- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Hyperactivation of the complement and coagulation systems is recognized as part of the clinical syndrome of COVID-19. Here we review systemic complement activation and local
complement activation in response to the causative virus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and their currently known relationships to hyperinflammation and
thrombosis. We also provide an update on early clinical findings and emerging clinical trial evidence that suggest potential therapeutic benefit of complement inhibition in severe COVID-19.
SIMILAR CONTENT BEING VIEWED BY OTHERS ASSOCIATION OF COVID-19 INFLAMMATION WITH ACTIVATION OF THE C5A–C5AR1 AXIS Article 29 July 2020 IMMUNITY, ENDOTHELIAL INJURY AND COMPLEMENT-INDUCED
COAGULOPATHY IN COVID-19 Article 19 October 2020 THE SIGNAL PATHWAYS AND TREATMENT OF CYTOKINE STORM IN COVID-19 Article Open access 07 July 2021 INTRODUCTION COVID-19, caused by severe
acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is the largest pandemic disease of humans in the past century. Thus far, approximately 225 million cases and more than 4.5 million
deaths have been attributed to COVID-19 globally1. The clinical manifestations of COVID-19 can be highly variable, ranging from mild upper respiratory tract symptoms in most cases to severe,
life-threatening, multi-organ disease2. Up to one-third of patients affected by COVID-19 will develop persistent symptoms3, and some will have permanent end-organ dysfunction pursuant to
tissue fibrosis4. Despite the emerging epidemiological data, the underlying pathogenesis of COVID-19 and its optimal treatment are still poorly understood. Undoubtedly, detailed mechanistic
understanding of this disease will enable more effective therapies for its treatment, save lives and preserve healthy tissues. The complement system is an ancient, evolutionarily conserved
and non-redundant component of immunity. It is classically viewed as a liver-derived and plasma-operative system constantly scanning the blood and interstitial fluids for invading pathogens
and self-derived noxious antigens. Pathogen sensing triggers one or several complement activation pathways (the lectin pathway, the classical pathway or the alternative pathway) and
cleavage-mediated activation of the key components C3 and C5 by C3 and C5 convertases, respectively, into bioactive C3a and C3b and C5a and C5b. C3a and C5a are anaphylatoxins that mediate
the general inflammatory reaction, C3b is a major opsonin that induces tagging and phagocytic uptake of pathogens and C5b seeds the formation of the membrane attack complex (MAC; C5b–9),
which directly lyses pathogens5. Owing to its central role in the detection and removal of pathogens, complement should intuitively be protective during SARS-CoV-2 infection. However,
several lines of evidence implicate this system as a key component in the pathogenesis of COVID-19, and emerging trial data suggest clinical benefits from targeting this system
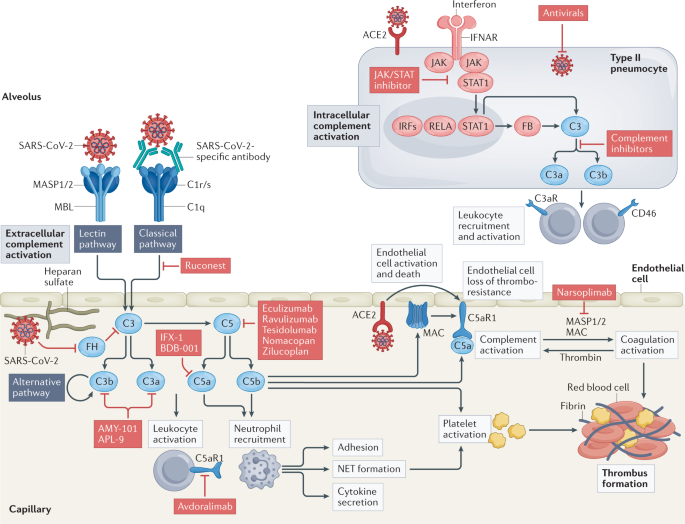
therapeutically (Fig. 1). Thus, complement, like many components of the immune system, could be viewed as a ‘double-edged sword’, with its dysregulation leading to harmful effects. Of
course, how and why the normally protective complement system becomes pathogenic in COVID-19 is not yet known, but one could speculate that, consistent with the Goldilocks principle, some
complement activation is beneficial in the initial response to the virus, but too much, sustained for too long, propagates disease. Through parallels with other coronaviruses, through
systems biology approaches and through detailed pathological and clinical observations, researchers and clinicians now realize the importance of complement in pathophysiology and see the
potential therapeutic benefit of complement inhibition6,7,8,9. Here we review the emerging roles of complement in COVID-19-associated thromboinflammation, propose a key contribution from
lung intracellular complement as well as systemic complement activation, and finally provide an update on eagerly awaited clinical trial results. COMPLEMENT IN SEVERE COVID-19 Multiple lines
of evidence implicate hyperactivation of the complement system in the pathophysiology of COVID-19. Mechanistically, SARS-CoV-2 itself can activate the complement system either directly
through the lectin pathway, the classical pathway and/or the alternative pathway or cause endotheliopathy (endothelial cell injury and dysfunction) and thromboinflammation (inflammation
associated with coagulation and thrombosis), which in turn activate the complement system (see later and Fig. 1). Specifically, the spike and nucleocapsid proteins of SARS-CoV-2 can be
directly recognized by lectin pathway components, leading to complement activation10, and IgG and IgM antibodies directed against the receptor-binding domain of the spike protein initiate
classical pathway activation11. Furthermore, SARS-CoV-2 spike protein may directly dysregulate the alternative pathway of complement activation by binding heparan sulfate and competing with
factor H, which is a negative regulator of complement activity12,13,14. These observations are consistent with the long-known observation that complement activation is a frequent feature of
the pathophysiology of acute respiratory distress syndrome, induced by infection or other triggers15,16,17,18. For example, increased anaphylatoxin concentrations are found in serum from
patients with influenza virus H1N1 infection of the lungs19. Similarly, patients with severe COVID-19 have high circulating levels of C5a and of the terminal activation fragments, soluble
C5b–9, which correlate with clinical severity11,20,21,22, as well as high levels of processed fragments of C3, itself an independent risk factor for death23. Likewise, data from the UK
Biobank support genetic single-nucleotide variants in genes encoding the complement regulators decay accelerating factor (DAF; also known as CD55), complement factor H and complement
component 4-binding protein-α (C4BPα) as risk factors for morbidity and death from SARS-CoV-2 infection24. Significant microvascular deposition of mannan-binding lectin serine protease 2
(MASP2; the primary enzymatic initiator of the lectin pathway of complement activation), C4d and C5b–9 has been reported in the skin and lungs of patients, the last two complement components
co-localizing with SARS-CoV-2 spike protein25,26. Reports of children with vasculitic lesions and hypercoagulability in the context of COVID-19 (refs27,28) (the so-called multisystem
inflammatory syndrome in children (MIS-C)) also suggest complement involvement through co-triggering of the complement and coagulation cascades. A key study has shown that elevated plasma
levels of C5b–9 are common in MIS-C29, suggesting systemic complement activation in COVID-19. Characteristic endothelial cytomorphological changes observed in patients30,31,32 are also
pathognomonic of complement-mediated injury induced by C5b–9 (ref.33). Finally, early clinical findings and emerging trial evidence also indicate potential benefits of complement-targeting
therapies in COVID-19, as reviewed later. The findings of studies of patients align with findings in animals responding to related pandemic betacoronaviruses. For example, Middle East
respiratory syndrome coronavirus infection in mice causes extensive complement deposition in lung tissues, where the damage can be reduced by inhibition of distal complement components34. In
the same model, C5aR blockade limits the thickening of alveolar septa and prevents interstitial oedema and haemorrhage in the lungs34. Similarly, the complement components _C3_, _C1r_ and
_Cfb_ are all part of a pathogenic pulmonary gene signature associated with lethality in mouse models of SARS-CoV infection35, and deletion of _C3_ in these mice protects against disease36.
Indeed, primary infection with SARS-CoV upregulates complement genes in ferrets, including the lectin pathway components MASP1 and ficolin 1 (ref.37). SARS-CoV itself interacts with
mannose-binding lectin (MBL) to activate complement C3 proximally via the lectin pathway on virus-infected cells38 in a similar manner to SARS-CoV-2, the nucleocapsid protein of which
aggravates lung injury through MASP2-mediated complement hyperactivation10. The lectin pathway contributes to the excessive inflammatory response associated with other pandemic viral
infections, as MBL-deficient mice develop less severe disease when infected with influenza A virus strain H1N1 or strain H9N2/G1 compared with wild-type mice39. Although the detrimental
contributions of complement to COVID-19 are now recognized, the underlying molecular mechanisms driving pathology are less clear. Recent data indicate that the long-acknowledged but still
poorly understood functional connection between systemic complement and the coagulation cascade and also the intrinsic production of complement by lung cells are involved and may thus be
promising novel therapeutic targets. THROMBOINFLAMMATION IN COVID-19 SARS-CoV-2 infection of the respiratory epithelium triggers an inflammatory response driven by the host innate immune
system that in 20–30% of hospitalized patients results in endothelial injury and the activation of coagulation and thrombosis40, a phenomenon called ‘thromboinflammation’41. These events
start in the lung, but may extend to several other organs, and can clinically manifest themselves as renal, hepatic and heart dysfunction and eventually multi-organ failure42,43. Thrombosis
(venous thrombosis, pulmonary embolism and arterial thrombosis) is indeed a major complication of COVID-19. Despite routine thromboprophylaxis, almost 10% of patients hospitalized with
COVID-19 have thrombotic complications44, and clotting-associated complications are associated with death45,46,47,48,49,50. Indeed, pulmonary artery thrombosis and microangiopathy in
pulmonary tissue were observed in up to 79% of patients who died with COVID-19 as the primary cause and were associated with the presence of diffuse alveolar damage51, which supports the
role of thrombotic phenomena in the development of COVID-19-related lung damage. Patients with COVID-19-associated coagulopathy have highly elevated levels of plasma D-dimer52, a fibrin
degradation product that is present in the blood after activation of fibrinolysis. In a retrospective study of patients from Wuhan, China, D-dimer levels greater than 1 μg ml−1 at admission
to hospital predicted an 18-fold increase in the odds of dying following SARS-CoV-2 infection2. Endothelial cell injury and dysfunction (also termed ‘endotheliopathy’) play a central role in
COVID-19-associated thrombosis and coagulopathy. Endothelial cells and microvascular pericytes express entry receptors for SARS-CoV-2 (refs53,54,55), and endothelial tropism of SARS-CoV-2
has been suggested by autopsy studies and single-cell RNA sequencing atlases53,56. Here, SARS-CoV-2 binding to its host entry receptor, angiotensin-converting enzyme 2 (ACE2), blocks
ACE2-mediated production of angiotensin (1–7), thus preventing vasorelaxation and antithrombotic and anti-inflammatory effects on endothelial cells and enhancing the harmful effects of
angiotensin II, exacerbating endothelial cell activation and injury57. However, the evidence for direct SARS-CoV-2 infection of endothelial cells remains a matter of debate58,59,60,61,62.
Nevertheless, the levels of markers of endothelial cell activation and injury, including von Willebrand factor (vWF), angiopoietin 2, soluble P-selectin and soluble thrombomodulin, are
elevated in the circulation of patients with severe COVID-19 in intensive care units63,64,65; and vWF and thrombomodulin levels correlate with mortality63,66,67. Extensive endothelialitis
with inflammatory cells adhering to the endothelium and platelet-rich fibrin thrombi have been reported in large and small arterial vessels in the lungs, heart, liver, small bowel, kidneys
and other organs of patients who have succumbed to COVID-19 (refs6,30,43). Furthermore, diffuse microvascular thrombosis with endothelial cell injury and rarefaction have been observed most
often in pulmonary alveolar capillaries53, as well as in kidney glomerular and peritubular capillaries6,68,69 and in myocardial capillaries43, accompanied by deposits of complement
activation products within damaged microvasculature6,43,69. Histological examination of lungs, hearts and kidneys from patients with severe COVID-19 shows fibrin and intense C5b–9
immunostaining in the glomeruli and cardiac microthrombi, respectively6,43,69. The pattern of microvascular lesions and C5b–9 deposits, along with elevated levels of lactate dehydrogenase
and low levels of haptoglobin and schistocytes in peripheral blood, that have all been observed both in adults and children with severe COVID-19 (refs51,53,70), are similar to
complement-mediated thrombotic microangiopathy (also known as atypical haemolytic uraemic syndrome), a prototypic disease of complement C5-mediated endothelial injury71,72. The report of two
cases of atypical haemolytic uraemic syndrome relapse triggered by COVID-19, one of which was in a patient with a heterozygous missense variant in the _C3_ gene (an R139W gain-of-function
point mutation resulting in formation of a hyperactive C3 convertase), and cured by the anti-C5 antibody eculizumab73,74, indirectly supports a pathogenetic role of the complement terminal
pathway in COVID-19-associated thromboinflammation. The terminal complement components C5a and the MAC can promote endothelial injury and dysfunction through multiple processes75. C5a
recruits and activates neutrophils, monocytes and macrophages76. Furthermore, both C5a and the MAC induce chemokine release and upregulation of adhesion molecules on endothelial cells,
events that support transmigration of neutrophils and macrophages75,77,78. C5a and the MAC also promote platelet adhesion by stimulating the exocytosis of P-selectin and vWF multimers from
endothelial cells, tissue factor expression and shedding of thrombomodulin from cell surfaces76,79,80, which trigger the coagulation cascade. Activated platelets and C3 subsequently activate
the recruited neutrophils to release neutrophil extracellular traps (NETs), generating extracellular DNA and histones that are highly toxic to endothelial cells and accumulate in
microthrombi of patients with COVID-19 (refs8,81). The release of NETs (NETosis) is a unique form of neutrophil cell death that is intimately linked with the complement and coagulation
cascades82. C5a receptor 1 (C5aR1) and C5aR2 activation generates extracellular histones, which are major components of NETs and activate the intrinsic coagulation pathway83,84,85. NETs act
as a scaffold for thrombus formation by inducing platelet aggregation, while platelets in turn induce NET formation86,87,88. The cumulative results of these events are vascular injury and
loss of endothelial antithrombogenic properties, with massive formation of blood clots. Consistent with this, exposure of cultured microvascular endothelial cells to SARS-CoV-2 spike protein
induced leukocyte recruitment, C3 and C5b–9 deposition and platelet adhesion and thrombus formation on cell surfaces. Thrombus formation was halted by complement inhibitors89. The
observation that the signature of complement deposition across different organs in patients with severe COVID-19 mimics the signatures found in complement-driven thrombotic disease90
supports the notion that complement is a driving pathogenic force in severe COVID-19. Furthermore, it is possible that complement also contributes to SARS-CoV-2 vaccine-induced thrombotic
complications (Box 1). Thus, collectively, the available evidence places the consequences of C5 activation among the drivers of COVID-19-associated thromboinflammation and highlights the
potential of inhibitors targeting C5, C5a or C5aR1 to prevent and control thromboembolic complications in this debilitating infectious disease. BOX 1 SARS-COV-2 VACCINE-INDUCED THROMBOTIC
COMPLICATIONS The development of vaccines against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has occurred with unprecedented speed, with at least three vaccines currently
approved in most countries123,124 and more than six billion doses administered worldwide so far125. Overall, the risk of serious adverse effects has been remarkably low. However, several
cases of unexpected thrombotic events and thrombocytopenia have been observed in individuals vaccinated with the adenovector-based vaccines ChAdOx1 nCoV-19 (AstraZeneca, University of Oxford
and Serum Institute of India)126,127 and Ad26.COV2.S (Janssen; Johnson & Johnson)128,129,130. Most affected patients developed acute disseminated intravascular coagulation, which can
evolve into life-threatening cerebral venous thrombosis and thrombocytopenia following the first vaccination dose. The syndrome has been termed ‘vaccine-induced immune thrombotic
thrombocytopenia’ (VITT) and is caused by high titres of circulating IgG autoantibodies that recognize platelet-bound platelet factor 4 (PF4; also known as CXCL4) and induce uncontrolled,
FcγIIa-dependent platelet activation and uninhibited (local) coagulation126. Treatment of VITT includes use of non-heparin anticoagulants in combination with high-dose intravenous
immunoglobulins (to competitively inhibit the FcγIIa receptor)131. VITT-related venous thrombosis is not confined to classical sites such as the lungs and deep veins of the leg but is often
disseminated to the splanchnic, cerebral and ophthalmic veins132. The reasons for thrombosis at these uncommon locations in VITT are currently unclear. Although a role for complement in VITT
has not been explored, there are several intriguing features of VITT that are suggestive of potential complement involvement. For example, as the main driver of VITT is a platelet-bound
anti-PF4 IgG antibody, recognition of the IgG–platelet immune complexes by C1q can initiate activation of the classical pathway. This could trigger subsequent activation of the alternative
pathway and the coagulation system (Fig. 1) and create the thrombotic environment seen in VITT. Furthermore, the unusual locations of thrombotic precipitations observed in VITT are similar
to those seen in paroxysmal nocturnal haemoglobinuria, a condition caused by overactive complement due to deficiency in the complement regulator CD59 (refs133,134). Congruent with this, two
patients with VITT improved after treatment with the anti-C5 antibody eculizumab135. However, although plausible, the potential contributions of complement to VITT are currently hypothetical
and require further clinical and experimental exploration. INTRACELLULAR COMPLEMENT Complement circulating in plasma is produced and secreted by the liver91. However, complement components
are also produced by both immune92,93 and non-immune94,95,96 cells. In T cells, complement is produced both on activation through the T cell receptor and via ICAM1–LFA1 interactions during
diapedesis into tissues92,93. The latter scenario is relevant to COVID-19 as it represents a source of local complement in tissues, where plasma complement is absent, and is a requirement
for the bioenergetic reprogramming necessary for optimal effector function of immune cells92,97,98,99. Airway epithelial cells (AECs) also produce C3 and have the capacity to take up some C3
from exogenous sources96. In the context of cellular stress, for example from serum starvation, intracellular C3 protects AECs from apoptosis96, although the mechanism is currently unknown.
In the context of SARS-CoV-2 infection of AECs, expression of complement genes is among the biological pathways most highly induced by the virus100. Here, virus sensing triggers type I
interferon signalling through the JAK–STAT machinery and induces direct regulation of complement genes, notably including _C3_ and _CFB_ (encoding complement factor B)100. Factor B drives
the assembly of an inducible, intracellular alternative complement activation convertase, which processes C3 to C3a. Accordingly, SARS-CoV-2-infected cells produce C3a, and this can be
normalized by culture with ruxolitinib, an inhibitor of JAK1 and JAK2, or a cell-permeable inhibitor of factor B100. Importantly, immune cells close to SARS-CoV-2-infected AECs respond to
the milieu of heightened local C3 activation and upregulate genes induced by C3 fragment receptors (C3aR and CD46), generating proinflammatory mediators, a signature not detected in
circulating immune cells100. Whether increased C3 production and intracellular C3a generation by AECs triggered by SARS-CoV-2 infection has a cell-intrinsic effect has not yet been explored.
Collectively, AEC-derived complement is likely to represent a substantial local source of complement in the context of SARS-CoV-2 and is unlikely to be normalized by drugs targeting
complement components in the circulation. Given the contributions of canonical (plasma-circulating) and non-canonical (intracellular) complement to human immunological health101,102, it may
be worth exploring whether changes in complement activity with age may be a risk factor for severe COVID-19 (Box 2). BOX 2 RISK FACTORS FOR SEVERE COVID-19 One of the major unanswered
questions of the COVID-19 pandemic is why some people succumb to the disease while others experience only mild symptoms following infection. Surveillance data indicate that older adults
(over 70 years of age) and those with underlying health conditions have an increased risk of progression to severe COVID-19 (ref.136). Specifically, co-morbidities such as diabetes, heart
disease and hypertension (which are progressively more prevalent with age), obesity and smoking are associated with severe disease or fatal outcomes137. The underlying molecular mechanisms
that render these subgroups of patients more vulnerable are a focus of ongoing research — and complement perturbations should be considered as potential candidates. Complement plays an
active role in the development of hypertension138 and cardiovascular disease139, while smoking and obesity are both associated with chronically increased complement activation, which
perpetuates the associated diseases140,141. Thus, complement contributes to the pathological conditions priming individuals to development of severe COVID-19. More importantly, complement
may also contribute to the changes associated with the ageing immune system, referred to as ‘immunosenescence’142, and that augment vulnerability towards severe COVID-19 in otherwise healthy
individuals143. Specifically, the continued presence of low-grade inflammation (inflammageing) over a lifetime can fuel the onset or progression of age-related pathological inflammation and
autoimmunity. Indeed, the protein levels of classical and alternative pathway complement components in the circulation increase progressively with age144, and increases in the amounts of
circulating C3 and C1q proteins are considered biomarker candidates of ageing and age-related diseases145. Furthermore, increases in complement production in the brain contribute to
age-related synaptic loss and microglia dysfunction146, whereas an age-related increase in C1q protein levels in circulation impairs muscle regeneration over time147. Thus, it is feasible
that age-related changes in the amounts of specific complement proteins contribute to the detrimental imbalance in proinflammatory versus repair responses disproportionately observed in
older patients (over 70 years of age) with COVID-19. THERAPEUTIC PERSPECTIVES Early in the first wave of the COVID-19 pandemic, a number of case reports and small-scale clinical
interventions that were not randomized nor properly controlled advocated the use of complement-inhibiting therapies103,104,105,106,107,108,109, very similar to reports advocating the
wide-scale use of cytokine antagonists110. What has become clear over the past 18 months is that initial enthusiasm needs to be followed by rigorously executed clinical trials of appropriate
sample size across many countries before conclusions can be reached and clinical practice is altered. For COVID-19 therapy, it is emerging that timing is everything. The disease runs a
bimodal course with an early viral phase comprising mainly upper airway symptoms, followed in approximately 20% of patients by dyspnea and hypoxia, sometimes progressing to severe acute
respiratory distress syndrome. Despite the considerable numbers of patients who are eligible and willing to participate in clinical COVID-19 trials, only 20% of patients progress to severe
disease. Thus, the biggest hurdle to obtaining definitive answers with various intervention trials in severe COVID-19 has been to select the right timing and the right patient who might
benefit from the intervention, on the basis of either clinical or biochemically easy to measure (point of care) biomarkers. It is very likely that the hyperinflammation that characterizes
severe disease is driven by several molecular and cellular pathways that tend to self-amplify111, so in advanced disease, targeting only one pathway and hoping for improvement is likely to
be futile. Early intervention without causing immunosuppression is probably key. Currently, however, biomarkers predicting those who will go on to develop life-threatening COVID-19
(prognostic biomarkers) among the less ill and that could predict those who will respond to therapy (theragnostic biomarkers) are still lacking. One notable exception is the serum
concentration of soluble urokinase plasminogen activator receptor, which strongly predicts clinical responsiveness to blockade with the IL-1 receptor antagonist anakinra112. In the specific
case of complement inhibition studies, it is still complicated in most hospitals to get point-of-care assessments of the complement cascade and in a timely manner. Clinical practice is also
rapidly changing during this pandemic, and use of the broad anti-inflammatory drug dexamethasone has become the standard of care, potentially making it harder to replicate earlier findings
regarding new compounds in small-scale studies in larger randomized trials that now include dexamethasone as the standard of care. As an example, despite many negative clinical outcome
trials, the effectiveness of interfering with IL-6 in COVID-19 became clear only from a meta-analysis of more than 30 randomized intervention trials, in which it was shown that IL-6 reduces
28-day mortality mainly in the most severely ill patients113. Counterintuitively, the clinical benefit was, however, seen mainly in those receiving dexamethasone or other steroids,
potentially because these patients had a worse prognosis114,115. Specifically, with regard to complement, other considerations include whether components of the complement system themselves
should be targeted (extracellularly or within cells) or whether mechanisms inducing complement production and/or activation should be targeted instead (for example, using JAK inhibitors or
factor B inhibitors; see earlier). These considerations are important because AEC-derived complement is likely to represent a substantial local source of complement in the context of
SARS-CoV-2 and is unlikely to be normalized by drugs targeting complement components in the circulation. Cell-permeating drugs targeting complement intracellularly (such as factor B
inhibitors) or upstream components inducing complement gene transcription (such as JAK inhibitors) could potentially be more efficacious in severe COVID-19. Indeed, although many trials are
ongoing, some data already support beneficial efficacy of JAK inhibitors in the context of severe COVID-19116,117,118,119. Finally, although the proliferation of preprint publications
somewhat mitigates publication bias, it still remains likely that negative data may be under-reported. Strategies to target complement in COVID-19 have mainly used available drugs that were
already undergoing clinical testing or were already approved for clinical use in complement-mediated diseases, such as thrombotic microangiopathy associated with transplantation, atypical
haemolytic uraemic syndrome, paroxysmal nocturnal haemoglobinuria, macular degeneration, complement-mediated nephropathies and myasthenia gravis. As a result, most of these have focused on
targeting the C5 system. Initially, a few case series of C5-targeting therapies (eculizumab, a monoclonal antibody targeting C5, by Alexion Pharmaceuticals; BDB-001, a monoclonal antibody
against C5a, by Staidson Biopharmaceuticals) were trialled in a total of 16 patients with severe to critical COVID-19, 14 of whom recovered26,106,109. A subsequent proof-of-concept
non-randomized trial in 40 patients with severe COVID-19 and controls showed a survival benefit in the group treated in an intensive care unit setting with eculizumab120. On the basis of
these studies, several randomized controlled trials (RCTs) have been initiated, and some have completed enrolment. Targeting C5 in COVID-19 has been trialled using the monoclonal antibodies
eculizumab (NCT04288713 and NCT04346797), ravulizumab (Ultomiris; NCT04570397 and NCT04369469) and tesidolumab (LFG316, Novartis)108, or using small protein or peptide antagonists of C5 such
as nomacopan (Akari Therapeutics; trial about to start in the UK and Brazil) or zilucoplan (ZILU-COV trial (NCT04382755, UCB Pharma). Interim analysis of a large phase III trial with
ravulizumab (NCT04369469) in patients with COVID-19 already receiving mechanical ventilation at randomization could not demonstrate clinical benefit, leading to a pause in the study. In the
ZILU-COV trial, in contrast, 81 patients with COVID-19 and signs of hypoxia not yet requiring mechanical ventilation were randomized to receive 32 mg of zilucoplan subcutaneous daily for 14
days in addition to prophylactic broad-spectrum antibiotics (to cover the risk of meningococcal disease) or standard-of-care treatment and the same antibiotic regimen121. Patients receiving
zilucoplan had improved oxygenation parameters at day 15, accompanied by a lowering of circulating cytokine levels. At day 28 there was also lower mortality in zilucoplan-treated patients,
although the study was underpowered to detect effects on mortality. Importantly, there was no risk of opportunistic infections in those receiving zilucoplan. This indicates that early timing
of C5 blockade may be crucial. Trials are also studying the impact of blocking either C5a or C5aR. An initial RCT using the C5a-targeting antibody vilobelimab (NCT04333420, InflaRx)
reported minimal improvement in oxygenation in 30 patients and controls, although there was a trend for increased survival in the vilobelimab arm122. This study has now been extended to a
large multicentre placebo-controlled RCT involving 390 patients receiving mechanical ventilation (NCT04333420) and is currently recruiting patients. Similarly, a large phase II/III RCT
involving 368 participants is addressing the C5a-specific antibody BDB-001 (NCT 04449588, Staidson Biopharmaceuticals) and is in progress. Despite promising results of C5aR blockade in a
preclinical model20, development of the C5aR1-specific monoclonal antibody avdoralimab (Innate Pharma, NCT04371367) was discontinued after disappointing effects of a phase II RCT in patients
with various degrees of COVID-19 severity. One explanation is that blockade of C5aR1 alone is not sufficient to halt the detrimental effects of complement in COVID-19 because the MAC is
still formed or because C5 can still signal through alternative receptors, such as C5aR2. Targeting the C3 pathway is also an attractive scenario. Use of the C3 inhibitor AMY-101 (Amyndas
Pharma) was initially reported in two case reports for treatment of four patients with severe COVID-19, all of whom subsequently recovered105,106. A larger phase II study with 144 patients
(NCT04395456) is planned but is not yet recruiting. The C3 antagonist APL9 (Apellis, NCT04402060) was discontinued after no difference in mortality was found in a phase I/II RCT in 65
patients with severe COVID-19. Other strategies of which the outcome is not yet known include blocking complement early in the cascade, such as by using the recombinant human C1 inhibitor
Ruconest (Pharming, NCT04705831, NCT0530136 and NCT04414631) or the MASP2-specific antibody narsoplimab (Omeros, NCT04488081) to block the lectin pathway of complement activation. Similarly,
given the potential for SARS-CoV-2 spike protein to activate complement via the alternative pathway12,13,14, a phase II clinical trial of remdesivir, with or without a factor D inhibitor,
is currently under way (Alexion, NCT04988035). An important cautionary note that also needs to be considered in the context of strategies that inhibit a non-redundant component of normal
immunity is the risk of life-threatening, invasive infections that are distinct from COVID-19 itself. As examples, a fatal case of _Klebsiella pneumoniae_ infection following compassionate
use of a C5-blocking monoclonal antibody in one of five patients108 and a more than doubling of infectious complications, including bacteraemia and ventilator-associated pneumonia, in a
clinical trial of eculizumab have been reported120. These infectious complications occurred despite adequate prior antibiotic prophylaxis and vaccination. Thus, the potential gains of
complement inhibition need also to be balanced against the potential risks and harms that may also ensue. CONCLUSIONS Both clinical and basic science studies suggest that uncontrolled
activity of the complement system may be a central player in the pathogenesis of COVID-19. Clinical trial evidence is now beginning to substantiate this suspicion. This clearly is an
exciting development; however, it should still be viewed with caution because, similarly to IL-6-targeting therapies, it is likely that clinical trials will indicate that the benefits of
complement-targeting therapies will depend on several confounders, namely severity of disease, timing of initiation and co-administered drugs. Thus, at present there is no compelling
evidence to strongly advocate complement-targeting treatments for all patients presenting with severe COVID-19. This will likely change in the future as the niche for complement targeting
becomes clearer and as we gain a better understanding of the distinct spatio-temporal contributions of complement following SARS-CoV-2 infection, its crosstalk with the coagulation system
and the role of genetic polymorphisms in both systems. REFERENCES * Dong, E., Du, H. & Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. _Lancet Infect.
Dis._ 20, 533–534 (2020). CAS PubMed PubMed Central Google Scholar * Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a
retrospective cohort study. _Lancet_ 395, 1054–1062 (2020). CAS PubMed PubMed Central Google Scholar * Whitaker, M. et al. Persistent symptoms following SARS-CoV-2 infection in a random
community sample of 508,707 people. Preprint at _medRxiv_ https://doi.org/10.1101/2021.06.28.21259452 (2021). Article Google Scholar * Vasarmidi, E., Tsitoura, E., Spandidos, D. A.,
Tzanakis, N. & Antoniou, K. M. Pulmonary fibrosis in the aftermath of the COVID-19 era (review). _Exp. Ther. Med._ 20, 2557–2560 (2020). CAS PubMed PubMed Central Google Scholar *
Noris, M. & Remuzzi, G. Overview of complement activation and regulation. _Semin. Nephrol._ 33, 479–492 (2013). CAS PubMed PubMed Central Google Scholar * Noris, M., Benigni, A.
& Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. _Kidney Int._ 98, 314–322 (2020). CAS PubMed PubMed Central Google Scholar * Lo, M. W., Kemper, C.
& Woodruff, T. M. COVID-19: complement, coagulation, and collateral damage. _J. Immunol._ 205, ji2000644 (2020). Google Scholar * Skendros, P. et al. Complement and tissue
factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. _J. Clin. Invest._ 130, 6151–6157 (2020). CAS PubMed PubMed Central Google Scholar * Perico,
L. et al. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. _Nat. Rev. Nephrol._ 17, 46–64 (2021). PubMed Google Scholar * Ali, Y. M. et al. Lectin pathway
mediates complement activation by SARS-CoV-2 proteins. _Front. Immunol._ 12, 714511 (2021). THIS STUDY PROVIDES THE FIRST DEMONSTRATION THAT SARS-COV-2 DIRECTLY ACTIVATES COMPLEMENT VIA
MASP1 AND MASP2. CAS PubMed PubMed Central Google Scholar * Holter, J. C. et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients.
_Proc. Natl Acad. Sci. USA_ 117, 25018–25025 (2020). CAS PubMed PubMed Central Google Scholar * Yu, J. et al. Complement dysregulation is associated with severe COVID-19 illness.
_Haematologica_ https://doi.org/10.3324/haematol.2021.279155 (2021). Article PubMed PubMed Central Google Scholar * Yu, J. et al. Direct activation of the alternative complement pathway
by SARS-CoV-2 spike proteins is blocked by factor D inhibition. _Blood_ 136, 2080–2089 (2020). PubMed Google Scholar * Satyam, A. et al. Activation of classical and alternative complement
pathways in the pathogenesis of lung injury in COVID-19. _Clin. Immunol._ 226, 108716 (2021). CAS PubMed PubMed Central Google Scholar * Robbins, R. A., Russ, W. D., Rasmussen, J. K.
& Clayton, M. M. Activation of the complement system in the adult respiratory distress syndrome1–4. _Am. Rev. Respir. Dis._ 135, 651–658 (1987). CAS PubMed Google Scholar * Zilow, G.,
Sturm, J. A., Rother, U. & Kirschfink, M. Complement activation and the prognostic value of C3a in patients at risk of adult respiratory distress syndrome. _Clin. Exp. Immunol._ 79,
151–157 (1990). CAS PubMed PubMed Central Google Scholar * Zilow, G., Joka, T., Obertacke, U., Rother, U. & Kirschfink, M. Generation of anaphylatoxin C3a in plasma and
bronchoalveolar lavage fluid in trauma patients at risk for the adult respiratory distress syndrome. _Crit. Care Med._ 20, 468–473 (1992). CAS PubMed Google Scholar * Duchateau, J. et al.
Complement activation in patients at risk of developing the adult respiratory distress syndrome1–3. _Am. Rev. Respir. Dis._ 130, 1058–1064 (1984). CAS PubMed Google Scholar * Ohta, R. et
al. Serum concentrations of complement anaphylatoxins and proinflammatory mediators in patients with 2009 H1N1 influenza. _Microbiol. Immunol._ 55, 191–198 (2011). CAS PubMed Google
Scholar * Carvelli, J. et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. _Nature_ 588, 146–150 (2020). THIS IS A KEY PUBLICATION DEMONSTRATING THAT
INCREASED C5A LEVELS IN THE CIRCULATION AND HYPEREXPRESSION AND HYPERACTIVATION OF C5AR1 ON BLOOD AND PULMONARY IMMUNE CELLS CONTRIBUTE TO COVID-19 SEVERITY. CAS PubMed PubMed Central
Google Scholar * Cugno, M. et al. Complement activation in patients with COVID-19: a novel therapeutic target. _J. Allergy Clin. Immun._ 146, 215–217 (2020). CAS PubMed Google Scholar *
Shen, B. et al. Proteomic and metabolomic characterization of COVID-19 patient sera. _Cell_ 182, 59–72.e15 (2020). THIS IS A FUNDAMENTAL DATA SOURCE ALLOWING NOVEL INSIGHTS INTO DISEASE
MECHANISMS OF COVID-19 BEYOND THE BATTERY OF SINGLE-CELL RNA-SEQUENCING DATA AVAILABLE. CAS PubMed PubMed Central Google Scholar * Sinkovits, G. et al. Complement overactivation and
consumption predicts in-hospital mortality in SARS-CoV-2 infection. _Front. Immunol._ 12, 663187 (2021). CAS PubMed PubMed Central Google Scholar * Ramlall, V. et al. Immune complement
and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. _Nat. Med._ 26, 1609–1615 (2020). CAS PubMed PubMed Central Google Scholar * Magro, C. et al. Complement
associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. _Transl. Res._ 220, 1–13 (2020). CAS PubMed PubMed Central Google
Scholar * Gao, T. et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. Preprint at _medRxiv_
https://doi.org/10.1101/2020.03.29.20041962 (2020). Article PubMed PubMed Central Google Scholar * Riphagen, S., Gomez, X., Gonzalez-Martinez, C., Wilkinson, N. & Theocharis, P.
Hyperinflammatory shock in children during COVID-19 pandemic. _Lancet_ 395, 1607–1608 (2020). CAS PubMed PubMed Central Google Scholar * Verdoni, L. et al. An outbreak of severe
Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. _Lancet_ 395, 1771–1778 (2020). THIS IS AMONG THE FIRST STUDIES SHOWING THAT
SARS-COV-2 CAN INDUCE A SPECTRUM OF MULTISYSTEM INFLAMMATORY SYNDROMES. CAS PubMed PubMed Central Google Scholar * Syrimi, E. et al. The immune landscape of SARS-CoV-2-associated
multisystem inflammatory syndrome in children (MIS-C) from acute disease to recovery. _iScience_ 24, 103215 (2021). CAS PubMed PubMed Central Google Scholar * Varga, Z. et al.
Endothelial cell infection and endotheliitis in COVID-19. _Lancet_ 395, 1417–1418 (2020). CAS PubMed PubMed Central Google Scholar * Su, H. et al. Renal histopathological analysis of 26
postmortem findings of patients with COVID-19 in China. _Kidney Int._ 98, 219–227 (2020). CAS PubMed PubMed Central Google Scholar * Bryce, C. et al. Pathophysiology of SARS-CoV-2:
targeting of endothelial cells renders a complex disease with thrombotic microangiopathy and aberrant immune response. The Mount Sinai COVID-19 autopsy experience. Preprint at _medRxiv_
https://doi.org/10.1101/2020.05.18.20099960 (2020). Article Google Scholar * Timmermans, S. A. M. E. G. et al. C5b9 formation on endothelial cells reflects complement defects among
patients with renal thrombotic microangiopathy and severe hypertension. _J. Am. Soc. Nephrol._ 29, 2234–2243 (2018). CAS PubMed PubMed Central Google Scholar * Jiang, Y. et al. Blockade
of the C5a–C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. _Emerg. Microbes Infec_ 7, 77 (2018). Google Scholar * Rockx, B. et al. Early upregulation of
acute respiratory distress syndrome-associated cytokines promotes lethal disease in an aged-mouse model of severe acute respiratory syndrome coronavirus infection. _J. Virol._ 83, 7062–7074
(2009). CAS PubMed PubMed Central Google Scholar * Gralinski, L. E. et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. _mBio_ 9,
e01753–18 (2018). THIS STUDY PROVIDES THE FIRST EVIDENCE IN A SMALL ANIMAL IN VIVO MODEL THAT COMPLEMENT ACTIVATION CONTRIBUTES TO SARS-COV-2-INDUCED AIRWAY PATHOLOGY BY DEMONSTRATING THAT
C3-DEFICIENT, VIRUS-INFECTED MICE HAVE LESS PATHOLOGY COMPARED WITH WILD-TYPE MICE. PubMed PubMed Central Google Scholar * Cameron, M. J. et al. Lack of innate interferon responses during
SARS coronavirus infection in a vaccination and reinfection ferret model. _PLoS ONE_ 7, e45842 (2012). CAS PubMed PubMed Central Google Scholar * Ip, W. K. E. et al. Mannose-binding
lectin in severe acute respiratory syndrome coronavirus infection. _J. Infect. Dis._ 191, 1697–1704 (2005). CAS PubMed Google Scholar * Ling, M. T. et al. Mannose-binding lectin
contributes to deleterious inflammatory response in pandemic H1N1 and avian H9N2 infection. _J. Infect. Dis._ 205, 44–53 (2012). CAS PubMed Google Scholar * Middeldorp, S. et al.
Incidence of venous thromboembolism in hospitalized patients with COVID-19. _J. Vasc. Surg. Venous Lymphatic Disord._ 9, 536 (2021). Google Scholar * Iffah, R. & Gavins, F. N. E.
Thromboinflammation in coronavirus disease 2019: the clot thickens. _Brit J. Pharmacol._ https://doi.org/10.1111/bph.15594 (2021). Article Google Scholar * Wadman, M., Couzin-Frankel, J.,
Kaiser, J. & Matacic, C. A rampage through the body. _Science_ 368, 356–360 (2020). CAS PubMed Google Scholar * Pellegrini, D. et al. Microthrombi as a major cause of cardiac injury
in COVID-19: a pathologic study. _Circulation_ 143, 1031–1042 (2021). CAS PubMed Google Scholar * Al-Samkari, H. et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of
SARS-CoV-2 infection. _Blood_ 136, 489–500 (2020). CAS PubMed Google Scholar * Pujhari, S., Paul, S., Ahluwalia, J. & Rasgon, J. L. Clotting disorder in severe acute respiratory
syndrome coronavirus 2. _Rev. Med. Virol._ 31, e2177 (2021). CAS PubMed Google Scholar * Lax, S. F. et al. Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a
prospective, single-center, clinicopathologic case series. _Ann. Intern. Med._ 173, 350–361 (2020). PubMed Google Scholar * Shi, J. et al. Coagulation dysfunction in ICU patients with
coronavirus disease 2019 in Wuhan, China: a retrospective observational study of 75 fatal cases. _Aging_ 13, 1591–1607 (2020). PubMed PubMed Central Google Scholar * Violi, F. et al.
Arterial and venous thrombosis in coronavirus 2019 disease (Covid-19): relationship with mortality. _Intern. Emerg. Med._ 16, 1231–1237 (2021). PubMed PubMed Central Google Scholar *
Pérez-García, F. et al. Age-adjusted endothelial activation and stress index for coronavirus disease 2019 at admission is a reliable predictor for 28-day mortality in hospitalized patients
with coronavirus disease 2019. _Front. Med._ 8, 736028 (2021). Google Scholar * Contou, D. et al. Causes and timing of death in critically ill COVID-19 patients. _Crit. Care_ 25, 79 (2021).
PubMed PubMed Central Google Scholar * Romanova, E. S. et al. Cause of death based on systematic post-mortem studies in patients with positive SARS-CoV-2 tissue PCR during the COVID-19
pandemic. _J. Intern. Med._ 290, 655–665 (2021). CAS PubMed Google Scholar * Spiezia, L. et al. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for
acute respiratory failure. _Thromb. Haemost._ 120, 998–1000 (2020). PubMed PubMed Central Google Scholar * Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis, and
angiogenesis in Covid-19. _N. Engl. J. Med._ 383, 120–128 (2020). CAS PubMed PubMed Central Google Scholar * Aimes, R. et al. Endothelial cell serine proteases expressed during vascular
morphogenesis and angiogenesis. _Thromb. Haemost._ 89, 561–572 (2003). CAS PubMed Google Scholar * Nicin, L. et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2
in human hearts. _Eur. Heart J._ 41, 1804–1806 (2020). CAS PubMed Google Scholar * Delorey, T. M. et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. _Nature_
595, 107–113 (2021). CAS PubMed PubMed Central Google Scholar * Vinci, R. et al. From angiotensin-converting enzyme 2 disruption to thromboinflammatory microvascular disease: a paradigm
drawn from COVID-19. _Int. J. Cardiol._ 326, 243–247 (2021). CAS PubMed Google Scholar * Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using
clinical-grade soluble human ACE2. _Cell_ 181, 905–913.e7 (2020). CAS PubMed PubMed Central Google Scholar * Wang, P. et al. A cross-talk between epithelium and endothelium mediates
human alveolar–capillary injury during SARS-CoV-2 infection. _Cell Death Dis._ 11, 1042 (2020). CAS PubMed PubMed Central Google Scholar * Bryce, C. et al. Pathophysiology of SARS-CoV-2:
the Mount Sinai COVID-19 autopsy experience. _Mod. Pathol._ 34, 1456–1467 (2021). CAS PubMed PubMed Central Google Scholar * Sia, S. F. et al. Pathogenesis and transmission of
SARS-CoV-2 in golden hamsters. _Nature_ 583, 834–838 (2020). WITH THIS PUBLICATION, THE AUTHORS PROVIDE A HAMSTER IN VIVO MODEL OF MILD SARS-COV-2 INFECTION WHICH MIMICS HUMAN DISEASE BETTER
THAN COMPARABLE MOUSE MODELS. CAS PubMed PubMed Central Google Scholar * Schimmel, L. et al. Endothelial cells are not productively infected by SARS-CoV-2. _Clin. Transl. Immunol._ 10,
e1350 (2021). CAS Google Scholar * Goshua, G. et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. _Lancet Haematol._ 7,
e575–e582 (2020). PubMed PubMed Central Google Scholar * O’Sullivan, J. M., Gonagle, D. M., Ward, S. E., Preston, R. J. S. & O’Donnell, J. S. Endothelial cells orchestrate COVID-19
coagulopathy. _Lancet Haematol._ 7, e553–e555 (2020). PubMed PubMed Central Google Scholar * Ma, L. et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2
infection. _Sci. Immunol._ 6, eabh2259 (2021). PubMed Central Google Scholar * Cotter, A. H., Yang, S.-J. T., Shafi, H., Cotter, T. M. & Palmer-Toy, D. E. Elevated von Willebrand
factor antigen is an early predictor of mortality and prolonged length of stay for coronavirus disease 2019 (COVID-19) inpatients. _Arch. Pathol. Lab. Med._
https://doi.org/10.5858/arpa.2021-0255-sa (2021). Article Google Scholar * Marchetti, M. et al. Endothelium activation markers in severe hospitalized COVID-19 patients: role in mortality
risk prediction. _TH Open_ 5, e253–e263 (2021). PubMed PubMed Central Google Scholar * Fox, S. E. et al. Pulmonary and cardiac pathology in African American patients with COVID-19: an
autopsy series from New Orleans. _Lancet Respir. Med._ 8, 681–686 (2020). CAS PubMed PubMed Central Google Scholar * Pfister, F. et al. Complement activation in kidneys of patients with
COVID-19. _Front. Immunol._ 11, 594849 (2021). PubMed PubMed Central Google Scholar * Diorio, C. et al. Evidence of thrombotic microangiopathy in children with SARS-CoV-2 across the
spectrum of clinical presentations. _Blood Adv._ 4, 6051–6063 (2020). CAS PubMed PubMed Central Google Scholar * Noris, M., Mescia, F. & Remuzzi, G. STEC-HUS, atypical HUS and TTP
are all diseases of complement activation. _Nat. Rev. Nephrol._ 8, 622–633 (2012). CAS PubMed Google Scholar * Noris, M. et al. Dynamics of complement activation in aHUS and how to
monitor eculizumab therapy. _Blood_ 124, 1715–1726 (2014). CAS PubMed PubMed Central Google Scholar * Mat, O. et al. Kidney thrombotic microangiopathy after COVID-19 associated with C3
gene mutation. _Kidney Int. Rep._ 6, 1732–1737 (2021). PubMed PubMed Central Google Scholar * Ville, S. et al. Atypical HUS relapse triggered by Covid-19. _Kidney Int._ 99, 267–268
(2020). PubMed PubMed Central Google Scholar * Riedl, M. et al. Complement activation induces neutrophil adhesion and neutrophil-platelet aggregate formation on vascular endothelial
cells. _Kidney Int. Rep._ 2, 66–75 (2017). PubMed Google Scholar * Skeie, J. M., Fingert, J. H., Russell, S. R., Stone, E. M. & Mullins, R. F. Complement component C5a activates ICAM-1
expression on human choroidal endothelial cells. _Invest. Ophth Vis. Sci._ 51, 5336–5342 (2010). Google Scholar * Kilgore, K. S., Ward, P. A. & Warren, J. S. Neutrophil adhesion to
human endothelial cells is induced by the membrane attack complex: the roles of P-selectin and platelet activating factor. _Inflammation_ 22, 583–598 (1998). THIS IS PIONEERING WORK
CONNECTING MAC FORMATION WITH ENDOTHELIAL CELL AND PLATELET ACTIVATION. CAS PubMed Google Scholar * Foreman, K. E. et al. C5a-induced expression of P-selectin in endothelial cells. _J.
Clin. Invest._ 94, 1147–1155 (1994). CAS PubMed PubMed Central Google Scholar * Ikeda, K. et al. C5a induces tissue factor activity on endothelial cells. _Thromb. Haemost._ 77, 394–398
(1997). CAS PubMed Google Scholar * Bettoni, S. et al. Interaction between multimeric von Willebrand factor and complement: a fresh look to the pathophysiology of microvascular
thrombosis. _J. Immunol._ 199, 1021–1040 (2017). CAS PubMed Google Scholar * Leppkes, M. et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. _Ebiomedicine_ 58, 102925
(2020). PubMed PubMed Central Google Scholar * Bont, C. M., de, Boelens, W. C. & Pruijn, G. J. M. NETosis, complement, and coagulation: a triangular relationship. _Cell Mol.
Immunol._ 16, 19–27 (2019). PubMed Google Scholar * Xu, J. et al. Extracellular histones are major mediators of death in sepsis. _Nat. Med._ 15, 1318–1321 (2009). CAS PubMed PubMed
Central Google Scholar * Semeraro, F. et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. _Blood_ 118,
1952–1961 (2011). CAS PubMed PubMed Central Google Scholar * Bosmann, M. et al. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation
in acute lung injury. _FASEB J._ 27, 5010–5021 (2013). THIS IS THE FIRST STUDY TO CONNECT LOCAL HISTONE H3 AND H4 RELEASE AS DAMPS THAT DRIVE C5 ACTIVATION AND SUBSEQUENT INDUCTION OF TISSUE
PATHOLOGIES IN RAT AND MOUSE MODELS OF ACUTE LUNG INJURY. CAS PubMed PubMed Central Google Scholar * Caudrillier, A. et al. Platelets induce neutrophil extracellular traps in
transfusion-related acute lung injury. _J. Clin. Invest._ 122, 2661–2671 (2012). CAS PubMed PubMed Central Google Scholar * Middleton, E. A. et al. Neutrophil extracellular traps
contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. _Blood_ 136, 1169–1179 (2020). THIS STUDY IS AMONG THE FIRST TO DEMONSTRATE THAT NEUTROPHILS AND SPECIFICALLY
NEUTROPHIL NETS CONTRIBUTE TO COVID-19 PATHOLOGY. CAS PubMed Google Scholar * Barnes, B. J. et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. _J. Exp. Med._
217, e20200652 (2020). CAS PubMed PubMed Central Google Scholar * Perico, L. et al. SARS-CoV-2 spike protein 1 activates microvascular endothelial cells and complement system leading to
thrombus formation. _SSRN Electron. J._ https://doi.org/10.2139/ssrn.3864027 (2021). Article Google Scholar * Macor, P. et al. Multiple-organ complement deposition on vascular endothelium
in COVID-19 patients. _Biomed_ 9, 1003 (2021). CAS Google Scholar * Walport, M. J. Complement. First two parts. _N. Engl. J. Med._ 344, 1058–1066 (2001). CAS PubMed Google Scholar *
Kolev, M. et al. Diapedesis-induced integrin signaling via LFA-1 facilitates tissue immunity by inducing intrinsic complement C3 expression in immune cells. _Immunity_ 52, 513–527.e8 (2020).
CAS PubMed PubMed Central Google Scholar * Liszewski, M. K. et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. _Immunity_ 39,
1143–1157 (2013). CAS PubMed PubMed Central Google Scholar * Friščić, J. et al. The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial
fibroblasts. _Immunity_ 54, 1002–1021.e10 (2021). PubMed Google Scholar * Pratt, J. R., Abe, K., Miyazaki, M., Zhou, W. & Sacks, S. H. In situ localization of C3 Synthesis in
experimental acute renal allograft rejection. _Am. J. Pathol._ 157, 825–831 (2000). CAS PubMed PubMed Central Google Scholar * Kulkarni, H. S. et al. Intracellular C3 protects human
airway epithelial cells from stress-associated cell death. _Am. J. Resp. Cell Mol._ 60, 144–157 (2019). THIS STUDY SUPPORTS THE EMERGING CONCEPT OF INTRACELLULAR COMPLEMENT AS KEY DRIVER OF
HEALTHY CELL HOMEOSTASIS BY SHOWING THAT INTRACELLULAR C3 INDUCES CELL LIFE-SAVING AUTOPHAGY IN EPITHELIAL CELLS EXPOSED TO OXIDATIVE STRESS. CAS Google Scholar * Arbore, G. et al.
Complement receptor CD46 co-stimulates optimal human CD8+ T cell effector function via fatty acid metabolism. _Nat. Commun._ 9, 4186 (2018). PubMed PubMed Central Google Scholar * Kolev,
M. et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. _Immunity_ 42, 1033–1047 (2015). CAS PubMed PubMed Central Google Scholar * Arbore,
G. et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. _Science_ 352, aad1210 (2016). PubMed PubMed Central Google Scholar * Yan, B. et al.
SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. _Sci. Immunol._ 6, eabg0833 (2021). THIS IS THE FIRST STUDY SHOWING THAT SARS-COV-2 INDUCES PATHOLOGICAL C3
HYPERACTIVATION IN INFECTED TYPE II PNEUMOCYTES AND THIS RESULTS IN A SHIFT IN THE BALANCE FROM C3 HOMEOSTATIC ACTIVITY TO PATHOLOGICAL ACTIVITY THAT CONTRIBUTES TO COVID-19 PATHOLOGY.
PubMed PubMed Central Google Scholar * Hajishengallis, G., Reis, E. S., Mastellos, D. C., Ricklin, D. & Lambris, J. D. Novel mechanisms and functions of complement. _Nat. Immunol._
18, 1288–1298 (2017). CAS PubMed PubMed Central Google Scholar * Hess, C. & Kemper, C. Complement-mediated regulation of metabolism and basic cellular processes. _Immunity_ 45,
240–254 (2016). CAS PubMed PubMed Central Google Scholar * Kulasekararaj, A. G. et al. Terminal complement inhibition dampens the inflammation during COVID-19. _Brit J. Haematol._ 190,
e141–e143 (2020). CAS Google Scholar * Laurence, J. et al. Anti-complement C5 therapy with eculizumab in three cases of critical COVID-19. _Clin. Immunol._ 219, 108555 (2020). CAS PubMed
PubMed Central Google Scholar * Mastaglio, S. et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. _Clin. Immunol._ 215, 108450 (2020). CAS PubMed PubMed
Central Google Scholar * Mastellos, D. C. et al. Complement C3 vs C5 inhibition in severe COVID-19: early clinical findings reveal differential biological efficacy. _Clin. Immunol._ 220,
108598 (2020). CAS PubMed PubMed Central Google Scholar * Urwyler, P. et al. Treatment of COVID-19 with conestat alfa, a regulator of the complement, contact activation and
kallikrein-kinin system. _Front. Immunol._ 11, 2072 (2020). CAS PubMed PubMed Central Google Scholar * Zelek, W. M. et al. Complement inhibition with the C5 blocker LFG316 in severe
COVID-19. _Am. J. Resp. Crit. Care_ 202, 1304–1308 (2020). CAS Google Scholar * Diurno, F. et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL
Napoli 2 Nord experience. _Eur. Rev. Med. Pharm._ 24, 4040–4047 (2020). CAS Google Scholar * Cavalli, G. et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19,
acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. _Lancet Rheumatol._ 2, e325–e331 (2020). PubMed PubMed Central Google Scholar * Merad, M.,
Subramanian, A. & Wang, T. T. An aberrant inflammatory response in severe COVID-19. _Cell Host Microbe_ 29, 1043–1047 (2021). CAS PubMed PubMed Central Google Scholar *
Kyriazopoulou, E. et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. _Nat.
Med._ 27, 1752–1760 (2021). CAS PubMed PubMed Central Google Scholar * WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group. Association between administration of
IL-6 antagonists and mortality among patients hospitalized for COVID-19. _JAMA_ 326, 499–518 (2021). Google Scholar * RECOVERY Collaborative Group. Dexamethasone in hospitalized patients
with Covid-19. _N. Engl. J. Med._ 384, 693–704 (2020). THIS SEMINAL TRIAL SHOWS THAT ANTI-INFLAMMATORY DRUG APPLICATION CAN AMELIORATE COVID-19. Google Scholar * REMAP-CAP Investigators.
Interleukin-6 receptor antagonists in critically ill patients with Covid-19. _N. Engl. J. Med._ 384, 1491–1502 (2021). Google Scholar * Kalil, A. C. et al. Baricitinib plus remdesivir for
hospitalized adults with Covid-19. _N. Engl. J. Med._ 384, 795–807 (2021). CAS PubMed Google Scholar * Cao, Y. et al. Ruxolitinib in treatment of severe coronavirus disease 2019
(COVID-19): a multicenter, single-blind, randomized controlled trial. _J. Allergy Clin. Immun._ 146, 137–146.e3 (2020). CAS PubMed Google Scholar * Cantini, F. et al. Baricitinib therapy
in COVID-19: a pilot study on safety and clinical impact. _J. Infect._ 81, 318–356 (2020). CAS PubMed PubMed Central Google Scholar * Bronte, V. et al. Baricitinib restrains the immune
dysregulation in severe COVID-19 patients. _J. Clin. Invest._ 130, 6409–6416 (2020). CAS PubMed PubMed Central Google Scholar * Annane, D. et al. Eculizumab as an emergency treatment for
adult patients with severe COVID-19 in the intensive care unit: a proof-of-concept study. _Eclinicalmedicine_ 28, 100590 (2020). PubMed PubMed Central Google Scholar * Declercq, J. et
al. Zilucoplan in patients with acute hypoxic respiratory failure due to COVID-19 (ZILU-COV): A structured summary of a study protocol for a randomised controlled trial. _Trials_ 21, 934
(2020). CAS PubMed PubMed Central Google Scholar * Vlaar, A. P. J. et al. Anti-C5a antibody IFX-1 (vilobelimab) treatment versus best supportive care for patients with severe COVID-19
(PANAMO): an exploratory, open-label, phase 2 randomised controlled trial. _Lancet Rheumatol._ 2, e764–e773 (2020). PubMed PubMed Central Google Scholar * FDA. COVID-19 vaccines.
https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/covid-19-vaccines (2021). * EMA. COVID-19 vaccines.
https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/vaccines-covid-19/covid-19-vaccines-authorised (2021). *
Bloomberg. Bloomberg vaccine tracker. https://www.bloomberg.com/graphics/covid-vaccine-tracker-global-distribution/#global (2021). * Greinacher, A. et al. Thrombotic thrombocytopenia after
ChAdOx1 nCov-19 vaccination. _N. Engl. J. Med._ 384, 2092–2101 (2021). CAS PubMed Google Scholar * Choi, P. Y.-I. Thrombotic thrombocytopenia after ChAdOx1 nCoV-19 vaccination. _N. Engl.
J. Med._ 385, e11 (2021). PubMed Google Scholar * Muir, K.-L., Kallam, A., Koepsell, S. A. & Gundabolu, K. Thrombotic thrombocytopenia after Ad26.COV2.S vaccination. _N. Engl. J. Med._
384, 1964–1965 (2021). CAS PubMed Google Scholar * Sadoff, J., Davis, K. & Douoguih, M. Thrombotic thrombocytopenia after Ad26.COV2.S vaccination — response from the manufacturer.
_N. Engl. J. Med._ 384, 1965–1966 (2021). CAS PubMed Google Scholar * See, I. et al. US case reports of cerebral venous sinus thrombosis with thrombocytopenia after Ad26.COV2.S
vaccination, March 2 to April 21, 2021. _JAMA_ 325, 2448–2456 (2021). CAS PubMed Google Scholar * Graf, T. et al. Immediate high-dose intravenous immunoglobulins followed by direct
thrombin-inhibitor treatment is crucial for survival in Sars-Covid-19-adenoviral vector vaccine-induced immune thrombotic thrombocytopenia VITT with cerebral sinus venous and portal vein
thrombosis. _J. Neurol._ 268, 4483–4485 (2021). CAS PubMed PubMed Central Google Scholar * Pomara, C. et al. Post-mortem findings in vaccine-induced thrombotic thombocytopenia.
_Haematologica_ 106, 2291–2293 (2021). CAS PubMed PubMed Central Google Scholar * Hill, A., DeZern, A. E., Kinoshita, T. & Brodsky, R. A. Paroxysmal nocturnal haemoglobinuria. _Nat.
Rev. Dis. Prim._ 3, 17028 (2017). PubMed Google Scholar * Scheuerle, A. F., Serbecic, N. & Beutelspacher, S. C. Paroxysmal nocturnal hemoglobinuria may cause retinal vascular
occlusions. _Int. Ophthalmol._ 29, 187–190 (2009). PubMed Google Scholar * Tiede, A. et al. Prothrombotic immune thrombocytopenia after COVID-19 vaccine. _Blood_ 138, 350–353 (2021). CAS
PubMed PubMed Central Google Scholar * CDC COVID-19 Response Team. Preliminary estimates of the prevalence of selected underlying health conditions among patients with coronavirus disease
2019 — United States, February 12–March 28, 2020. _MMWR Morb. Mortal. Wkly Rep._ 69, 382–386 (2020). PubMed Central Google Scholar * Jordan, R. E., Adab, P. & Cheng, K. K. Covid-19:
risk factors for severe disease and death. _BMJ_ 368, m1198 (2020). PubMed Google Scholar * Wenzel, U. O., Kemper, C. & Bode, M. The role of complement in arterial hypertension and
hypertensive end organ damage. _Br. J. Pharmacol._ 178, 2849–2862 (2021). CAS PubMed Google Scholar * Lappegård, K. T. et al. A vital role for complement in heart disease. _Mol. Immunol._
61, 126–134 (2014). PubMed Google Scholar * Arason, G. J. et al. Smoking and a complement gene polymorphism interact in promoting cardiovascular disease morbidity and mortality. _Clin.
Exp. Immunol._ 149, 132–138 (2007). CAS PubMed PubMed Central Google Scholar * Copenhaver, M., Yu, C.-Y. & Hoffman, R. P. Complement components, C3 and C4, and the metabolic
syndrome. _Curr. Diabetes Rev._ 15, 44–48 (2018). Google Scholar * Goronzy, J. J. & Weyand, C. M. Understanding immunosenescence to improve responses to vaccines. _Nat. Immunol._ 14,
428–436 (2013). CAS PubMed PubMed Central Google Scholar * Pietrobon, A. J., Teixeira, F. M. E. & Sato, M. N. Immunosenescence and inflammaging: risk factors of severe COVID-19 in
older people. _Front. Immunol._ 11, 579220 (2020). CAS PubMed PubMed Central Google Scholar * da Costa, M. G. et al. Age and sex-associated changes of complement activity and complement
levels in a healthy Caucasian population. _Front. Immunol._ 9, 2664 (2018). Google Scholar * Cardoso, A. L. et al. Towards frailty biomarkers: candidates from genes and pathways regulated
in aging and age-related diseases. _Ageing Res. Rev._ 47, 214–277 (2018). CAS PubMed Google Scholar * Shi, Q. et al. Complement C3 deficiency protects against neurodegeneration in aged
plaque-rich APP/PS1 mice. _Sci. Transl. Med._ 9, eaaf6295 (2017). PubMed PubMed Central Google Scholar * Naito, A. T. et al. Complement C1q activates canonical Wnt signaling and promotes
aging-related phenotypes. _Cell_ 149, 1298–1313 (2012). CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS Research described in this article was supported (in
part) by the Intramural Research Program of the US National Institute of Diabetes and Digestive and Kidney Diseases (project number ZIA/DK075149 (B.A).) and the US National Heart, Lung, and
Blood Institute (project number ZIA/Hl006223 (C.K.)), by an FWO COVID-19 grant to B.N.L., and by a COVID-19 grant from the Fondazione Italcementi Cav. Lav. CARLO PESENTI to M.N. AUTHOR
INFORMATION AUTHORS AND AFFILIATIONS * Immunoregulation Section, Kidney Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health,
Bethesda, MD, USA Behdad Afzali * Istituto di Ricerche Farmacologiche “Mario Negri”, Clinical Research Center for Rare Diseases “Aldo e Cele Daccò”, Ranica, Italy Marina Noris * “Centro Anna
Maria Astori”, Bergamo, Italy Marina Noris * Laboratory of Immunoregulation and Mucosal Immunology, VIB-UGent Center for Inflammation Research, Ghent, Belgium Bart N. Lambrecht * Department
of Internal Medicine and Pediatrics, Ghent University, Ghent, Belgium Bart N. Lambrecht * Department of Pulmonary Medicine, Erasmus University Medical Center, Rotterdam, Netherlands Bart N.
Lambrecht * Complement and Inflammation Research Section (CIRS), National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA Claudia Kemper * Institute for
Systemic Inflammation Research, University of Lübeck, Lübeck, Germany Claudia Kemper Authors * Behdad Afzali View author publications You can also search for this author inPubMed Google
Scholar * Marina Noris View author publications You can also search for this author inPubMed Google Scholar * Bart N. Lambrecht View author publications You can also search for this author
inPubMed Google Scholar * Claudia Kemper View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS The authors contributed equally to all aspects of
the article. CORRESPONDING AUTHORS Correspondence to Behdad Afzali, Marina Noris, Bart N. Lambrecht or Claudia Kemper. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Reviews Immunology_ thanks M. Bosmann and the other, anonymous, reviewer(s) for their contribution to the peer
review of this work. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Afzali, B., Noris, M., Lambrecht, B.N. _et al._ The state of complement in COVID-19. _Nat Rev Immunol_ 22, 77–84 (2022).
https://doi.org/10.1038/s41577-021-00665-1 Download citation * Accepted: 25 November 2021 * Published: 15 December 2021 * Issue Date: February 2022 * DOI:
https://doi.org/10.1038/s41577-021-00665-1 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative