimode Plate Reader (PerkinElmer). Overnight bacterial
cultures were diluted 100-fold in LB medium with appropriate antibiotics and grown at 37 °C in 10 ml of LB supplemented with 1 mM CaCl2. At OD600 0.6, 200 μl aliquots were transferred to
96-well plates and infected at the indicated MOI. Optical density was monitored for 10 h. All experiments were performed in three biological replicates.
Solid medium toxicity assayPARIS toxicity in the presence of T7 Ocr or Ptr1 and Ptr2 was measured using a spot-test assay. PARIS+ (pFR85) and PARIS− (pFR66) cultures carrying the pBAD vector-encoding-indicated trigger
were grown overnight in 10 ml of LB at 37 °C. Stationary cultures were diluted to OD600 0.6 and plated on LB or minimal-medium (M9 with 5% v/v LB) agar plates supplemented with 0.2% l-ara
by serial tenfold dilution. Control plates without induction contained 0.2% glucose to prevent leakage of the araBAD promoter. To assess the toxicity of the PARIS AriA D401N/E404Q mutant, we
used either pFR85 (PARIS expression driven from native promoter, BW25113 E. coli strain) or pRAW 464 vector (PARIS overexpression from T7 promoter, BL-21 (DE3) E. coli strain). The cultures
were plated on M9 as described above, and PARIS expression was induced with 1 mM IPTG. Ocr expression was driven from pBAD with 0.2% l-ara.
Liquid culture toxicity assayTo assess the toxicity of AriB expression alone compared with activated PARIS, E. coli BL-21 AI cells were transformed with plasmids encoding PARIS (pRAW 464) and AriB (pRAW 466) or PARIS
(pRAW 464) and Ocr (pRAW 472). Following overnight growth of cultures in LB supplemented with the appropriate antibiotic, cells were diluted to OD600 0.1 in LB + antibiotics before growing
to OD600 0.5. After cells had reached OD600 0.5, 200 μl of culture was added to 800 μl of medium supplemented with antibiotics and either 0.4% glucose, 0.2% l-ara and 0.1 mM IPTG or 0.5%
l-ara and 0.1 mM IPTG. Next, 200 μl of each culture was aliquoted into a 96-well plate (Evergreen, no. 222-8030-01F). Growth was monitored using an Agilent BioTek Cytation5 plate reader set
to 30 °C with continuous linear shaking, and OD600 readings taken every 5 min for 15 h.
Escherichia coli MG1655 strains carrying plasmids pFR85 (Ptet ariAB), pFR85∆AriA (pFR99), pFR85∆AriB (pFR100) or pFR85-AriA(R212*) (pFD294), in which a stop codon was introduced in AriA,
truncating the protein before the domain that interacts with AriB, were diluted 1:100 from an overnight culture in LB + kanamycin (50 µg ml−1) and arrayed in a 96-well plate. Growth was then
monitored every 10 min for 30 h at OD600 in an infinite M200 Pro plate reader (Tecan) at 37 °C with shaking. Absorbance values of three biological replicates were analysed using GraphPad
Prism v.10.1.2. Standard two-way analysis of variance was used to calculate P values for growth rates relative to WT PARIS.
Fluorescence microscopyPARIS+ (pFR85) and PARIS− (pFR66) carrying pBAD Ocr were diluted 1:100 and cultivated in LB supplemented with appropriate antibiotics at 37 °C. When OD600 reached 0.4, an aliquot was mixed
with DAPI (Invitrogen) at 1 mg ml−1 final concentration and propidium iodide (Invitrogen) at 1 μg ml−1 final concentration and incubated at room temperature in the dark for 5 min. LB + 1.5%
agarose slabs (approximately 0.2 mm thick) were prepared on a 75 × 25 mm2 microscopy slide (Fisher Scientific). Agarose slabs contained staining dyes at the same concentration were
optionally supplemented with 0.2% l-ara, to induce Ocr expression. Roughly 1 μl of stained cells was placed on an agarose slab and, following 1 min of drying, the slab was covered by a small
(22 × 22 mm2) coverslip (Fisher Scientific). Imaging was performed on a Nikon Eclipse Ti-E inverted epifluorescence microscope. Each field of view was imaged in the transmitted light
channel (200 ms exposure) and in the DAPI (filterset Semrock DAPI-50LP-A, 200 ms exposure) and propidium iodide (filterset TxRed-4040C, 200 ms exposure) channels.
Extraction of total DNATotal DNA was extracted at different time points (15, 30, 60 and 120 min) with or without DAPG to induce the expression of Ocr in the presence of PARIS. DNA was extracted using the Wizard
genomic DNA purification kit (Promega) according to the manufacturer’s instructions and treated with RNase at 0.1 mg ml−1. Each sample of total DNA was loaded on 1% agarose 1× TAE
gel.
TUNEL assayTUNEL assays was performed to estimate in vivo the accumulation of dsDNA breaks, according to methods described previously51. PARIS+ (pFR85) culture carrying the pBAD Ocr vector was grown to
OD600 0.3, followed by Ocr induction with 0.2% l-ara for 2 h. As a positive control, cells were treated with 0.1% H2O2 to induce accumulation of DNA breaks, then 2 ml of cells was harvested
by centrifugation and treated according to the standard protocol (TUNEL Assay Kit – FITC, no. ab66108, abcam). Measurement of fluorescein isothiocyanate fluorescence was performed with the
CytoFLEX cytometer (Beckman Coulter) in the fluorescein isothiocyanate channel. A total of 100,000 events were collected for each sample in three biological replicates. Data were analysed
and visualized in FlowJo v.10 (ref. 52).
Hi-C procedure and sequencingEscherichia coli MG1655 carrying plasmids pFR85 (Ptet ariAB) and pFD250 (PPhlF ocr), or control plasmids pFR66 (sfGFP) and pFD245 (PhlF sfGFP), was diluted 1:100 from an overnight culture in
LB + kanamycin (50 µg ml−1) + chloramphenicol (20 µg ml−1) and grown to OD600 0.3. DAPG 50 µM was then added, followed by incubation for 15 and 30 min. To compare the effect of PARIS
activation with that of treatment with chloramphenicol, E. coli MG1655 harbouring pFR66 was diluted 1:100 from an overnight culture in LB + kanamycin (50 µg ml−1) to OD600 0.3, followed by
the addition of chloramphenicol (20 µg ml−1) and incubation for 15 and 30 min. Cell fixation was performed with 4% formaldehyde (Sigma-Aldrich, catalogue no. F8775) as described in ref. 53.
Quenching of formaldehyde with 300 mM glycine was performed at 4 °C for 20 min. Hi-C experiments were performed with the Arima kit. Samples were sonicated using Covaris (DNA 300 base pairs).
Preparation of samples for paired-end sequencing was performed using the Invitrogen TM Colibri TM PS DNA Library Prep Kit for Illumina according to the manufacturer’s instructions; the
detailed protocol is available in ref. 53. Reads were aligned with bowtie2 v.2.4.4, and Hi-C contact maps were generated using hicstuff v.3.0.3 (https://github.com/koszullab/hicstuff) with
default parameters and using the HpaII enzyme for digestion. Contacts were filtered as described in ref. 54, and PCR duplicates (defined as paired reads mapping at exactly the same position)
were discarded. Matrices were binned at 4 kb. Balanced normalizations were performed using the ICE algorithm55. For all comparative analyses, matrices were downsampled to the same number of
contacts. The Hi-C signal was computed as contacts between adjacent 5 kb bins, as described in ref. 56. For comparison of this signal with other genomics tracks, we binned it at the desired
resolution.
Metabolic labellingMetabolic labelling with 3H-met, 3H-u and 3H-t in experiments was performed as described previously57, with minor modifications. E. coli BW25113 PARIS+ (pFR85) strain was transformed with
the pBAD plasmid carrying the Ocr trigger (for l-ara-inducible expression). Transformed cells were initially plated on LB plates supplemented with 100 μg ml−1 ampicillin, 25 μg ml−1
kanamycin and 0.2% glucose. Using individual E. coli colonies for inoculation, 2 ml liquid cultures were prepared in defined Neidhardt MOPS minimal medium (supplemented with 100 μg ml−1
ampicillin, 25 μg ml−1 kanamycin, 0.1% casamino acids and 1% glucose) and grown overnight at 37 °C with shaking. Subsequently, experimental 20 ml cultures were prepared in 125 ml conical
flasks in MOPS medium, supplemented with 0.5% glycerol, 100 μg ml−1 ampicillin and 25 μg ml−1 kanamycin, as well as a set of 19 amino acids (lacking methionine), each at a final
concentration of 25 μg ml−1. These cultures were inoculated at OD600 0.05 and grown at 37 °C with shaking to OD600 0.3. At this point, 1 ml aliquots (designated as the preinduction zero time
point) were transferred to 1.5 ml Eppendorf tubes containing 10 μl of the respective radioisotope (3H-met (0.77 μCi, PerkinElmer), 3H-u (0.1 μCi, PerkinElmer) or 3H-t (0.32 μCi,
PerkinElmer)) preincubated at 37 °C. Concurrently, Ocr expression in the remaining 19 ml culture was induced by the addition of l-ara to a final concentration of 0.2%. Throughout the Ocr
induction time course, 1 ml aliquots were taken from the 20 ml culture and transferred to 1.5 ml Eppendorf tubes containing 10 μl of the appropriate radioisotope. Radioisotope incorporation
was halted after 8 min of incubation at 37 °C by the addition of 200 μl of ice-cold 50% trichloroacetic acid (TCA) to the 1 ml cultures. In addition, 1 ml aliquots were sampled for OD600
measurement during the induction time course. The resultant 1.2 ml TCA-halted culture samples were loaded onto GF/C filters (Whatman) prewashed with 5% TCA, and the unincorporated label was
removed by washing the filter twice with 5 ml of ice-cold TCA followed by two 5 ml washes with 95% EtOH. The filters were placed in scintillation vials and dried for 2 h at room temperature,
followed by the addition of 5 ml of EcoLite-scintillation cocktail (MP Biomedicals). Following shaking for 15 min, radioactivity was quantified using the automatic TDCR Liquid Scintillation
Counter (HIDEX). Isotope incorporation was quantified by normalization of radioactivity counts (CPM) to OD600, with the preinduction zero time point serving as the reference (set to 100%).
All experiments were performed as biological triplicates using three independent liquid starter cultures inoculated with different colonies.
In vitro translationIn vitro translation was monitored by the production of luciferase signal in a PURExpress in vitro protein synthesis kit (NEB), using firefly luciferase mRNA as an input. pT7-Fluc, encoding
the firefly luciferase (fluc) gene under the control of the T7 promoter, was used as a template for T7 in vitro transcription with the MEGAscript kit (Thermo Scientific). mRNA was treated
with DNase I (Thermo Scientific) and purified with the Monarch PCR & DNA cleanup kit (NEB). The PURExpress in vitro translation reaction was assembled in RNase-free tubes according to the
manufacturer’s instructions, with minor modifications. Reaction mixtures were adjusted to a total volume of 5 μl (2 μl of solution A, 1.5 μl of solution B, 0.3 μl of RiboLock (40 U μl−1,
Thermo Scientific) and 0.2 μl of d-luciferin (10 mM, Sigma)), supplemented with either 0.5 μl of activated AriB or AriB (E26A) (100 μg ml−1 final concentration) or the same volume of buffer
A (50 mM NaCl, 1 mM EDTA, 100 mM Tris-HCl and 5 mM β-ME, pH 8.0) as a control. The reaction was started by the addition of 0.5 μl of purified fluc mRNA (500 ng), immediately placed in a
384-well white plate and covered with optically clear film. Accumulation of luminescent signal was measured with the EnSpire Multimode Plate Reader (PerkinElmer) for 3 h at 37 °C. AriB
concentration was determined with the Qubit Protein Broad Range Assay Kit (Invitrogen).
Isolation and sequencing of T5 escaper mutantsFor the selection of PARIS-escape mutants, phage T5Mos was continuously incubated with PARIS+ (pFR85) culture for 3 days. In brief, after reaching OD600 0.6, cells were infected with T5Mos
at an approximate MOI of 0.1 and the culture incubated overnight at 37 °C. Phage was collected the following day and 1 ml of lysate was used to initiate the next round of infection with
fresh PARIS+ (pFR85) culture. Escaper mutants were purified from single plaques obtained from PARIS+ (pFR85) culture and produced on the PARIS− (pFR66) culture. Eight millilitres of of
high-titre lysate (around 1010 plaque-forming units per millilitre) was PEG precipitated, and phage genomic DNA was purified as described previously58. DNA libraries were prepared according
to a standard protocol and sequenced on the MiniSeq platform (Illumina) with paired-end 150 cycles (75 + 75). Genome assemblies were performed with SPAdes implemented in Unicycler59. To
identify mutations, genomes of T5Mos escapers were aligned with the sequenced genomes of T5Mos and T5wt from initial stocks. T5 strains reported by Glukhov et al.33 were sequenced on the
DNBSeq-G400 platform (BGI) to validate boundaries of deletions.
Extraction of total RNATotal RNA was isolated from E. coli strain MG1655 harbouring either pFR85 (Ptet ariAB) or control plasmid pFR66 (Ptet sfGFP). Cells also carried pFD250 (PPhlF ocr) with Ocr under the control
of an inducible DAPG PPhlF promoter (Fig. 5g and Extended Data Fig. 8b), as well as plasmid pFD287 carrying the tRNALys of phage T5 under the control of the araBAD promoter inducible by
l-ara (Fig. 5h).
Cells were diluted 1:100 in LB with the appropriate antibiotics from overnight cultures and grown with agitation at 37 °C. When cultures reached OD600 0.2, aTc was added to a final
concentration of 0.5 µg ml−1 to induce expression of the PARIS system. For the experiment shown in Fig 5h, l-ara was added to a final concentration of 0.2% to induce T5 tRNALys from plasmid
pFD287. Growth continued to OD600 0.4, and the Ocr trigger was induced with DAPG added to a final concentration of 50 µM, followed by incubation for an additional 15 and 30 min post
induction.
Cells were centrifuged at 4,000g for 10 min, resuspended with 0.2 ml of lysozyme buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 1% Triton X-100 and 20 mg ml−1 lysozyme) and lysed for 30 min at 37
°C. One millilitre of TRIzol reagent (Zymo Research) was added to the samples, with incubation for 5 min at room temperature. Lysates were extracted with 0.2 ml of cold chloroform and
centrifuged at 12,000g for 15 min at 4 °C. The aqueous phase of the sample was precipitated with 0.5 ml of 100% cold isopropanol, incubated for 10 min at room temperature and centrifuged at
12,000g for 10 min at 4 °C. RNA pellets were washed with 1 ml of 75% ethanol, dried at room temperature and dissolved in 50 µl of nuclease-free water. Total RNA concentration was measured
with a nanodrop spectrophotometer, and samples were then resolved in 7 M urea 10% acrylamide gel before visualization with SYBR Gold.
Preparation of enriched small RNA fractions fromtotal RNA samples
Cells were grown in 20 ml of medium under the same conditions as those used for the extraction of total RNA. When the induced cultures reached OD600 0.8, cells were centrifuged at 5,000g for
25 min, washed in 5 ml of 0.9% NaCl and centrifuged again at 5,000g for 25 min. The following is adapted from ref. 60. The pellets were suspended in 2 ml of 50 mM sodium acetate (NaOAc) and
10 mM MgOAc pH 5.0, with the addition of 1.9 ml of commercial acidic phenol pH 4.5 (Ambion). The resulting mixture was shaken at 200 rpm for 30 min at 37 °C then centrifuged at 5,000g for
15 min at 4 °C. The upper aqueous phases were collected and subjected to precipitation of total nucleic acids in 0.1 M NaCl, supplemented with an equal volume of 2-propanol, for overnight
incubation at room temperature. Following centrifugation at 14,500g for 15 min at 4 °C, pellets were washed with 80% ethanol and air-dried.
Removal of rRNA from samples was achieved through precipitation: pellets were suspended in 0.8 ml of cold 1 M NaCl and spun at 9,500g for 20 min at 4 °C. Supernatants were collected, mixed
with 1.7 ml of ethanol, incubated for 30 min at −20 °C and then centrifuged at 14,500g for 5 min at 4 °C. The pellets were then washed with 80% ethanol and air-dried.
Removal of DNA from samples was achieved through precipitation: pellets were resuspended in 0.6 ml of 0.3 M NaOAc pH 5.0. The mixture was then heated for 5 min at 60 °C with regular
pipetting, followed by the addition of 0.34 ml of 2-propanol and incubation for 10 min at room temperature; the solution was then centrifuged at 14,500g for 5 min at room temperature and
supernatants collected.
The resulting small RNA fractions were precipitated by the addition of 0.23 ml of ethanol to the supernatant, centrifuging at 14,500g for 15 min at 4 °C, washing the pellet with 80% ethanol
and air-drying for 5–10 min. Pellets were dissolved in 0.35 ml of RNase-free water.
For deacylation, the fraction was incubated in 0.1 M Tris pH 9.0 for 45 min at 37 °C. Final precipitation in 0.3 M NaOAc pH 5.0 was achieved by the addition of 2.7 volumes of ethanol,
incubation for 30 min at −80 °C and centrifugation at 16,000g for 25 min at 4 °C. Pellets were washed with 80% ethanol, air-dried and dissolved in 80 µl of 1 mM sodium citrate.
Cleavageassay by AriB
Either 50 nM activated AriB or nuclease-deficient AriB (mutant E26A) dimer was incubated for 30 min at 37 °C with 90 ng of enriched small RNAs, extracted as described above, in a 10 µl
reaction mix containing 20 mM Tris pH 8.0 and 200 mM NaCl. MgCl2, when added, was at 2 mM final concentration. Reactions were stopped by the addition of 10 µl of 100% formamide, followed by
loading on a 15% acrylamide/7 M urea denaturing medium gel run at 22 W constant. SYBR Gold-stained gels were imaged using the ChemiDoc MP imaging system (Bio-Rad).
Mapping of the AriBcleavage site by primer extension
For precise identification of the AriB cleavage site, DNA probe F693 (5′-/FAM/TGGTGGTGGGTCGTGCAGGATTCGAACCTGCGACC-3′, harbouring a fluorescein at the 5′-end) was used in a reaction conducted
at room temperature on small RNAs that were incubated with or without activated AriB. In a 12 µl reaction, 2 µl of 1 µM probe was incubated with 750 ng of RNA on a heat block at 95 °C,
which was then switched off and left to cool down. Next, 8 µl of PCR with reverse transcription mix (Superscript III RT, Invitrogen) was added to the reaction mix and loaded onto a
thermocycler with the following conditions: 5 min at 25 °C, 60 min at 55 °C and 15 min at 70 °C. Following the addition of 20 µl of 100% formamide, reactions were loaded on a 15%
acrylamide/7 M urea denaturing medium gel run at 22 W constant. Fluorescein signal was recorded using the ChemiDoc MP imaging system (Bio-Rad). The ladder was generated by performing an
extension reaction using fluorescent probe F693 annealed to a 106 nt DNA template
(5′-CGATTGAGGCCGGTAATACGACTCACTATAGGGTCGTTAGCTCAGTTGGTAGAGCAGTTGACTTTTAATCAATTGGTCGCAGGTTCGAATCCTGCACGACCCACCA-3′) containing the 76 bases corresponding to tRNALys. Extension was performed
in a Taq DNA polymerase PCR mix incubated under the following conditions: five cycles of 3 min at 95 °C (30 s at 95 °C, 30 s at 66 °C, 2 min at 72 °C), followed by 5 min at 72 °C.
Mappingthe AriB cleavage site by specific reverse transcription and Sanger sequencing
AriB-cleaved small RNAs (750 ng) were incubated for 1 h at 25 °C in a 30 µl ligation reaction with 3 µl of 5′ Ligation Reaction Buffer (10X), 2.5 µl of 5′ Ligation Enzyme Mix and 1 µl of 5′
SR Adaptor for Illumina (denatured) from the NEBNext Multiplex Small RNA Library Prep Set (no. E7300S). A tRNALys-specific reverse transcription was then performed on 15 µl of the ligation
reaction by the addition of 1.5 µl of 10 mM dNTPs, 6 µl of 5× First-Strand Buffer, 1.5 µl of 0.1 M DTT, 1.5 µl of SuperScript III RT from Invitrogen′s SuperScript III kit (no. 18080-093) and
50 pmol of the reverse transcription primer (5′-TGTGCTCTTCCGATCT GGTGGGTCGTGCAGGATTCGAACCTG-3′) in a total volume of 30 µl, with incubation at 55 °C for 1 h. PCR was performed using the
Thermo Scientific DreamTaq Green PCR Master Mix (2×) kit with the following oligos: 5′-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3′ and
5′-CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′. A band of approximately 120 base pairs was gel purified and sent for Sanger sequencing with the primer
5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′.
RNA blotRNAs were run for 1 h at 180 V in a 7 M urea 10% acrylamide gel and then transferred to a nylon membrane (Invitrogen) using the XCell SureLock Mini-Cell system with the XCell II Blot Module
(Invitrogen). The membrane was then cross-linked for 5 min with ultraviolet radiation. Following prehybridization with 30 ml of solution containing 6× SSC (saline sodium citrate buffer),
0.5% SDS and 0.05% casein for 1 h at 45 °C, the membrane was hybridized overnight with 2 nM FAM (final concentration) (6-carboxyfluorescein) or IR (infrared dye IRDye 800 or IRDye 700)
labelled oligonucleotide probes (Supplementary Table 3) in 10 ml of prehybridization solution at 45 °C. Finally, the membrane was washed twice with 2× SSC and 0.1% SDS for 10 min at room
temperature and twice with 0.2× SSC and 0.1% SDS for 10 min at 60 °C. Images were captured using the ChemiDoc MP Imaging system (Bio-Rad).
The sequences of the E. coli tRNALys (B803) and T5 tRNALys (B806) probes, as well as their homology to E. coli and T5 tRNALys, are shown in Extended Data Fig. 11a. To confirm the specificity
of the T5 tRNALys probe (B806), a RNA blot was performed with this probe in the absence of T5 tRNALys, showing that our conclusion could not have been affected by non-specific binding of
this probe to E. coli tRNALys or another tRNA (Extended Data Fig. 11b). We can also confirm that the B803 probe used to detect E. coli tRNALys does not detect T5 tRNALys, because the signal
of this probe was lost when PARIS was activated in the presence of T5 tRNALys (Fig. 5h).
Defence system detectionDefence system detection was performed using DefenseFinder v.1.1.1 (ref. 61) and DefenseFinder model v.1.2.3 on all RefSeq complete genomes from July 2022.
Domain annotationDefence system domain annotation was done using the HHpred62 webserver with PFAM63 database v.33.1 and standard settings. For PARIS, the trigger-binding and coiled-coil domains were
annotated using the structure information and ESMfold64 structure prediction for AriAB.
AriA and AriB treesThe PARIS systems detected by DefenseFinder v.1.1.1 on RefSeq complete genomes from July 2022 were used to build phylogenetic trees. Only hits to AriA and AriB with a coverage of more than
75% were retained, to avoid pseudogenes. Hits were then clustered using Mmseqs2 (ref. 65) v.13.45111 on AriA and AriAB with an identity threshold of 80% and coverage threshold of 90%. A
single representative of each cluster was then used to build the AriA tree, and the cognate AriB was used to build the AriB tree. The AriA tree was built using the AAA15 ATPase domain only
and the AriB tree using the DUF4435 domain only. Domains were detected by HMMsearch (from HMMER 3.3.2) on the E. coli B185 PARIS system, and a first alignment using Mafft v.7.505 (ref. 66)
(default parameters) was used to extract the corresponding sequences in other PARIS variants. The final alignment used to make the tree was carried out using MUSCLE v.5.1.linux64 (ref. 67)
using the model super5, and trim using clipkit v.1.3.0 with the option seq-gap. The DUF4435 tree using M5 ribonuclease as outgroup was done using selected representative hits from different
clades and aligned using muscle v.5.1.linux64 with the model super5. All trees were built using Iqtree v.2.0.6 (refs. 68,69) using ModelFinder and Ultrafast Bootstrap 1000.
Defence systemATPase AAA15/21 tree
The ATPase AAA15/21 defence system containing detection was performed using hmmsearch from HMMER 3.3.2 and Pfam Hidden Markov Model 33.1 on defence systems previously detected. Random
selection of 20 proteins from the detection results with a coverage of more than 75% was used, in addition to experimentally validated homologues, to build the tree. ATPase alignment was
carried out using Mafft v.7.505, and the tree was built using Iqtree v.2.0.6 with ModelFinder and Ultrafast Bootstrap 1000.
Statistics and reproducibilityThe pulldown experiments presented in Fig. 3a,b,e,f,i were reproduced in two or three biological replicates. The experiments presented in Fig. 5g,i,j were reproduced three times and in Fig.
5h two times.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availabilitySequencing data have been deposited in NCBI database under BioProject ID PRJNA1040033 and GEO accession no. GSE270519. EM maps of PARIS and the atomic model for the asymmetric unit of the
complex were deposited to the Electron Microscopy Data Bank (EMDB) and Protein Databank (PDB) databases. Accession codes can be found in Extended Data Table 1. The PDB code for the
experimentally determined structure of the PARIS asymmetric unit is 8UX9. EMDB accession code nos. are EMD-42719, EMD-43103, EMD-43104 and EMD-43105. All constructs (WT and mutants) used in
this study can be obtained on request to the lead contacts. Source data are provided with this paper.
Code availabilityNo custom code was used.
Change history04 December 2024A Correction to this paper has been published: https://doi.org/10.1038/s41586-024-08427-4
References Chevallereau, A. & Westra, E. R. Bacterial immunity: mobile genetic elements are hotspots for defence systems. Curr. Biol. 32, R923–R926 (2022).
Article CAS PubMed Google Scholar
Rousset, F. et al. Phages and their satellites encode hotspots of antiviral systems. Cell Host Microbe 30, 740–753 (2022).
Article CAS PubMed PubMed Central Google Scholar
Zhang, T. et al. Direct activation of a bacterial innate immune system by a viral capsid protein. Nature 612, 132–140 (2022).
Article ADS CAS PubMed PubMed Central Google Scholar
Wein, T. & Sorek, R. Bacterial origins of human cell-autonomous innate immune mechanisms. Nat. Rev. Immunol. 22, 629–638 (2022).
Article CAS PubMed Google Scholar
Makarova, K. S., Wolf, Y. I., Snir, S. & Koonin, E. V. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 193, 6039–6056 (2011).
Article CAS PubMed PubMed Central Google Scholar
Rocha, E. P. C. & Bikard, D. Microbial defenses against mobile genetic elements and viruses: who defends whom from what? PLoS Biol. 20, e3001514 (2022).
Article CAS PubMed PubMed Central Google Scholar
Schiltz, C. J., Lee, A., Partlow, E. A., Hosford, C. J. & Chappie, J. S. Structural characterization of Class 2 OLD family nucleases supports a two-metal catalysis mechanism for cleavage.
Nucleic Acids Res. 47, 9448–9463 (2019).
Article CAS PubMed PubMed Central Google Scholar
Schiltz, C. J., Adams, M. C. & Chappie, J. S. The full-length structure of Thermus scotoductus OLD defines the ATP hydrolysis properties and catalytic mechanism of Class 1 OLD family
nucleases. Nucleic Acids Res. 48, 2762–2776 (2020).
Article CAS PubMed PubMed Central Google Scholar
Haggård-Ljungquist, E., Barreiro, V., Calendar, R., Kurnit, D. M. & Cheng, H. The P2 phage old gene: sequence, transcription and translational control. Gene 85, 25–33 (1989).
Article PubMed Google Scholar
Wilkinson, M. et al. Structural basis for the inhibition of RecBCD by Gam and its synergistic antibacterial effect with quinolones. eLife 5, e22963 (2016).
Dot, E. W., Thomason, L. C. & Chappie, J. S. Everything OLD is new again: how structural, functional, and bioinformatic advances have redefined a neglected nuclease family. Mol. Microbiol.
120, 122–140 (2023).
Article CAS PubMed Google Scholar
Cheng, R. et al. A nucleotide-sensing endonuclease from the Gabija bacterial defense system. Nucleic Acids Res. 49, 5216–5229 (2021).
Article ADS CAS PubMed PubMed Central Google Scholar
Antine, S. P. et al. Structural basis of Gabija anti-phage defence and viral immune evasion. Nature 625, 360–365 (2024).
Article ADS CAS PubMed Google Scholar
Mestre, M. R., González-Delgado, A., Gutiérrez-Rus, L. I., Martínez-Abarca, F. & Toro, N. Systematic prediction of genes functionally associated with bacterial retrons and classification of
the encoded tripartite systems. Nucleic Acids Res. 48, 12632–12647 (2020).
Article CAS PubMed PubMed Central Google Scholar
Ernits, K. et al. The structural basis of hyperpromiscuity in a core combinatorial network of type II toxin–antitoxin and related phage defense systems. Proc. Natl Acad. Sci. USA 120,
e2305393120 (2023).
Article CAS PubMed PubMed Central Google Scholar
Bryant, P., Pozzati, G. & Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 13, 1265 (2022).
Article ADS CAS PubMed PubMed Central Google Scholar
Hopfner, K. Rad50/SMC proteins and ABC transporters: unifying concepts from high-resolution structures. Curr. Opin. Struct. Biol. 13, 249–255 (2003).
Article CAS PubMed Google Scholar
Hopfner, K.-P. et al. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 101, 789–800 (2000).
Article CAS PubMed Google Scholar
Lammens, A. & Hopfner, K.-P. Structural basis for adenylate kinase activity in ABC ATPases. J. Mol. Biol. 401, 265–273 (2010).
Article CAS PubMed Google Scholar
Xie, T., Zhang, Z., Fang, Q., Du, B. & Gong, X. Structural basis of substrate recognition and translocation by human ABCA4. Nat. Commun. 12, 3853 (2021).
Article ADS CAS PubMed PubMed Central Google Scholar
Krishnan, A., Burroughs, A. M., Iyer, L. M. & Aravind, L. Comprehensive classification of ABC ATPases and their functional radiation in nucleoprotein dynamics and biological conflict
systems. Nucleic Acids Res. 48, 10045–10075 (2020).
Article CAS PubMed PubMed Central Google Scholar
Gut, F. et al. Structural mechanism of endonucleolytic processing of blocked DNA ends and hairpins by Mre11-Rad50. Mol. Cell 82, 3513–3522 (2022).
Article CAS PubMed Google Scholar
Seifert, F. U., Lammens, K., Stoehr, G., Kessler, B. & Hopfner, K. Structural mechanism of ATP‐dependent DNA binding and DNA end bridging by eukaryotic Rad50. EMBO J. 35, 759–772 (2016).
Article CAS PubMed PubMed Central Google Scholar
Higgins, C. F. & Linton, K. J. The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11, 918–926 (2004).
Article CAS PubMed Google Scholar
Walkinshaw, M. D. et al. Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol. Cell 9, 187–194 (2002).
Article CAS PubMed Google Scholar
Isaev, A. et al. Phage T7 DNA mimic protein Ocr is a potent inhibitor of BREX defence. Nucleic Acids Res. 48, 5397–5406 (2020).
Article CAS PubMed PubMed Central Google Scholar
Deep, A. et al. The SMC-family Wadjet complex protects bacteria from plasmid transformation by recognition and cleavage of closed-circular DNA. Mol. Cell 82, 4145–4159 (2022).
Article CAS PubMed PubMed Central Google Scholar
Weiß, M. et al. The MksG nuclease is the executing part of the bacterial plasmid defense system MksBEFG. Nucleic Acids Res. 51, 3288–3306 (2023).
Article PubMed PubMed Central Google Scholar
Aravind, L., Leipe, D. D. & Koonin, E. V. Toprim—a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res.
26, 4205–4213 (1998).
Article CAS PubMed PubMed Central Google Scholar
Zimmerman, S. B. Toroidal nucleoids in Escherichia coli exposed to chloramphenicol. J. Struct. Biol. 138, 199–206 (2002).
Article CAS PubMed Google Scholar
Yirmiya, E. et al. Phages overcome bacterial immunity via diverse anti-defence proteins. Nature 625, 352–359 (2023).
Article ADS PubMed Google Scholar
Wang, J. et al. Complete genome sequence of bacteriophage T5. Virology 332, 45–65 (2005).
Article ADS CAS PubMed Google Scholar
Glukhov, A. S., Krutilina, A. I., Kaliman, A. V., Shlyapnikov, M. G. & Ksenzenko, V. N. Bacteriophage T5 mutants carrying deletions in tRNA gene region. Mol. Biol. (Mosk) 52, 3–9 (2018).
Article CAS PubMed Google Scholar
Jain, I. et al. tRNA anticodon cleavage by target-activated CRISPR-Cas13a effector. Sci. Adv. 10, eadl0164 (2024).
Article CAS PubMed PubMed Central Google Scholar
Jiang, Y., Meidler, R., Amitsur, M. & Kaufmann, G. Specific interaction between anticodon nuclease and the tRNA(Lys) wobble base. J. Mol. Biol. 305, 377–388 (2001).
Article CAS PubMed Google Scholar
Azam, A. H. et al. Viruses encode tRNA and anti-retron to evade bacterial immunity. Preprint at bioRxiv https://doi.org/10.1101/2023.03.15.532788 (2023).
Mogila, I. et al. Ribosomal stalk-captured CARF-RelE ribonuclease inhibits translation following CRISPR signaling. Science 382, 1036–1041 (2023).
Article ADS CAS PubMed Google Scholar
Bingham, R., Ekunwe, S. I. N., Falk, S., Snyder, L. & Kleanthous, C. The major head protein of bacteriophage T4 binds specifically to elongation factor Tu*. J. Biol. Chem. 275, 23219–23226
(2000).
Article CAS PubMed Google Scholar
Yang, J. Y. et al. Degradation of host translational machinery drives tRNA acquisition in viruses. Cell Syst. 12, 771–779 (2021).
Article CAS PubMed PubMed Central Google Scholar
van den Berg, D. F., van der Steen, B. A., Costa, A. R. & Brouns, S. J. Phage tRNAs evade tRNA-targeting host defenses through anticodon loop mutations. eLife 12, e85183 (2023).
Article PubMed PubMed Central Google Scholar
Albers, S. & Czech, A. Exploiting tRNAs to boost virulence. Life (Basel) 6, 4 (2016).
ADS PubMed Google Scholar
Zhang, F. et al. Human SAMD9 is a poxvirus-activatable anticodon nuclease inhibiting codon-specific protein synthesis. Sci. Adv. 9, eadh8502 (2023).
Article CAS PubMed PubMed Central Google Scholar
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Article CAS PubMed Google Scholar
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Article PubMed Google Scholar
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Article CAS PubMed Google Scholar
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Article CAS PubMed Google Scholar
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Article PubMed PubMed Central Google Scholar
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 74, 519–530 (2018).
Article ADS CAS PubMed PubMed Central Google Scholar
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Article ADS CAS PubMed PubMed Central Google Scholar
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Struct. Biol. 66, 486–501 (2010).
Article ADS CAS Google Scholar
Rohwer, F. & Azam, F. Detection of DNA damage in prokaryotes by terminal deoxyribonucleotide transferase-mediated dUTP nick end labeling. Appl. Environ. Microbiol. 66, 1001–1006 (2000).
Article ADS CAS PubMed PubMed Central Google Scholar
FlowJoTM Software v.10. (Becton, Dickinson and Company, 2023).
Cockram, C., Thierry, A. & Koszul, R. Generation of gene-level resolution chromosome contact maps in bacteria and archaea. STAR Protoc. 2, 100512 (2021).
Article CAS PubMed PubMed Central Google Scholar
Cournac, A., Marie-Nelly, H., Marbouty, M., Koszul, R. & Mozziconacci, J. Normalization of a chromosomal contact map. BMC Genomics 13, 436 (2012).
Article CAS PubMed PubMed Central Google Scholar
Imakaev, M. et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012).
Article CAS PubMed PubMed Central Google Scholar
Lioy, V. S. et al. Multiscale structuring of the E. coli chromosome by nucleoid-associated and condensin proteins. Cell 172, 771–783 (2018).
Article CAS PubMed Google Scholar
Kurata, T. et al. A hyperpromiscuous antitoxin protein domain for the neutralization of diverse toxin domains. Proc. Natl Acad. Sci. USA 119, e2102212119 (2022).
Article CAS PubMed PubMed Central Google Scholar
Sambrook, J. Molecular Cloning: a Laboratory Manual (Cold Spring Harbor Laboratory Press, 2001).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017).
Article ADS PubMed PubMed Central Google Scholar
Avcilar-Kucukgoze, I., Gamper, H., Hou, Y.-M. & Kashina, A. Purification and use of tRNA for enzymatic post-translational addition of amino acids to proteins. STAR Protoc. 1, 100207 (2020).
Article PubMed PubMed Central Google Scholar
Tesson, F. et al. Systematic and quantitative view of the antiviral arsenal of prokaryotes. Nat. Commun. 13, 2561 (2022).
Article ADS CAS PubMed PubMed Central Google Scholar
Söding, J., Biegert, A. & Lupas, A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005).
Article PubMed PubMed Central Google Scholar
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Article CAS PubMed Google Scholar
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Article ADS MathSciNet CAS PubMed Google Scholar
Mirdita, M., Steinegger, M. & Söding, J. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 35, 2856–2858 (2019).
Article CAS PubMed PubMed Central Google Scholar
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Article CAS PubMed PubMed Central Google Scholar
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Article CAS PubMed PubMed Central Google Scholar
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274
(2015).
Article CAS PubMed Google Scholar
Wiegand, T. et al. Functional and phylogenetic diversity of Cas10 proteins. CRISPR J. 6, 152–162 (2023).
Article CAS PubMed PubMed Central Google Scholar
Download references
AcknowledgementsWe thank members of the B.W. laboratory for feedback and discussions, C. Hophan-Nichols and the cyber security team at Montana State Univeristy for computational support, H. Callaway for
helpful suggestions with data processing and M. Lawrence and C. Gauvin for maintenance and operation of the cryo-EM Core Facility at Montana State University. The cryo-EM Core Facility at
Montana State University is supported by NSF no. 1828765 and the M. J. Murdock Charitable Trust. Research in the Wiedenheft laboratory is supported by the National Institutes of Health (no.
R35GM134867), the M. J. Murdock Charitable Trust, a young investigator award from Amgen and the Montana State University Agricultural Experimental Station (USDA NIFA). N.B. is supported by
an F31 from National Institutes of Health (NIH) (no. GM153146) and received support from Montana INBRE (no. P20GM103474). A.S.-F. is supported by NIH (no. K99GM147842) and the Burroughs
Wellcome Fund (no. G-1021106.01). A.B.G. is supported by Montana State University’s Undergraduate Scholars Program, and by the NIH NIGMS IDeA programme (no. P20GM103474). J.G., A.T. and R.K.
are supported by the European Research Council under the Horizon 2020 Programme (no. 771813). A.I. was supported by RSF grants (nos. 22-14-00004 and 24-74-10089). K.S. was supported by RSF
grant 24-14-140018. A.L. was supported by a Ministry of Science and Higher Education grant (no. 075-15-2019-1661). L.S., T.K. and V.H. are supported by the Swedish Research Council
(Vetenskapsrådet, no. 2021-01146). We thank A. Glukhov for sharing a collection of T5 deletion variants, T. Prince Maviza for help with in vitro translation assays and A. Demkina for help
with BGI sequencing. D.B., F.D., C.R. and B.S. are supported by the European Research Council (no. 101044479) and Agence Nationale de la Recherche (no. ANR-10-LABX-62-IBEID). A.B. and F.T.
are supported by a European Research Council Starting Grant (no. PECAN 101040529), MSD avenir (UNaDISC project) and the core funding of Institut Pasteur. Molecular graphics and analyses were
performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization and Informatics at the University of California, San Francisco, with support from NIH no.
R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. The funders had no role in the conceptualization,
designing, data collection, analysis, decision to publish or preparation of the manuscript.
Author informationAuthor notesThese authors contributed equally: Nathaniel Burman, Svetlana Belukhina, Florence Depardieu
Authors and Affiliations Department of Microbiology and Cell Biology, Montana State University, Bozeman, MT, USA
Nathaniel Burman, Royce A. Wilkinson, Andrew Santiago-Frangos, Ava B. Graham, Trevor Zahl, William S. Henriques, Murat Buyukyoruk & Blake Wiedenheft
The Center for Molecular and Cellular Biology, Moscow, Russia
Svetlana Belukhina, Mikhail Skutel, Anna Chechenina & Artem Isaev
Synthetic Biology, Institut Pasteur, Université Paris Cité, CNRS UMR 6047, Paris, France
Florence Depardieu, Christophe Rouillon, Baptiste Saudemont & David Bikard
Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology, Russian Academy of Sciences, Moscow, Russia
Alexei Livenskyi & Konstantin Severinov
Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow, Russia
Alexei Livenskyi
Peter the Great St Petersburg State Polytechnic University, St. Petersburg, Russia
Natalia Morozova
Department of Experimental Medical Science, Lund University, Lund, Sweden
Lena Shyrokova, Tatsuaki Kurata & Vasili Hauryliuk
Virus Centre, Lund University, Lund, Sweden
Vasili Hauryliuk
Science for Life Laboratory, Lund, Sweden
Vasili Hauryliuk
Waksman Institute, Rutgers, The State University of New Jersey, Piscataway, NJ, USA
Konstantin Severinov
Unité Régulation Spatiale des Génomes, Institut Pasteur, CNRS UMR 3525, Université Paris Cité, Paris, France
Justine Groseille, Agnès Thierry & Romain Koszul
Center for Interdisciplinary Research in Biology (CIRB), Collège de France, CNRS, INSERM, Université PSL, Paris, France
Justine Groseille
Collège Doctoral, Sorbonne Université, Paris, France
Justine Groseille
Molecular Diversity of Microbes, Institut Pasteur, CNRS UMR3525, Université Paris Cité, Paris, France
Florian Tesson & Aude Bernheim
AuthorsNathaniel BurmanView author publications You can also search for this author inPubMed Google Scholar
Svetlana BelukhinaView author publications You can also search for this author inPubMed Google Scholar
Florence DepardieuView author publications You can also search for this author inPubMed Google Scholar
Royce A. WilkinsonView author publications You can also search for this author inPubMed Google Scholar
Mikhail SkutelView author publications You can also search for this author inPubMed Google Scholar
Andrew Santiago-FrangosView author publications You can also search for this author inPubMed Google Scholar
Ava B. GrahamView author publications You can also search for this author inPubMed Google Scholar
Alexei LivenskyiView author publications You can also search for this author inPubMed Google Scholar
Anna ChecheninaView author publications You can also search for this author inPubMed Google Scholar
Natalia MorozovaView author publications You can also search for this author inPubMed Google Scholar
Trevor ZahlView author publications You can also search for this author inPubMed Google Scholar
William S. HenriquesView author publications You can also search for this author inPubMed Google Scholar
Murat BuyukyorukView author publications You can also search for this author inPubMed Google Scholar
Christophe RouillonView author publications You can also search for this author inPubMed Google Scholar
Baptiste SaudemontView author publications You can also search for this author inPubMed Google Scholar
Lena ShyrokovaView author publications You can also search for this author inPubMed Google Scholar
Tatsuaki KurataView author publications You can also search for this author inPubMed Google Scholar
Vasili HauryliukView author publications You can also search for this author inPubMed Google Scholar
Konstantin SeverinovView author publications You can also search for this author inPubMed Google Scholar
Justine GroseilleView author publications You can also search for this author inPubMed Google Scholar
Agnès ThierryView author publications You can also search for this author inPubMed Google Scholar
Romain KoszulView author publications You can also search for this author inPubMed Google Scholar
Florian TessonView author publications You can also search for this author inPubMed Google Scholar
Aude BernheimView author publications You can also search for this author inPubMed Google Scholar
David BikardView author publications You can also search for this author inPubMed Google Scholar
Blake WiedenheftView author publications You can also search for this author inPubMed Google Scholar
Artem IsaevView author publications You can also search for this author inPubMed Google Scholar
ContributionsThe biochemistry and structural data presented in Fig. 1 were generated by R.A.W., N.B., W.S.H. and A.B.G. The biochemistry and structural data presented in Fig. 2 were generated by R.A.W.,
N.B., W.S.H. and A.B.G. The plaque presented assays in Fig. 2 were performed by F.D. and S.B. The biochemistry data presented in Fig. 3a,b,e,f,h,i were generated by A.I., S.B., M.S. and A.C.
The biochemistry data presented in Fig. 3g were generated by T.Z. The plaque assays presented in Fig. 3,d,j were performed by F.D. The structural data presented in Fig. 3c were generated by
A.S.-F., R.A.W., N.B., W.S.H. and A.B.G. The efficiency of plating assay presented in Fig. 4a was performed by S.B. and F.D. The light microscopy presented in Fig. 4b was performed by N.M.
The metabolic labelling presented in Fig. 4c was performed by L.S., T.K. and V.H. In vitro translation presented in Fig. 4d was performed by S.B., M.S. and A.L. Plaque assays, efficiency of
plating and phage challenge assays presented in Fig. 5a–f were performed by A.I., S.B., M.S., F.D. and C.R. The data presented in Fig. 5g–k were performed by F.D. and C.R. Small RNA
extractions used in AriB cleavage assays (Fig 5i) and Sanger sequencing of the cleavage product (Extended Data Fig. 11) were performed by B.S. The evolutionary analysis presented in Fig. 6
was performed by A.B., F.T. and M.B. The Hi-C analysis presented in Extended Data Fig. 9 was performed by R.K., J.G. and A.T. Project conceptualization, administration and the original draft
were undertaken by B.W., A.I. and D.B., with review and editing by all authors.
Corresponding authors Correspondence to David Bikard, Blake Wiedenheft or Artem Isaev.
Ethics declarations Competing interestsThe authors declare no competing interests.
Peer review Peer review informationNature thanks Jinwei Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional informationPublisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tablesExtended Data Fig.1 AlphaFold2 structural predictions of AriA and AriB.
AlphaFold2-predicted structures of AriA and AriB oligomers colored by the predicted local distance difference test (pLDDT) score (0–100) with associated scale bars. Values greater than 90
indicate high confidence and values below 50 indicate low confidence. Predicted Aligned Error plots (PAE) are also shown for each structural prediction. a, AlphaFold2-predicted structure of
the AriA homodimer. Confidence is high (PLDDT >90, green) for the ATPase domain, and low for the insertion sequences (PLDDT<70, teal). b, AlphaFold2-predicted structure of the AriB homodimer
results in a high confidence model with more >98% of the residues above a PLDDT of 90. c, AlphaFold2-predicted structure of the AriA-B heterodimer and associated PAE plot. d, AlphaFold2
returned unreliable models when attempting to predict structures for higher ordered assemblies of AriA and AriB as demonstrated by the PLDDT plot below 50 for most of the molecule. e,
Clashing (red) between AriBs, prevents the assembly of two AriB subunits on a single AriA homodimer. Clashing is defined by atoms with overlap greater than 0.6 Å. f, A predicted homodimer of
AriB cannot associate with a homodimer of AriA without clashing (red). Atoms with an overlap greater than 0.6 Å are highlighted with red disks signifying the clash. g, Predicted structure
of the AriB dimer shown in panel b, highlighting residues involved in dimerization.
Extended Data Fig. 2 Structural comparison of PARIS homologs.a, Domain organization of phage defense systems where an ABC ATPase domain is associated with a TOPRIM nuclease domain. While the ATPase domain is highly conserved, the relative orientation
of the TOPRIM nuclease domains and dimerization domains vary between PARIS (PDB ID: 8UX9), Gabija (PDB ID: 8SM3), and the Thermus scotoductus Overcoming Lysogenization Defect (Ts OLD, PDB
ID: 6P74). b, AriB is the effector of PARIS defense. The AriB TOPRIM is homologous to the TOPRIM domains of Ts OLD (RMSD 1.1 Å, 20 C-alpha pairs) and GajA (RMSD 1.02 Å, 26 C-alpha pairs).
The TOPRIM domain of AriB is within the DUF4435, which plays a role in AriB dimerization. c, Structural comparison of AriA to Rad50, a universally conserved ATPase. AriA primarily differs
from Rad50 at Insertion Sequence (IS) 1 (residues 122-180) and IS2 (residues 242-289). IS1 introduces three alpha helices near the nucleotide binding domains of AriA that are predicted to be
involved in trigger recognition. IS2 expands the coiled coil domain with two alpha helices and enable the homohexameric assembly of AriA subunits in the PARIS complex. d, Structural
comparison of PARIS and related OLD systems highlights the unique assembly state of PARIS.
Extended Data Fig. 3 Workflow for structural determination of the PARIS complex.Image processing was performed using cryoSPARC v4.41. a, 7,340 movies were recorded. Micrographs with CTF-fits worse than 8 Å were removed prior to downstream processing (n = 5,988). Using a
de novo template generated from a 200-micrograph subset of this data, 4,078,384 particles were identified and extracted. From 17 selected classes, 1,643,515 particles remained, and a
3-class ab initio reconstruction yielded volumes shown in panel b. Particles belonging to junk classes (pink and red) were discarded. c, 25 representative 2-D Classes from 940,782 particles
in panel b. A total of 934,763 particles from 72 classes remained after final 2-D Classification (n = 100 classes). d, After successive rounds of ab initio and heterogenous refinements, a
532,010 particle volume was obtained, corresponding to the intact PARIS complex with compositional heterogeneity at two AriB attachment sites. Masks were generated for local refinement and
3-D classification to produce the reconstructions shown in e, g, and h, respectively. e, Local Refinement of one asymmetric unit of the PARIS immune complex with FSC and 3-D orientation plot
shown below. f, Close up of density map and model from E demonstrating map quality. g, C3 reconstruction of the PARIS complex at 3.71 Å-resolution with the experimental structure determined
in panel e fit into the density. h, Cryo-EM reconstruction of PARIS with AriB in the ‘trans’ arrangement at approximately 3.93 Å-resolution with experimental structure determined in panel
e, fit into the density.
Extended Data Fig. 4 AriB toxicity is dependent on association with AriA.a, Liquid culture toxicity assays of AriB alone or AriA:B together (PARIS). Genes are overexpressed using a T7 inducible promoter. AriB alone results in a mild growth defect, while
Ocr-triggered release of AriB (PARIS Ocr) is significantly more toxic. b, Growth of E. coli MG1655 carrying plasmid pFR85 (pSC101 with ariAB under the control of the native promoter), or
variants with ariA deleted, ariB deleted or a stop codon in ariA yielding an interrupted protein that lacks the interaction interface with AriB. c, Structure-guided mutations that disrupt
the AriA:AriB interface result in a loss of defense phenotype. d, Drop-test toxicity assay with PARIS (WT) or PARIS with AriA D401N/E404Q mutation. A modest growth defect can be observed
only for the PARIS AriA D401N/E404Q overexpressed from T7 promoter. e, The addition of pBAD Ocr to the PARIS AriA mutants does not increase the toxicity, indicating that AriB is not
functional when AriA interactions are disrupted. f, Close-up of the AriA:AriB interaction interface. Mutated residues are labeled with asterisks.
Source Data
Extended Data Fig. 5 Purification of activated AriB.a, Size exclusion chromatography calibration curve with estimated masses of AriA:Ocr and activated AriB complexes. b, Introduction of the Strep-tag at the C- or N- terminus of AriA, but not
on the C-terminus of AriB, interferes with PARIS anti-phage defense. c, Purification of activated AriB after mixing cell lysates containing strep-tagged AriB and non-tagged Ocr.
ExtendedData Fig. 6 Structure determination of AriA purified using a Ocr-Strep pulldown.
Image processing and analysis was performed in cryoSPARC v4.41. a, Representative micrograph from a total of 10,340 movies. Micrographs with CTF-fits worse than 8 Å-resolution were
discarded. Particle picking on 9,399 micrographs identified 4,415,782 particles which were extracted and subjected to 2-D Classification. b, Selected 2-D classes corresponding to the AriA
hexamer contained 667,479 particles. c, 2-Class ab initio reconstruction of particles from panel b. The gray class corresponds to the AriA homohexamer, while particles associated with the
orange class were discarded. Density for the ATPase domains is present, but one blade of the propeller is poorly resolved due to the increased flexibility of the AriA scaffold relative to
the fully assembled PARIS complex. d, After iterative rounds of sorting the 392,989 particles from panel C, a stack containing 62,732 particles was isolated and C3 refinement produced a 4.4
Å-resolution reconstruction of the AriA scaffold.
Extended Data Fig. 7 AriA ATPase activity.a, PARIS mediated hydrolysis of α32P ATP with PARIS alone or with 10-fold excess trigger (T7 Ocr). Reactions were run from 1 to 32 min and products were resolved via thin layer
chromatography. Reactions with α32P ATP incubated with T4 polynucleotide kinase were used as a positive control. Images are representative of reactions that were performed in triplicate. b,
Ocr alone was incubated with α32P ATP for 32 min indicating that the trigger alone does not hydrolyze ATP. c, Concentration of ADP formed at each time point was quantified and plotted. Data
points of three technical replicates are shown.
Extended Data Fig. 8 PARIS activation does not result in total RNA or DNA degradation.a, Total DNA was extracted from E. coli MG1655 carrying plasmids pFR85 (PARIS) and pFD250 (PPhlF-Ocr) at different time points after Ocr induction with or without DAPG (n = 3), or from a
non-induced control. Samples were run on a 1% agarose TAE gel. b, Total RNA was extracted from E. coli MG1655 carrying plasmid pFR85 (PARIS) or pFR66 (sfGFP), and pFD250 (PPhlF-Ocr) at
different time points after induction of Ocr expression with DAPG. Samples were run on a TBE Urea (7M) acrylamide (10%) gel and stained with SYBR-Gold. A representation of 3 replicates is
shown. c, TUNEL assay, demonstrating the lack of accumulation of dsDNA breaks in PARIS+ cells 2 h after Ocr induction, as measured through terminal deoxynucleotidyl transferase labeling of
free 3′-OH groups in DNA. As a positive control of DNA damage, cells were treated with 0.1% H2O2.
Extended Data Fig. 9 PARIS activation results in DNA compactization akin to inhibition oftranslation with chloramphenicol.
a, Hi-C contact maps (bin: 4kb). From left to right: WT (pFR66 + pFD245 control vectors), PARIS (pFR85) + PPhlF-ocr (pFD250), and chloramphenicol treated cells. Top and bottom rows
correspond to 15 and 30 min after induction with DAPG or treatment with chloramphenicol. b, Ratio between contact maps of WT at t = 30 min and contact maps 30 min after either PARIS
activation (left) or chloramphenicol treatment (right).
Extended Data Fig. 10 Identification of the T5 genomic region required for infection of PARIS+ cells.a, Growth of PARIS- or PARIS+ cells infected with PARIS-sensitive phage T5Mos at low or high MOI. The phage was added at t = 0. b, Phage T5 variants with non-essential deletions in the
region encoding tRNAs are sensitive to PARIS defense. Boundaries of the deletions are shown on the right, tRNA genes are highlighted in red. c, Overexpression of Fragment 1 (31885–32870)
derived from the deletion in the phage T5123 partially rescues T5123 infection of the PARIS+ culture. d, Comparison of E. coli and T5 tRNALys highlights substitution mutations near the
anticodon stem loop that we hypothesize prevents targeting by AriB. e, Swapping U-A for A-U (Mut1) on the E. coli tRNALys confers resistance to PARIS.
Extended Data Fig. 11 tRNALyscleavage product and phylogenetics of PARIS.
a, Alignment of the B803 and B806 Northern Blot probes to the E. coli and T5 tRNALys. b, Northern Blot with probe B806 in the presence or absence of the T5 tRNALys expressed from a pBAD. c,
Mapping of the tRNALys cleavage site by reverse-transcription, adapter ligation, PCR and Sanger sequencing. The cleavage site, identified as the junction between the tRNA sequence and the
ligated adapter, is marked with a vertical red line. d, Alignment of homologs of AriA representative of the different PARIS clades. Different domains of AriA are indicated. Grey scale
represents % of identity between homologs. e, Phylogenetic tree of DUF4435 domain of AriB using M5 Ribonuclease (TOPRIM) as an outgroup.
Extended Data Table 1 Cryo-EM data collection,processing, and model validationFull size tableSupplementary informationSupplementary Information
This file contains Supplementary Tables 1–3 and legends for Supplementary Videos 1 and 2.
Reporting SummarySupplementary Fig. 1Uncropped images.
Peer Review fileSupplemental Video 1Visualization of PARIS. Masked local refinement was used to generate a high-resolution map of one asymmetric unit (AriA2–AriB1). Density for ATPγS is evident in both NBDs of the AriA
homodimer. A series of electrostatic interactions link AriB to AriA. AriB is located directly above the NDBs of AriA. Catalytic residues in the TOPRIM domain are conserved. Focused
three-dimensional classification was used to isolate two isomers of the assembled complex, a cis arrangement and a trans arrangement. Viral triggers of PARIS, such as the T7 Ocr protein,
release AriB from the complex, which forms a homodimeric nuclease that cleaves host tRNALys.
Supplemental Video 2Ocr induces membrane permeability in cells expressing the PARIS immune system. Time-course video demonstrating PARIS-induced membrane permeability by propidium iodide staining. Cells turn
red within 30 min of Ocr induction.
Source dataSource Data Fig. 4Source Data Fig. 6Source Data Extended Data Fig. 4Rights and permissionsOpen Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution
and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you
modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in
this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Reprints and permissions
About this articleCite this article Burman, N., Belukhina, S., Depardieu, F. et al. A virally encoded tRNA neutralizes the PARIS antiviral defence system. Nature 634, 424–431 (2024).
https://doi.org/10.1038/s41586-024-07874-3
Download citation
Received: 22 December 2023
Accepted: 24 July 2024
Published: 07 August 2024
Issue Date: 10 October 2024
DOI: https://doi.org/10.1038/s41586-024-07874-3
Share this article Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article.
Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative






