- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Pathogenic mutations in LRRK2 cause Parkinson’s disease (PD). The G2019S variant is the most common, which results in abnormally high kinase activity. Compounds that target LRRK2 kinase
activity are currently being developed and tested in clinical trials. We recently found that G2019S LRRK2 causes mitochondrial DNA (mtDNA) damage and treatment with multiple classes of LRRK2
kinase inhibitors at concentrations associated with dephosphorylation of LRRK2 reversed mtDNA damage to healthy control levels. Because maintaining the normal function of LRRK2 in
heterozygous G2019S LRRK2 carriers while specifically targeting the G2019S LRRK2 activity could have an advantageous safety profile, we explored the efficacy of a G2019S mutant selective
LRRK2 inhibitor to reverse mtDNA damage in G2019S LRRK2 models and patient cells relative to non-selective LRRK2 inhibitors. Potency of LRRK2 kinase inhibition by EB-42168, a G2019S mutant
LRRK2 kinase inhibitor, and MLi-2, a non-selective inhibitor, was determined by measuring phosphorylation of LRRK2 at Ser935 and/or Ser1292 using quantitative western immunoblot analysis.
The Mito DNADX assay, which allows for the accurate real-time quantification of mtDNA damage in a 96-well platform, was performed in parallel. We confirmed that EB-42168 selectively inhibits
LRRK2 phosphorylation on G2019S LRRK2 relative to wild-type LRRK2. On the other hand, MLi-2 was equipotent for wild-type and G2019S LRRK2. Acute treatment with EB-42168 inhibited LRRK2
phosphorylation and also restored mtDNA damage to healthy control levels. We further investigated the relationship between LRRK2 kinase activity, mtDNA damage and mitophagy. Levels of mtDNA
damage caused by G2019S LRRK2 were fully re-established within 2 h of a LRRK2 inhibitor wash out and recovery experiment, indicating the mtDNA damage phenotype is highly dynamic. G2019S
LRRK2 mitophagy defects were not alleviated with LRRK2 kinase inhibition, suggesting that mitophagy is not mechanistically regulating LRRK2 kinase-mediated reversal of mtDNA damage in this
acute timeframe. Abrogation of mtDNA damage with the mutant selective tool inhibitor EB-42168 demonstrates the potential of a precision medicine approach for LRRK2 G2019S PD. Levels of mtDNA
damage may serve as a potential pharmacodynamic biomarker of altered kinase activity that could be useful for small molecule development and clinical trials.
Parkinson’s disease (PD) is the most common age-related movement neurodegenerative disorder. This chronic disease is characterized by progressive motor disability, non-motor symptoms and
decreased quality of life. Therapeutic strategies currently available rely on dopamine replacement, but do not slow or stop the progression of the disease and can lead to motor
complications. While these drugs may be useful in managing symptoms, there are no disease-modifying therapies that target the underlying pathogenic mechanisms of disease; thereby this
remains a significant and urgent unmet medical need for PD patients. In 2004, coding variants were first identified in Leucine-rich repeat kinase 2 (LRRK2), with missense mutations in LRRK2
now established as the most common genetic cause of autosomal-dominant PD1,2,3,4,5. LRRK2 is a large and complex protein, with two functional enzymatic domains—a Ras-like GTPase and a
serine-threonine kinase domain. The most frequent pathogenic LRRK2 mutation is the Gly2019Ser (G2019S) LRRK2 variant, which results in a modest increase in kinase activity6,7,8.
Interestingly, in addition to the LRRK2 G2019S variant, all pathogenic missense LRRK2 mutations also augment kinase activity, displaying higher levels of the autophosphorylation Ser1292
residue of LRRK29,10,11,12. Therefore, the toxic gain-of-function of LRRK2 kinase activity is strongly implicated as the cause of pathogenicity13. Consistent with these findings, a
neuroprotective effect of LRRK2 kinase inhibitors has been demonstrated in PD-relevant cell and rodent models14. Thus, LRRK2 represents a promising therapeutic target for disease
modification and several LRRK2 kinase inhibitors are in clinical development and/or trials.
Recently, the results of testing DNL201, a CNS-penetrant, selective, ATP-competitive, small-molecule LRRK2 kinase inhibitor in early phase human clinical trials were reported15. Importantly,
DNL201, in humans was able to cross the blood brain barrier, based on measurements of inhibitor concentrations in cerebrospinal fluid. Concomitant blood-based markers demonstrated a
dose-dependent inhibition of LRRK2 kinase by DNL20115. However, on-target safety liabilities for LRRK2 inhibitors have been identified in preclinical models. Several studies have shown lung
and/or kidney dyshomeostasis in rodent and nonhuman primate models either lacking LRRK2 or following treatment with LRRK2 kinase inhibitors, some of which may be
reversible16,17,18,19,20,21,22,23,24,25,26. Of note, lung function did not seem to be impacted at the DNL201 doses tested in single-ascending dose or multiple-ascending dose (10 days)
cohorts in healthy volunteers or in patients with PD. Longer-term monitoring will be critical to assess safety of chronic dosing with LRRK2 kinase inhibitors on lung and kidney function, as
well as in PD patients with clinically significant history of pulmonary and kidney disease, which are often co-morbidities in this population. Heterozygous loss-of-function variants at the
LRRK2 locus do not increase PD risk and have no apparent overt deleterious health consequences27,28. However, somatic LRRK2 mutations in breast cancer are associated with high-risk features
and reduced patient survival29, consistent with a potential risk for lung adenocarcinoma with reduced LRRK2 levels30, emphasizing the concern of targeting LRRK2 in humans. Additionally,
since the majority of PD patients carrying the G2019S LRRK2 variant are heterozygous, a precision medicine approach of selectively reducing G2019S LRRK2 kinase activity while sparing
wild-type LRRK2 physiological function could offer a potential safety advantage in this patient population.
Measuring endogenous LRRK2 kinase activity by monitoring LRRK2 autophosphorylation at serine 1292 (pSer1292) has been particularly challenging in endogenous expression systems, but can be
robustly detected in overexpressed cellular models or with a fractionation-based enrichment technique in G2019S LRRK2 but not wild-type tissue11,31,32. The phosphorylation of downstream
substrates, including Rab GTPase substrates, are difficult to measure reliably due to low stoichiometry4,5,33,34. The disease relevance of phospho-substrates of LRRK2, such as Rab10, is
unknown, and paradoxically, Rab10 phosphorylation is not elevated with the PD G2019S LRRK2 variant, a finding reported by multiple groups35,36,37,38,39. Thus, limited markers for evaluating
basal G2019S LRRK2-related kinase activity exist. The indirect but kinase conformation dependent phosphorylation site at serine 935 (pSer935) is most widely used for measuring LRRK2 kinase
inhibition, despite not faithfully reflecting LRRK2 protein kinase activity12,40,41. Further optimizing and developing tools to measure endogenous LRRK2 kinase activity and inhibition in
vivo, is a critical unmet need for future clinical trials. Mitochondrial function is significantly impacted in PD42. Importantly, mitochondrial DNA (mtDNA) homeostasis is disrupted in both
idiopathic and familial PD cases, including those with LRRK2 mutations, and are associated with mtDNA damage43,44,45,46,47,48,49,50. We found that increased mtDNA damage in PD
patient-derived immune cells heterozygous for the G2019S LRRK2 mutation was abrogated following treatment with multiple classes of LRRK2 kinase inhibitors and correlated with measures of
pSer935 LRRK2 dephosphorylation44,49. Therefore, measurement of mtDNA damage may serve as a surrogate for LRRK2 kinase activity and consequently of kinase inhibitor activity. Recently LRRK2
kinase inhibitors with significant selectivity for mutant (G2019S) LRRK2 compared to wild-type LRRK2 have been discovered, but it is unknown if these compounds have similar effects on G2019S
LRRK2 dependent mtDNA damage20,51,52,53,54,55,56. It is not known whether the accumulation of LRRK2 G2019S-dependent mtDNA damage is solely due to the pathogenic mutant allele, or the
differential stoichiometry between WT-mutant and mutant-mutant protein pairing in the LRRK2 dimer, which may impact structure and enzymatic activation. Thus, we have now extended these
studies and investigated whether using a selective LRRK2 kinase inhibitor was able to reverse mtDNA damage in heterozygous G2019S cells at inhibitor concentrations shown to preferentially
inhibit G2019S kinase while sparing wild-type kinase activity.
The aim of the present study was to compare the highly selective G2019S LRRK2 inhibitor (EB-42168) with the non-selective LRRK2 inhibitor (MLi-2) on mtDNA damage levels in LRRK2 models and
patient cells. LRRK2 phosphorylation was examined in parallel to establish target engagement and compare these biomarker outcomes in response to the selective G2019S LRRK2 inhibitor,
EB-42168. We further investigated the relationship between LRRK2 kinase activity, mtDNA damage and mitophagy. Measuring mtDNA damage together with LRRK2 phosphorylation is an innovative
biomarker approach to determine LRRK2 kinase activity and may be helpful in considering the drug efficacy of compounds targeting hyperactive kinase activity in G2019S LRRK2 PD patients in
the context of a clinical trial.
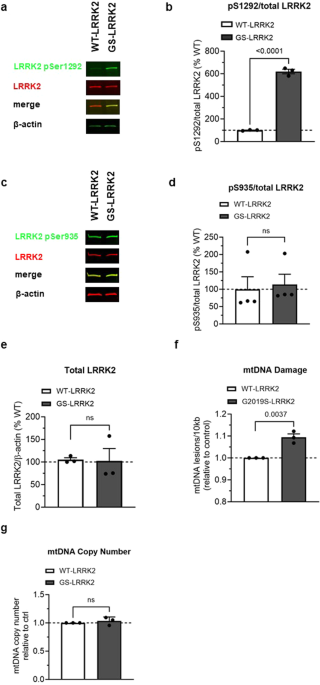
LRRK2 kinase activity was assessed by measuring the relative levels of autophosphorylation at pSer1292 by immunoblot in HEK293 cells stably transfected with human LRRK2 or the G2019S variant
of human LRRK2 (named WT-LRRK2 and G2019S-LRRK2, respectively)10. pSer1292 was increased ~6 fold in G2019S-LRRK2 cells compared to WT-LRRK2, consistent with previous reports (Fig. 1a, b20).
Levels of both pSer935 and total LRRK2 were similar between the WT-LRRK2 and G2019S-LRRK2 expressing cell lines (Fig. 1c–e). The similar expression levels of LRRK2 between the two cell
lines allowed for a direct comparison of the two genotypes on mtDNA damage phenotypes. G2019S-LRRK2 induced a higher level of mtDNA damage relative to WT-LRRK2 expressing cells (Fig. 1f),
without any changes in the steady state of mtDNA copy number (Fig. 1g). These results are consistent with our prior study showing that mtDNA damage was increased in primary midbrain neurons
overexpressing G2019S LRRK2 compared to wild-type LRRK2, a kinase dead LRRK2 mutant or the GFP expressing control44.
a Representative western blot of WT-LRRK2 or G2019S-LRRK2 overexpressing cells show expression of LRRK2 pSer1292 and full-length LRRK2. β-actin was blotted as a loading control. b
Quantification of western blots demonstrate ~6-fold increase of LRRK2 pSer1292 in G2019S-LRRK2 compared to WT-LRRK2 expressing cells. c Representative western blots of WT-LRRK2 or
G2019S-LRRK2 overexpressing cells show expression of LRRK2 pSer935 and full-length LRRK2. β-actin was blotted as a loading control. d Quantification of western blots demonstrate no
difference of LRRK2 pSer935 levels between G2019S-LRRK2 and WT-LRRK2 expressing cells. e Quantification of western blots demonstrate no difference of LRRK2 protein levels between the two
cell lines. f Mitochondrial DNA damage was increased in G2019S-LRRK2 relative to WT-LRRK2 expressing cells. g The differences in mtDNA damage between the cell lines were not attributable to
changes in steady state mtDNA levels. Data are presented as mean ± SEM. n = 3–4 replicates. Statistical significance was determined by unpaired t-test for all analyses. GS-LRRK2
G2019S-LRRK2, WT-LRRK2 wild-type LRRK2, ns non-significant.
The LRRK2 kinase inhibitor EB-42168 was previously shown to inhibit G2019S LRRK2 100-fold more potently than wild-type LRRK220. MLi‐2, by contrast, was equipotent in recombinant cell lines
expressing WT-LRRK2 or G2019S-LRRK2, and can therefore serve as a non-selective LRRK2 kinase inhibitor control20,24. We assessed the potency of EB-42168 and MLi-2 to inhibit pSer935 and
pSer1292 LRRK2 in WT or G2019S- LRRK2 expressing cells in compound titration experiments with concentrations ranging from 10 nM–1 μM. At the lowest concentration of MLi-2 tested (10 nM),
pSer935 was significantly decreased by ~75% in both the WT-LRRK2 and G2019S-LRRK2 cells (Fig. 2a, b). At concentrations higher than 100 nM, MLi-2 showed nearly complete ablation of
phosphorylation at Ser935, with 5000 nM in WT LRRK2 cells and 54 nM in G2019S LRRK2 cells. Therefore, EB-42168 was >90-fold more potent on G2019S LRRK2 which is in line with previous
reports20,51.
a Representative western blot of WT-LRRK2 or G2019S-LRRK2 cells treated with DMSO, 10 nM, 100 nM or 1 µM MLi-2 for 2 h and assessed for LRRK2 pSer935 and full-length LRRK2. b Representative
western blot of WT-LRRK2 or G2019S-LRRK2 cells treated with DMSO, 10 nM, 100 nM or 1 µM EB-42168 for 2 h and assessed for LRRK2 pSer935 and full-length LRRK2. c Quantification of western
blots demonstrate a 70%, 90% and 95% decrease, respectively, in LRRK2 pSer935 levels with increasing concentration of MLi-2 in WT-LRRK2 expressing cells. Quantification of western blots
demonstrate an 80%, 96% and 97% decrease respectively in LRRK2 pSer935 levels with increasing concentration of MLi-2 in G2019S-LRRK2 expressing cells. d Quantification of western blots
demonstrate no change in LRRK2 pSer935 levels with EB-42168 treatment in WT-LRRK2 expressing cells. Quantification of western blots demonstrate a 65% and 95% decrease respectively in LRRK2
pSer935 levels with 100 nM and 1 µM of EB-42168 in G2019S-LRRK2 expressing cells. Data are presented as mean ± SEM. n = 3 replicates. (*p







