- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Homozygous or compound heterozygous (biallelic) variants in _PRKN_ are causal for PD with highly penetrant symptom expression, while the much more common heterozygous variants may
predispose to PD with highly reduced penetrance, through altered mitochondrial function. In the presence of pathogenic heterozygous variants, it is therefore important to test for
mitochondrial alteration in cells derived from variant carriers to establish potential presymptomatic molecular markers. We generated lymphoblasts (LCLs) and human induced pluripotent stem
cell (hiPSC)-derived neurons from non-manifesting heterozygous _PRKN_ variant carriers and tested them for mitochondrial functionality. In LCLs, we detected hyperactive mitochondrial
respiration, and, although milder compared to a biallelic _PRKN_-PD patient, hiPSC-derived neurons of non-manifesting heterozygous variant carriers also displayed several phenotypes of
altered mitochondrial function. Overall, we identified molecular phenotypes that might be used to monitor heterozygous _PRKN_ variant carriers during the prodromal phase. Such markers might
also be useful to identify individuals at greater risk of eventual disease development and for testing potential mitochondrial function-based neuroprotective therapies before
neurodegeneration advances. SIMILAR CONTENT BEING VIEWED BY OTHERS GENOMIC, TRANSCRIPTOMIC, AND METABOLOMIC PROFILES OF HIPSC-DERIVED DOPAMINE NEURONS FROM CLINICALLY DISCORDANT BROTHERS
WITH IDENTICAL _PRKN_ DELETIONS Article Open access 29 June 2022 MULTIPLE ALTERATIONS IN GLUTAMATERGIC TRANSMISSION AND DOPAMINE D2 RECEPTOR SPLICING IN INDUCED PLURIPOTENT STEM CELL-DERIVED
NEURONS FROM PATIENTS WITH FAMILIAL SCHIZOPHRENIA Article Open access 25 October 2021 ORTHOGONAL ANALYSIS OF MITOCHONDRIAL FUNCTION IN PARKINSON’S DISEASE PATIENTS Article Open access 03
April 2024 INTRODUCTION Parkinson´s disease (PD) is a neurodegenerative movement disorder, with both genetic and environmental factors playing an important role in disease onset, and age
being the biggest risk factor for developing the disease1,2. PD is clinically characterized by cardinal motor manifestations3,4, which are often preceded by non-motor symptoms for years or
even decades2. Even though most PD cases are classified as sporadic, a minority of cases (~10%) have a clear monogenic, autosomal dominant, or recessive, inheritance pattern5. To date, rare
variants in more than 20 genes have been reported to cause PD, although only about half of them are unequivocally linked to PD5. Impairment of mitochondrial function has been highlighted as
a key mechanism of this pathology, and several genes implicated in familial forms of PD, including _PRKN (Parkin), PINK1, PARK7 (DJ-1)_, and _SNCA_ (alpha-synuclein) are involved in the
regulation of mitochondrial function6. _PRKN_-linked PD is the most common form of early-onset PD across all ethnic groups, representing about 10–20% of cases with the age of onset (AAO)
from 40–50 years7,8 and up to 42.2% of cases with an AAO ≤20 years9. Homozygous or compound heterozygous (biallelic) variants in _PRKN_ are causal for PD with highly penetrant symptom
expression and early AAO, but may manifest with late onset in a subset of cases8,10. The much more common heterozygous variants in this gene may predispose to PD with highly reduced
penetrance11,12. However, the role of heterozygous _PRKN_ variants as causative or susceptibility factors for PD is still under vivid debate: two studies have found an increased risk for PD
in carriers of heterozygous _PRKN_ variants as compared to controls13,14, while two other studies with almost complete genotyping of _PRKN_ for copy number and single nucleotide
polymorphisms found that heterozygous _PRKN_ variants do not contribute to overall disease risk15,16. In the latter papers, however, there was no possibility to assess subtle, subclinical
phenotypes that might be present in the carriers. Heterozygous variant carriers present an extremely variable phenotype expressivity ranging from no clinical signs to subtle motor signs,
which do not allow for a definitive clinical diagnosis of PD, to overt symptoms of PD10,17,18. Importantly, a dopaminergic deficit has been clearly observed by positron emission tomography
in studies with asymptomatic heterozygous _PRKN_ variant carriers19,20,21, with a mean annual reduction in the putamen and caudate of 0.56 and 0.62%, respectively, compared to 0.5 and 2% in
biallelic _PRKN_ mutation carriers22. Pathogenic variants in _PRKN_ can affect Parkin function through different mechanisms impairing its activity, stopping the translation of a functional
protein, rendering the protein insoluble and enhancing aggregation, hampering protein folding and stability, and/or affecting its ability to bind to cofactors and substrates23. Overall, it
is a complex protein exerting numerous effects, which undeniably contribute to cellular homeostasis and survival. In particular, Parkin deficiency was proven to disrupt mitochondrial
performance, demonstrating the crucial role of these organelles in the onset of _PRKN_-linked disease24,25. Parkin protein is an E3 ubiquitin ligase, which is localized in the cytoplasm and
translocates to the mitochondria upon specific stimulation26,27. It ubiquitinates many substrate proteins, including itself, for proteasomal degradation or signaling processes28,29.
Following mitochondrial membrane depolarization, Parkin is recruited to mitochondria and activated by PINK1, resulting in the ubiquitination of outer mitochondrial membrane proteins,
including Mitofusin-1 and Mitofusin-2, which are essential for mitochondrial fusion30. Concurrently with proteasomal degradation of mitochondrial substrate proteins, components of the
autophagy machinery are recruited, leading to the selective autophagic removal of the damaged organelle, a process known as mitophagy31,32. Additionally, Parkin is involved in the activation
of mitochondrial biogenesis through ubiquitination and subsequent degradation of PARIS (parkin interacting substrate), a transcriptional repressor of the master regulator of mitochondrial
biogenesis, PGC1-α (peroxisome proliferator-activated receptor gamma coactivator 1α)33 or via metabolic modulation of the Sirtuin1/PGC1-α pathway34. Furthermore, Parkin is involved in
mitochondrial DNA (mtDNA) maintenance34,35. Interestingly, after crossing the Mutator mouse, which possesses a proofreading-deficient form of POLG, the polymerase responsible for mtDNA
replication, resulting in accumulation of mtDNA deletions, with a _PRKN_ knock-out mouse, which both by themselves do not show neurodegeneration, the resulting double knockout displays
obvious mitochondrial dysfunction and PD pathology. This was linked to a greater predicted pathogenicity score for mtDNA variants35. Moreover, in iPSC-derived POLG-mutant neurons deficient
in Parkin, mtDNA dyshomeostasis coincided with the release of mtDNA molecules into the extracellular space, where they can propagate inflammation34. Considering that the frequency of
heterozygous _PRKN_ variant carriers in the population is up to 4%11,36, and that some of these carriers may eventually develop the disease, it is important to further elucidate the role of
pathogenic heterozygous variants in this recessive gene, by testing the causal link between the heterozygous variants and expressivity of molecular phenotypes. Therefore, in this study, we
sought to evaluate alterations of mitochondrial integrity, including disparities at the level of mtDNA and mitochondrial function, in blood and cellular models of non-manifesting
heterozygous _PRKN_ variant carriers. RESULTS ENHANCED MITOCHONDRIAL RESPIRATION IN INTACT LYMPHOBLASTS OF HETEROZYGOUS _PRKN_ EXON 7 DELETION CARRIERS From 20 individuals (11 carriers of a
heterozygous _PRKN_ exon 7 deletion and 9 controls) (Fig. 1 and Supplementary Table 1), lymphoblastoid cell lines (LCLs) (Fig. 2a) were derived, and protein levels of Parkin were estimated
using an antibody able to recognize only Parkin with an intact C-terminus (Supplementary Fig. 1a). In heterozygous mutation carriers, protein levels were ∼50% of those detected in controls.
Oxygen consumption was measured by means of high-resolution respirometry; representative traces of oxygen consumption in intact LCLs illustrate the applied multiple
substrate-uncoupler-inhibitor titration (SUIT) protocol (Fig. 2b). Under physiological conditions of non-permeabilized LCLs, individuals carrying a heterozygous exon 7 deletion in _PRKN_
present a significantly increased oxygen consumption, identified in the routine respiratory state (_p_ = 0.023, 3.03 vs 2.87) (Fig. 2c), suggesting hyperactive mitochondrial respiration in
LCLs derived from heterozygous variant carriers. For subsequent respiratory states, including proton leak and maximal respiratory capacity (Fig. 2d and Supplementary Table 2), group
differences were not statistically significant. Accordingly, ATP turnover, calculated by the decrease in oxygen consumption upon injection of the ATP synthase inhibitor oligomycin, suggests
an increased ATP production in the LCLs of the heterozygous variant carriers. These data indicate hyperactive mitochondrial respiration in blood-derived cells of carriers of a pathogenic
heterozygous _PRKN_ variant. LYMPHOBLASTS OF HETEROZYGOUS EXON 7 DELETION CARRIERS EXHIBIT ELEVATED MROS PRODUCTION AND UNALTERED MITOCHONDRIAL MEMBRANE POTENTIAL To measure mitochondrial
ROS (mROS) production, LCLs of heterozygous exon 7 deletion carriers and controls were stained with MitoSox Red dye, and fluorescence intensity was measured by flow cytometry. As depicted in
Fig. 2e, f, the quantification of the MitoSox Red signal showed an increase of mROS in the LCLs of heterozygous variant carriers, compared to the control group (_p_ = 0.031, 22.90 vs.
15.55, respectively). We further evaluated the mitochondrial membrane potential (MMP), which is considered an important parameter of mitochondrial integrity37, by using the cell-permeant dye
TMRM (tetramethylrhodamine methyl ester). Flow cytometry analysis of the fluorescence intensity showed no significant difference in the LCLs of the carriers compared to the control group
(_p_ = 0.604, 718.7 vs. 755.7) (Fig. 2h, i). Collectively, increased mROS production, but unchanged MMP, are in line with the enhanced mitochondrial respiratory activity and enhanced
electron transport observed in the heterozygous carrier group. HUMAN IPSC-DERIVED NEURONS OF HETEROZYGOUS VARIANT CARRIERS SUGGEST A REDUCED OXYGEN CONSUMPTION RATE We further investigated
mitochondrial phenotypes in DA neurons of heterozygous exon 7 deletion carriers as a second cell type, which might be more susceptible to changes induced by a _PRKN_ variant, since they
might rely predominantly on mitochondrial oxidative metabolism38,39,40. We reprogrammed peripheral blood mononuclear cells (PBMCs) of four individuals (two carriers of a heterozygous exon 7
deletion and two controls; for three of them, we also generated LCLs) to human induced pluripotent stem cells (hiPSCs)41 (Fig. 1 and Supplementary Table 1) and differentiated them to DA
neurons (Supplementary Fig. 1b, c). We also included a homozygous _PRKN_-PD hiPSC line (exon 3 deletion or 1-bp deletion in exon 9) and an additional control line in all experiments
(Supplementary Table 1). Protein levels of Parkin were estimated using an antibody able to recognize only Parkin with an intact C-terminus. In hiPSC-derived DA neurons from heterozygous
mutation carriers, protein levels were ∼50% of those detected in controls, and no Parkin protein was detectable in the homozygous _PRKN_ mutation carrier lacking the C-terminal part of
Parkin (Supplementary Fig. 1b). To measure oxidative phosphorylation activity in hiPSC-derived neurons (Fig. 3a), we quantified extracellular oxygen consumption rate (OCR) under basal
conditions (routine respiration) and upon treatment with the protonophore CCCP (carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone), leading to mitochondrial uncoupling (Fig. 3b). The two
non-manifesting individuals carrying a heterozygous _PRKN_ variant, and the homozygous _PRKN_-PD patient showed a trend towards a decreased basal OCR level compared to the control group
(controls 0.73 ± 0.1 vs. 0.55 ± 0.1 vs. 0.45 ± 0.17; _p_ = 0.48, _p_ = 0.29) (Fig. 3c). When challenging the neurons with CCCP to induce maximal respiration, both heterozygous variant
carriers and the homozygous _PRKN_-PD line revealed a significant decrease of oxygen consumption (controls 1.18 ± 0.13 vs. 0.79 ± 0.1 vs. 0.66 ± 0.15; _p_ = 0.044, _p_ = 0.02, respectively)
(Fig. 3d). Spare respiratory capacity indicates the mitochondrial capability to respond to a chronic mitochondrial insult, which might be the presence of a single _PRKN_ variant. This
parameter was significantly reduced for the homozygous _PRKN_-PD line (1.26 ± 0.15 vs. 0.70 ± 0.17; _p_ = 0.026) but also for the heterozygous _PRKN_ variant lines (1.26 ± 0.15 vs. 0.83 ±
0.15; _p_ = 0.049) (Fig. 3e). The decrease in OCR levels at maximal respiration as well as for the spare respiratory capacity was seemingly more pronounced in the PD patient line with
homozygous _PRKN_ variants as compared to the heterozygous variant carriers, although the difference between these two groups was not significant (_p_ = 0.57, _p_ = 0.65, respectively).
OXIDATIVE STRESS LEVELS IN HIPSC-DERIVED NEURONS OF HETEROZYGOUS VARIANT CARRIERS To explore oxidative stress in hiPSC-derived neurons of individuals carrying a heterozygous variant in
_PRKN_, we measured mitochondrial superoxide (mROS) by staining with the fluorogenic dye MitoSox Red. As depicted in Fig. 4a, neurons obtained from the heterozygous variant carriers as well
as from the PD patient carrying a homozygous _PRKN_ variant, displayed MitoSox-positive mitochondria with a significantly higher mean fluorescence intensity as compared to the control
neurons (0.97 ± 0.05 vs. 1.16 ± 0.05 vs. 1.22 ± 0.06; _p_ = 0.010, _p_ = 0.007, respectively) (Fig. 4b). When comparing mROS levels between the heterozygous variant carriers and the PD
patient, no statistically significant difference was detected (_p_ = 0.594. 1.16 ± 0.05 vs. 1.22 ± 0.06, respectively) but the direction of the effect observed for the respiration
experiments was confirmed. This suggests increased oxidative stress levels in the hiPSC-derived neurons carrying a heterozygous _PRKN_ variant compared to the controls, similar to the
phenotype observed in the homozygous _PRKN_-PD patient. mROS levels are partly regulated by the mitochondrial antioxidant enzyme superoxide dismutase 2 (SOD2, MnSOD)42,43. To investigate the
regulation of mROS in our lines, we quantified the protein levels of SOD2 by means of Western blot (Fig. 4c). Interestingly, SOD2 levels in the patient carrying a homozygous _PRKN_ variant
were significantly decreased when compared to the control group (1.00 ± 0.07 vs. 0.61 ± 0.09, _p_ = 0.005) (Fig. 4d). For the individuals carrying a heterozygous variant, a trend towards
reduction was observed compared to the control group (0.84 ± 0.07 vs. 1.00 ± 0.07; _p_ = 0.169), which might suggest deficiencies in the defense mechanism against oxidative stress in
individuals either lacking Parkin or showing reduced Parkin levels. MITOCHONDRIAL MEMBRANE POTENTIAL ALTERATIONS IN HIPSC-DERIVED NEURONS OF HETEROZYGOUS VARIANT CARRIERS MMP was assessed
using the cell-permeant dye TMRM and live imaging confocal microscopy (Fig. 4e). Mean fluorescence intensity analysis revealed a significant reduction of MMP for the heterozygous _PRKN_
variant carriers (_p_ = 0.049) as well as for the homozygous _PRKN_-PD patient neurons (_p_ = 0.008), compared to the control group (1.04 ± 0.04 vs. 0.93 ± 0.03 vs. 0.85 ± 0.05) (Fig. 4f).
Taken together, these results indicate an altered mitochondrial functionality in the presence of disease caused by homozygous _PRKN_ variants, which is detectable also in non-manifesting
carriers of a heterozygous _PRKN_ variant. MITOCHONDRIAL NETWORK MORPHOLOGY IS UNAFFECTED IN HIPSC-DERIVED NEURONS OF HETEROZYGOUS VARIANT CARRIERS IN _PRKN_ Considering the functional
mitochondrial impairments observed so far in the neuronal model, we performed a morphological analysis of the mitochondrial network by staining with the fluorescent dye Mitotracker green
(MTG), which specifically localizes to mitochondria regardless of their membrane potential (Fig. 5a). hiPSC-derived neurons from the patient carrying a homozygous _PRKN_ variant revealed an
increased mitochondrial fragmentation (quantified as the total length of the modeled mitochondrial network divided by the number of unconnected parts), when compared to the control group
(0.96 ± 0.11 vs. 0.59 ± 0.13; _p_ = 0.044), while heterozygous _PRKN_ variant carriers showed a trend towards increased mitochondrial fragmentation without a statistically significant group
change (0.96 ± 0.11 vs. 0.78 ± 0.11; _p_ = 0.313) (Fig. 5b). The direction of the effect of these data coincides with the altered mitochondrial functionality observed in hiPSC-derived
neurons of diseased homozygous and non-manifesting single variant carriers in _PRKN_. ABUNDANCE OF OUTER MITOCHONDRIAL MEMBRANE PROTEINS IN HIPSC-DERIVED NEURONS OF HETEROZYGOUS _PRKN_
VARIANT CARRIERS The general ubiquitination of outer mitochondrial membrane (OMM) proteins, such as Mitofusin-2 (MFN2), by Parkin is one of the early steps in mitophagy30,44,45,46, and
Mitofusin-2 abundance was found to be decreased upon mitochondrial depolarization in induced neurons with endogenous Parkin levels47. We, therefore, evaluated MFN2 protein levels upon
induction of mitochondrial depolarization with CCCP in hiPSC-derived neurons (Fig. 6a), which were revealed to be significantly higher in the homozygous _PRKN_-PD line and the heterozygous
variant carrier lines, compared to the controls (90.2 ± 8.4 vs. 69.14 ± 8.4 vs. 41.9 ± 8.4; _p_ = 0.016, _p_ = 0.0007, respectively) (Fig. 6b), while there was no difference at basal
conditions (data not shown). Moreover, it was possible to observe a band above the non-modified MFN2 in the controls and heterozygous variant carriers, possibly corresponding to the
monoubiquitinated form of MFN2 (Fig. 6a). As an additional OMM protein, we further analyzed the levels of the mitochondrial import receptor subunit TOM70 (Fig. 6a), which upon depolarization
revealed a trend towards increased levels for the heterozygous carrier group compared to the control group (p = 0.101; 59.55 ± 12 vs. 31.03 ± 12.2 respectively), and a significant increase
for the homozygous _PRKN_-PD patient neurons (_p_ = 0.002), as compared to the controls (100.2 ± 14.1 vs. 31.03 ± 12.2) (Fig. 6c). These results indicate that carrying a variant in _PRKN_
leads to elevated outer membrane protein levels under stress conditions, thus possibly suggesting reduced ubiquitination, which might negatively impact mitophagy initiation or a general
increase in mitochondrial mass. MITOCHONDRIAL DNA ANALYSES IN NON-MANIFESTING INDIVIDUALS CARRYING A PATHOGENIC HETEROZYGOUS VARIANT IN _PRKN_ We evaluated mitochondrial DNA (mtDNA) copy
number in the blood of non-manifesting carriers (_n_ = 341) of 17 pathogenic heterozygous _PRKN_ variants, identified previously11, compared to controls (_n_ = 8445, PD gene
mutation-negative). This analysis suggests a trend towards higher levels in the group of variant carriers (_p_ = 0.067, median carriers 142.2 vs. median of controls 134.95, respectively)
(Supplementary Fig. 2a), while in the derived LCLs of a subgroup of 11 variant carriers and nine controls, no difference was detected (_p_ = 0.602, median carriers 1970 vs. median controls
2104, respectively) (Supplementary Fig. 2d). The same was true for the mtDNA copy number of a subset of hiPSC-derived neurons of three controls, two variant carriers, and two _PRKN_-PD
patients (5140 vs. 3309 vs. 3523, respectively) (Supplementary Fig. 2e). Comparison of blood samples of also a small group of 29 idiopathic PD (iPD) patients showed a trend for reduced mtDNA
copy number levels (_p_ = 0.053, median iPD 112.08 vs. median heterozygous carriers 142.2). Additionally, mtDNA-associated 7S DNA levels and mtDNA major arc deletion levels were assessed in
a smaller group of heterozygous _PRKN_ mutation carriers (_n_ = 79) and controls (_n_ = 77), where no differences were detected between groups (_p_ = 0.53, 1.055 vs. 1.048, respectively and
_p_ = 0.73, 1.054 vs. 1.048, respectively) (Supplementary Fig. 2b, c). DISCUSSION In the present study, we show that non-manifesting individuals carrying a pathogenic heterozygous variant
in _PRKN_ present molecular phenotypes related to mitochondrial function. In peripheral blood of unaffected carriers of 17 heterozygous _PRKN_ variants and controls, identified previously in
the population-based CHRIS study11, a group comparison suggested a possible trend towards increased mtDNA copy number in heterozygous _PRKN_ variant carriers. Several markers of
mitochondrial functionality reveal significant differences between controls, non-manifesting carriers of a single pathogenic variant, and affected carriers of two variants in two cellular
models derived from the investigated individuals. These mitochondrial alterations might, therefore, further support an increased vulnerability in heterozygous variant carriers and might be
useful for identifying individuals at possibly greater risk of (subclinical) health problems or at worse, disease development. Although PD is principally a neurodegenerative disorder,
mounting data reveal molecular phenotypes in peripheral tissues suggesting the initiation of the pathology in the periphery. Of relevance, several studies show clear functional impairments
in blood cells of iPD patients48,49,50,51. Furthermore, abnormalities in mitochondrial function have also been reported in peripheral tissues of biallelic _PRKN_-linked PD
patients52,53,54,55. Our data collected in LCLs are characterized by enhanced mitochondrial respiration in non-manifesting individuals carrying a heterozygous _PRKN_ exon 7 deletion in the
routine (basal) respiratory state. Of note, a previous study reported a dramatically elevated basal mitochondrial respiration in iPD LCLs, which was observed independently of patient age,
disease progression, and disease severity, suggesting an early and stable switch to a hyperactive state56. More recently, whole-cell transcriptomes and proteomes of iPD and healthy control
LCLs showed downregulated transcripts for mitochondrial respiratory chain complexes but upregulated proteins, thereby suggesting that the elevated mitochondrial respiration observed in iPD
LCLs is translationally regulated57. Interestingly, two studies performed in _PRKN_-mutant skin fibroblasts from PD patients also reported a higher mitochondrial respiratory rate compared to
controls58,59. Our findings indicating an upregulation of basal mitochondrial respiration in LCLs of heterozygous _PRKN_ variant carriers are suggestive of a cellular compensatory mechanism
to overcome the changes occurring upon the presence of a _PRKN_ variant, aiming to preserve mitochondrial function. Furthermore, these results may suggest mitochondrial hyperactivity in
peripheral tissues as an early feature of mitochondrial dysfunction in PD. As expected, the heterozygous carrier group also presented an increased production of mROS, supporting the theory
that elevated mROS may result from increased electron transport rates. Likewise, elevated ROS coupled with increased mitochondrial respiration was reported in iPD LCLs56. Conversely, a
recent study conducted in _PRKN_-mutant LCLs reported an oxidative stress phenotype only upon treatment with toxins60. This suggests that a variant in _PRKN_ may confer vulnerability to the
system when subjected to environmental insults. In a new study61, an analysis of a larger set of _PRKN_/_PINK1_ biallelic carrier PD patients and heterozygous _PRKN_/_PINK1_ carriers both
with and without PD symptoms, mtDNA heteroplasmy along with major arc deletions and 7S DNA were identified as potential disease manifestation markers for _PINK1_/_PRKN_ PD. As mtDNA
heteroplasmy is reliably measurable, and as the results support the direct correlation between overt phenotype and mtDNA alterations, performing blood-based deep mtDNA sequencing may be an
important additional test to consider, especially in asymptomatic carriers. Increased mtDNA mutation load has functional consequences, which may provide additional functional assays
alongside mtDNA heteroplasmy measurements. In the study sample reported here, while for mtDNA-associated 7S DNA levels and mtDNA major arc deletions, there was no difference between groups,
the direction of the effect for both traits was in the same direction as that observed in this larger study sample61. Furthermore, we also detected a trend for increased levels of mtDNA copy
number in non-manifesting carriers of heterozygous _PRKN_ variants. This may suggest an accumulation of mtDNA molecules as a consequence of an impaired mitochondrial clearance process or a
compensatory increase in mitochondrial biogenesis due to the presence of a single variant in _PRKN_. In hiPSC-derived neurons of non-manifesting heterozygous _PRKN_ exon 7 deletion carriers,
which might be more susceptible to the genetic insult due to their predominant reliance on oxidative phosphorylation38,40,62,63,64, we found an overall decreased OCR, indicating a
significant difference when cells were forced to perform at maximal capacity. This goes in parallel with reduced OCR observed in a homozygous _PRKN-_PD line, confirming previous reports on
deficits in mitochondrial respiration and complex I activity in Parkin-deficient human neurons65,66,67. These findings suggest a respiratory impairment in neurons of heterozygous _PRKN_
variant carriers, possibly indicating that a disease phenotype may be unmasked when stressors are added to the single genetic variant. Furthermore, heterozygous carriers, as well as a
patient carrying two _PRKN_ variants, exhibited increased levels of mitochondrial-derived oxidants, thus corroborating a mitochondrial phenotype in the absence or partial deficiency of
Parkin. Increased oxidative stress levels were reported previously in Parkin-deficient iPSC-derived neurons64,68,69,70 and might give rise to a compensatory cellular response of antioxidant
effectors such as catalase and SOD1/271. Interestingly, our results suggest a reduction of mitochondrial SOD2 in complete or partial deficiency of Parkin. Accordingly, a decreased activity
of SOD2 in fibroblasts of PD patients with _PRKN_ variants, paired with increased mROS levels, was shown previously72. Furthermore, we observed a trend toward an increased mitochondrial
network fragmentation in the neurons of the heterozygous variant carriers, and an increased fragmentation in the homozygous _PRKN_-PD line, which confirms previous data65,66,70,73,74,75.
These data coincide with the altered mitochondrial function observed for the heterozygous _PRKN_ carrier group as well as for the _PRKN_-PD line. The role of _PRKN_ variants in mitophagy was
studied broadly in cells overexpressing Parkin but was also confirmed in hiPSC-derived neurons with endogenous Parkin levels64,65,69,73,76. As expected, upon mitochondrial membrane
depolarization, we found the levels of OMM proteins and Parkin substrates significantly increased in the neurons of the homozygous _PRKN_-PD line compared to the control neurons, consistent
with impaired Parkin-dependent degradation, as observed previously for both _PRKN_ and _PINK1_ knockout animals and mutant human fibroblasts45,46. Remarkably, however, we were able to also
observe an increase of OMM proteins in the hiPSC-derived neurons of non-manifesting individuals carrying a heterozygous _PRKN_ variant, compatible with reduced Parkin activity.
Interestingly, comparable results were obtained in fibroblasts of affected heterozygous _PRKN_ variant carriers, excluding a second cryptic pathogenic variant15. Altogether, when assessing
mitochondrial function in the hiPSC-derived DA neuron model, we were able not only to confirm an impaired mitochondrial phenotype observed previously in hiPSC-derived DA neurons of
homozygous _PRKN_-PD patients, but also to observe changes in mitochondrial homeostasis in clinically unaffected individuals carrying a heterozygous variant in _PRKN_, although these data
need further confirmation since they are based on only two variant carriers. In addition, all measurements were performed on a mixed population of neurons, hence subtle differences in TH+
neurons may be lost. Based on the results in LCLs and hiPSC-derived neurons, we postulate that a beginning disease process could be characterized by hyperactive respiration in peripheral,
rapidly turned over LCLs, where mitochondria seem to react to the insult, possibly as a compensatory mechanism, while in post-mitotic neurons, the damage of the insult might accumulate over
time, contributing eventually to disease manifestation. An additional difference between the LCL and iPSC-derived neuronal models might be based on their preferential reliance upon the
glycolysis and oxidative phosphorylation pathways for energy production. Although EBV strongly induces oxidative phosphorylation upon B-cell infection and transformation into LCLs77, these
cells might rely more on glycolysis78, and defects in the glycolysis pathway, providing pyruvate for the tricarboxylic acid cycle, might force a shift from glycolysis to mitochondrial
respiration in the heterozygous _PRKN_ variant carrier group. Overall, we consider this study as a proof of concept for detecting molecular phenotypes in cellular models derived from
heterozygous _PRKN_ variant carriers. In additional studies, it will be interesting to correlate molecular phenotypes with subclinical (prodromal) phenotypes. For this, larger sample sizes
will be needed79,80. In summary, two different cellular models of non-manifesting individuals carrying a heterozygous exon 7 deletion in _PRKN_ present detectable mitochondrial defects,
albeit with small effects. Notably, the altered molecular phenotype in the unaffected heterozygous variant carriers is seemingly milder as compared to PD patients lacking Parkin, possibly
suggesting a gene dosage effect. This likely mirrors the clear dosage effect observed in mtDNA heteroplasmy, major arc deletions, and 7S DNA reported recently61. However, questions as to
what extent these alterations can affect the overall function of the mitochondria to the point of translating into disease remain to be investigated. Additional factors, including age and
environment, may push single variant carriers over a gradual threshold, whereby carriers might convert from presenting mild, barely observable subclinical phenotypes to becoming symptomatic.
The gradient of mitochondrial function markers detected between heterozygous and homozygous _PRKN_ variant carriers suggests that there may indeed be a threshold of disrupted mitochondrial
function, past which a _PRKN_ variant carrier may convert to a disease state. It will therefore be important to study the degree to which mitochondrial protection factors may represent a
strategy to rescue mitochondrial function in the cell models of heterozygous variant carriers81,82. Such a strategy may delay, or possibly prevent, the conversion to clinical phenotype. In
conclusion, our findings show an altered molecular phenotype in non-manifesting carriers of a pathogenic, heterozygous _PRKN_ variant, characterized by disparities at the level of
mitochondrial function in two different cellular models derived from the variant carriers. To adequately understand the individual risk for disease conversion, there is a need for more
longitudinal studies with putatively healthy individuals carrying single _PRKN_ variants, more in-depth clinical phenotyping, and measurements of the molecular markers described in this
study. METHODS ETHICS STATEMENT The CHRIS study is a joint initiative with the South Tyrolean healthcare system conducted under the leadership of the Institute for Biomedicine, Eurac
Research, and was approved by the Ethics Committee of the Healthcare System of the Autonomous Province of Bozen/Bolzano (Prot. 0043339-BZ, 28/04/2011). The generation of iPSCs was approved
by the Ethics Committee of the Healthcare System of the Autonomous Province of Bozen/Bolzano (Prot. 0042955-BZ, 06/04/2018). Study participants provided their written informed consent to
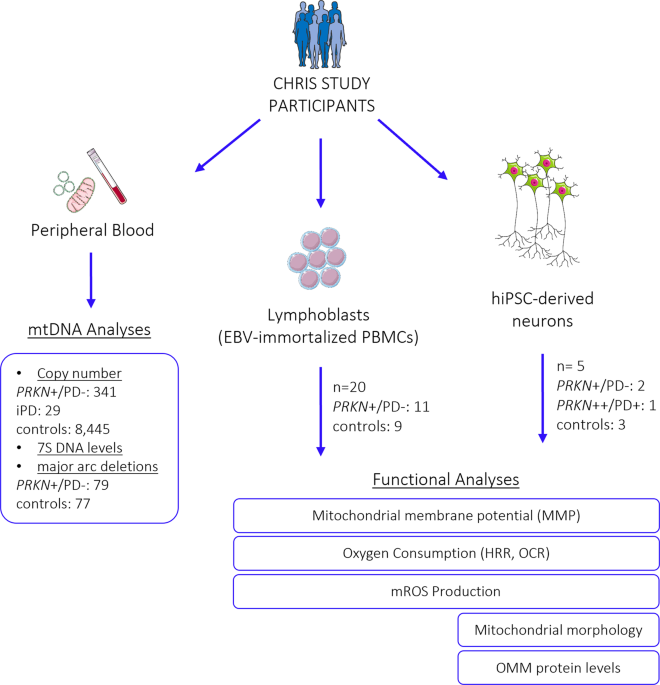
participate in this study. STUDY PARTICIPANTS Study participants were enrolled in the Cooperative Health Research in South Tyrol (CHRIS) study83. mtDNA copy number was quantified in 341
non-manifesting carriers of 17 pathogenic heterozygous _PRKN_ variants (_PRKN_+/PD−), identified previously11, 29 idiopathic PD (iPD) patients, and 8445 controls. Analyses for mtDNA major
arc deletions and mtDNA transcription initiation were conducted in a subset of 79 heterozygous _PRKN_ variant carriers and 77 controls. Furthermore, mitochondrial performance was assessed in
cellular models derived from 21 CHRIS study participants originating from four families, who were recalled and deep-phenotyped based on their genetic profile18. For these 21 individuals,
lymphoblastoid cell lines (LCLs) and/or human induced pluripotent stem cell (hiPSC)-derived neurons were generated (Supplementary Table 1). From these, 11 carried a heterozygous _PRKN_
variant (exon 7 deletion, _PRKN_+/PD−), which is among the most frequent mutations identified, and ten were control individuals from the same families (matched by close age), without _PRKN_
variants. Family 1 included six variant carriers and five controls, family 2 one carrier and one control, and families 3 and 4 included two carriers and two controls each. None of the
heterozygous _PRKN_ mutation carriers fulfilled the diagnostic criteria for PD, which was assessed with a screening questionnaire for parkinsonism84 and by clinical examination of the
individuals, for which cell lines were established18. In the neurological assessment, no difference between groups was observed for motor or non-motor symptoms and _substantia nigra_
hyperechogenicity, but sensor-based quantification of movements under specific task challenge allowed discrimination of unaffected heterozygous mutation carriers from mutation-free controls
(larger sample set including the 21 individuals with cell lines generated in this study)18. hiPSCs were generated for two heterozygous _PRKN_ mutation carriers and two controls, all from the
same family41; both mutation carriers (_PRKN_+/PD− 1, _PRKN_+/PD− 2) presented with _substantia nigra_ hyperechogenicity and subtle motor signs, one control (Control 2) did not show any
subclinical or prodromal signs, and the other control (Control 3) was positive for _substantia nigra_ hyperechogenicity. Additionally, hiPSC lines from a _PRKN_-PD patient with a homozygous
mutation (_PRKN_++/PD+)66,85 and a control individual (https://cells.ebisc.org/STBCi033-B/) were included in the functional analyses for comparison. Demographic and basic clinical data for
individuals with cell lines are summarized in Supplementary Table 1. An overview of all individuals included in the study is depicted in Fig. 1. GENERATION OF LYMPHOBLASTOID CELL LINES AND
CULTURE Peripheral blood was collected in EDTA-buffered collection tubes, and peripheral blood mononuclear cells (PBMCs) were isolated using Histopaque—1077 cell separation medium (Sigma
Aldrich) and Leucosep tubes (Greiner Bio-one), according to manufacturer’s instructions. Lymphoblastoid cell lines (LCLs) were established by infecting B-cells from the PBMCs with Epstein
Barr Virus (EBV). Briefly, PBMCs were incubated with 2 mL of culture supernatant from B95.8 cells (marmoset cell line) expressing EBV, at 37 °C in a 5% CO2 atmosphere for 2.5 h. Then, cells
were kept in RPMI 1640 (Roswell Park Memorial Institute) culture medium (Biowest), supplemented with 10% Fetal bovine serum (FBS) (Sigma Aldrich) and 1% penicillin-streptomycin (Thermo
Fisher Scientific), containing phytohemagglutinin (PHA) (Sigma Aldrich) to induce the secretion of T-cell-assisted B-cell growth factors and the removal of T-cells86,87. One week after
infection, clusters were visible, and PHA was withdrawn. For experiments, LCLs were seeded at a concentration of 3 × 105 cells/mL and passaged at intervals of three days. Cells were kept in
culture in a saturated, humidified atmosphere at 37 °C and 5% CO2 for a maximum of 10 passages. FLOW CYTOMETRY Flow cytometry was used to assess mitochondrial membrane potential (MMP) and
the production of mitochondrial reactive oxygen species (mROS) in LCLs. The potentiometric dye tetramethylrhodamine methyl ester (TMRM Image-iT™; Thermo Fisher Scientific) was used as an MMP
indicator with a final concentration of 100 nM. To exclude dead cells from the analyzed samples, VivaFix™ Cell Viability Assay (Biorad) was used together with TMRM. Labeled cells were
counted based on TMRM intensity, which corresponds to the levels of their MMP. As a positive control, to represent the background activity of TMRM staining, cells were treated with CCCP (50
µM, 1 h), which completely dissipates the MMP. To assess mROS production (mitochondrial superoxide), MitoSOX™ Red reagent (Thermo Fisher Scientific) was used. Cells were co-stained with 2.5
µM MitoSox Red and 200 nM Mito Tracker™ Green (MTG, Thermo Fisher Scientific) to select the cell population of interest for the acquisition and analysis. Stained LCLs were interrogated using
an S3e Cell Sorter (Biorad), operated by the ProSort™ Software, version 1.6. The fluorescence intensity of 20,000 events was quantified and analyzed using the FlowJo™ v10.6.1 software.
Fluorescence intensity was determined based on the gating strategy described in the supplementary information (Supplementary Fig. 3). HIGH-RESOLUTION RESPIROMETRY IN LCLS Mitochondrial
oxygen consumption was measured in LCLs by means of high-resolution respirometry (HRR) with the Oxygraph-2K (O2K, Oroboros Instruments, Innsbruck, Austria). Experiments were carried out
under normoxic conditions (O2 concentration at and below air saturation). Air calibration was performed according to the manufacturer´s instructions, and the temperature was set at 37 ±
0.001 °C (electronic Peltier regulation) under constant stirring at 750 rpm. Data recording was performed in real-time using the DatLab7 software (Oroboros Instruments), which was also used
for post-experimental analysis. Experiments were performed in MiR05 respiration medium (Oroboros Instruments), and manual titration of mitochondrial inhibitors and uncouplers was performed
using Hamilton syringes (customized by Hamilton®). Briefly, 5 ×106 LCLs were harvested and added to each O2K chamber. Mitochondrial respiration activity of intact LCLs was characterized by
using the following multiple substrate-uncoupler-inhibitor titration protocol (SUIT-003, BioBlast): (1) ROUTINE corresponds to physiological respiration (depending on endogenous substrates),
(2) LEAK respiration is induced by adding the ATP synthase inhibitor oligomycin (Omy, 10 nM; Sigma Aldrich) and corresponds to the non-phosphorylating resting state, when oxygen flux is
compensating for the proton leak, (3) maximal capacity of the electron transfer system determined after uncoupling by stepwise titration (usually 1–2 steps) of the protonophore carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, U, 0.5 μM each step; Sigma Aldrich), (4) residual oxygen consumption achieved after the injection of complex I inhibitor rotenone (Rot, 0.5
μM; Sigma Aldrich), (5) addition of succinate (S, 10 mM; Acros Organics) to test the intactness of the cell membrane, and (6) residual oxygen consumption after the injection of the complex
III-inhibitor antimycin A (Ama, 2.5 μM; Sigma Aldrich). Respiratory fluxes were corrected for instrumental background considering the oxygen consumption by the sensors and the oxygen
diffusion of the chamber. Respiration related to ATP turnover was calculated as the difference between ROUTINE and LEAK respiration (R-L), and spare respiratory capacity was determined as
the difference between maximal respiration and routine respiration (ET-R). Absolute respiration values were normalized for the total number of cells per chamber and citrate synthase
activity. CITRATE SYNTHASE (CS) ACTIVITY CS was assayed spectrophotometrically at 412 nm and 30 °C on the EnVision 2105 Multimode Plate Reader (Perkin-Elmer), according to a published
protocol88 with slight modifications, where the final concentrations for the reaction were acetyl-CoA 100 µM, oxalacetate 100 µM and 5,5′-dithiobis- (2-dinitrobenzoic acid) 100 µM in tris
chloride buffer, pH 8 (100 mM). Following respirometry in the O2K, LCLs were removed from the chamber, and an aliquot was snap-frozen. CS activity measurements were performed in duplicates
for each respirometry experiment, and averaged values were used for the normalization of respirometry results. DIFFERENTIATION OF HIPSCS INTO DOPAMINERGIC NEURONS The direct differentiation
of hiPSCs into dopaminergic (DA) neurons was conducted as described previously89,90, with minor modifications66. Human iPSC colonies were disaggregated into single cells using accutase and
replated onto matrigel (BD)-coated dishes in mTeSR™ 1 complete medium, supplemented with 10 μM ROCK inhibitor Y-27632 (Miltenyi Biotech) at a density of 57,000–62,000 cells/cm2.
Differentiation was started once hiPSCs reached a confluence of 90% by adding knockout serum replacement (KSR) medium supplemented with SMAD pathway inhibitors SB431542 (SB, Miltenyi
Biotech) and LDN-193189 (LDN, StemMACS). On days 1 to 5, KSR medium was added to the cells in the presence of SB, LDN, recombinant Human Sonic Hedgehog (SHH, R&D System), recombinant
Human FGF-8a (FGF8, R&D System), and Purmorphamine (Pu, StemMACS). The Wnt pathway activator molecule CHIR99021 (CH, StemMACS) was included from days 3–12. During days 6–10 of
differentiation, increasing amounts of Neurobasal medium plus B27 supplement (NB-B27 medium, Thermo Fisher Scientific) was added to the KSR medium (25, 50, and 75%), and upon day 7, SHH,
FGF8, and Pu were withdrawn. On day 11, maturation of DA neurons was initiated by adding recombinant Human BDNF (Peprotech), ascorbic acid (Sigma Aldrich), recombinant Human TGF-ß3
(Peprotech), cyclic-AMP (EnzoLifescience) and DAPT (Tocris). Cells were passaged between days 12 and 15 _en bloc_90, and between days 20 and 25 of differentiation, they were replated as
single cells onto plastic dishes or live imaging chamber slides, previously coated with poly-D-lysine (Sigma Aldrich) and laminin (Sigma Aldrich), for western blot and imaging experiments.
For oxygen consumption rate (OCR) assay experiments, neurons were replated en bloc between days 12 and 15 and replated at day 30 on the 96-well plates for the OCR assay, where they were
stabilized and further differentiated until day 34. By using this differentiation protocol, a yield of ~40% of the DA neuron marker tyrosine hydroxylase (TH)-expressing neurons is obtained,
as shown previously66. Detailed experimental procedures are available in the supplementary material. MITOCHONDRIAL OXYGEN CONSUMPTION RATE IN HIPSC-NEURONS Mitochondrial oxygen consumption
rate (OCR) was measured using the Extracellular O2 Consumption Assay Kit (Abcam, ab197243) according to the manufacturer’s instructions. At day 31 of differentiation, 2 × 105 hiPSC-derived
neurons were seeded on a 96-well cell culture plate, previously coated with poly-d-lysine (Sigma Aldrich) and laminin (Sigma Aldrich) and incubated in a CO2 incubator at 37 °C with NB-B27
medium supplemented with neuronal differentiation factors. Cells recovered for 72 h prior to performing the OCR measurements. For the assay, the medium was replaced with 150 µl of fresh
NB-B27 medium for routine (basal) OCR measurements, or with 150 µl of NB-B27 medium containing 2.5 µM FCCP for maximal respiration measurements. Next, 10 μL of Extracellular O2 consumption
reagent were added to each well, except to the blank control, and 100 µl of high-sensitivity mineral oil (pre-heated at 37 °C) were added to limit back diffusion of ambient oxygen.
Fluorescence intensities were measured using the EnVision 2105 Multimode Plate Reader (Perkin-Elmer), pre-heated at 37 °C, in 2-min- intervals for a total of 200 min, at excitation/emission
wavelengths = 355/642 nm. Respiration of cells results in oxygen depletion from the surrounding environment, causing an increase of fluorescence signal. Immediately after performing the
Extracellular O2 Consumption Assay, the CyQuant™ proliferation assay (Thermo Fisher Scientific) was employed to determine the number of live cells in each well, according to the
manufacturer´s instructions. Fluorescence intensity was corrected with the blank control, and OCR was determined by selecting the linear portion of the signal profile (avoiding any initial
lag or subsequent plateau) and applying the linear regression to determine the slope. The OCR calculated for each well was normalized to the cell number determined by CyQuant fluorescence
dye. Fluorescence intensities detecting OCR are expressed as relative fluorescence units (RFU) versus time (min). LIVE IMAGING To assess MMP, mROS production, and mitochondrial morphology in
hiPSC-derived neurons under live imaging conditions, 2 × 105 cells per well were seeded as single cells on an eight-well live imaging chamber slide (ibidi GmBH) previously coated with
poly-d-lysine (Sigma Aldrich) and laminin (Sigma Aldrich). Neurons were imaged at day 40 of differentiation. Cells were incubated with a staining solution containing either 100 nM TMRM to
assess MMP, 2.5 µM MitoSox Red to evaluate ROS production, or 100 nM MTG to evaluate mitochondrial morphology, at 37 °C and 5% CO2 for 30 min. As a positive control, to represent the
background activity of TMRM staining with the concentration used, cells were treated with CCCP (10 µM, 45 min), which completely dissipates the MMP. After washing with pre-warmed HBSS 1X,
cells were directly imaged in HBSS at 37 °C and 5% CO2 using a Leica SP8-X confocal microscope (Leica Microsystems, LAS-X acquisition software), with hybrid detectors and a 63X oil immersion
objective. For mean intensity measurements of TMRM and MitoSox Red in thresholded images, Image J software was used (https://imagej.nih.gov/ij/index.html). Mitochondrial fragmentation,
assessed based on MTG staining, was calculated using the filament function in Imaris software (version 9.6.0, Bitplane) (Oxford Instruments): for each field, the total length of the modeled
mitochondrial network was calculated and divided by the number of unconnected parts. IMMUNOFLUORESCENCE For immunocytochemical analysis, differentiated neurons were grown on a 16-well format
chamber slide System Nunc™ Lab-Tek™ (Thermo Fisher Scientific), previously coated with poly-d-lysine (Sigma Aldrich) and laminin (Sigma Aldrich), and fixed in a 4% paraformaldehyde
solution. Next, cells were permeabilized in 0.5% Triton X-100—PBS and blocked with a blocking solution (3% BSA in PBS) at RT. Immunostaining was performed by the addition of primary
antibodies to co-stain the DA neuronal marker TH (rabbit anti-TH, Merck Millipore MAB318) and the class III member of the beta-tubulin family (mouse anti-TUJ1, Biolegend). Subsequently,
cells were incubated with the appropriate fluorescently labeled secondary antibodies (goat anti-mouse Alexa Fluor 488-conjugated, goat anti-rabbit Alexa Fluor 555-conjugated, Thermo Fisher
Scientific), nuclei were stained by adding NucBlue™ Fixed Cell Stain (Thermo Fisher Scientific), and the slides were mounted with Dako Fluorescence mounting medium. Images were acquired
using a Leica SP8-X confocal microscope (Leica Microsystems, LAS-X acquisition software), with hybrid detectors and a 63X oil immersion objective. TREATMENT CONDITIONS In order to assess
protein levels of MFN2 (Mitofusin-2) and TOM70 (mitochondrial import receptor subunit) in hiPSC-derived neurons upon dissipation of the mitochondrial membrane potential, treatment with 20 µM
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (CCCP) (Sigma Aldrich) was performed for 15 h. WESTERN BLOT ANALYSIS For Western blot analysis, whole-cell homogenates were used.
Harvested cells were resuspended in cold RIPA buffer (Thermo Fisher Scientific) supplemented with protease inhibitors cOmplete™ Protease Inhibitor Cocktail (Roche) and phosphatase inhibitors
PhosSTOP EASYpack phosphatase inhibitors (Roche). Protein concentrations were assessed using a BCA Protein Assay Kit (Thermo Fisher Scientific), and 10 µg of total protein lysates were
loaded per well on a NuPAGE 4–12% Bis-Tris SDS-PAGE gel (Thermo Fisher Scientific). After electrophoresis, proteins were transferred onto a PVDF membrane (BioRad) and probed with antibodies
raised against MFN2 (Abcam, ab56889), TOM70 (Abcam, ab135602), SOD2 (Cell Signaling, 13141 S), Parkin (Abcam, 77924), and GAPDH (Millipore, MAB374). Subsequently, the blots were incubated
with the corresponding secondary antibodies, and target proteins were detected by enhanced chemiluminescence using Clarity™ ECL Western Kit (BioRad). The chemiluminescence signal was
detected using the ChemiDoc™ Touch Imaging System (BioRad) and quantified by densitometry using Image Lab 6.0 analyzer software (BioRad). Optical density values assessed for target proteins
were normalized by the indicated loading control. For each figure, all blots were processed in parallel and originated from the same experiment. MITOCHONDRIAL DNA ANALYSES mtDNA was
extracted from whole blood and cultured cells (LCLs and hiPSC-derived neurons) using a standard procedure for extraction of genomic DNA. mtDNA copy number was measured using a duplex
quantitative real-time PCR assay to simultaneously detect the mtDNA gene _tRNA-Leu_ and the single-copy nuclear gene _beta-2-microglobulin_ (_B2M_), as previously described in ref. 91. mtDNA
copy number data of the CHRIS study participants were available from a previous study91. mtDNA transcription-associated 7S DNA levels and major arc deletions were assessed using a real-time
PCR assay based on TaqMan chemistry34. Probes were used for the quantification of mtDNA sequences in the minor arc gene _NADH-dehydrogenase 1_ (_MT-ND1_), the major arc gene _NADH
dehydrogenase 4_ (_MT-ND4_), and the D-loop, located within the non-coding region (NCR) of mtDNA. A region of mtDNA-7S was amplified using the forward primer 5′-CCCACACGTTCCCCTTAAATAA-3′ and
the reverse primer 5′-CGTGAGTGGTTAATAGGGTGATAGAC-3′; a region of _MT-ND1_ was amplified using the forward primer 5′-CCCTAAAACCCGCCACATCTAC-3′ and the reverse primer
5′-GAGCGATGGTGAGAGCTAAGGT-3′; and a region of _MT-ND4_ was amplified using the forward primer 5′-CCATTCTCCTCCTATCCCTCAAC-3′ and the reverse primer 5′-CACAATCTGATGTTTTGGTTAAACTATATTT-3′.
Probe sequences were: ALEXA594-5′-ACATCACGATGGATCAC-3′ -MGB for D-Loop, VIC-5′ -CCATCACCCTCTACATCACCGCCC-3′ -QSY for _MT-ND1_ and FAM-5′ -CCGACATCATTACCGGGTTTTCCTCTTG-3′ -QSY for _MT-ND4_.
The real-time PCR was performed using the following conditions: 95 °C for 10 s, 45 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 3 sec. STATISTICAL ANALYSES Statistical analyses
were performed using R version 4.1.2 and GraphPad Prism 9. One-way ANOVA was used in experiments comparing three groups, followed by Tukey’s post hoc test to correct for multiple
comparisons. For analyzing differences between the two experimental groups, the non-parametric Mann–Whitney _U_-test was utilized. The threshold for significance was set at _p_ < 0.05.
All experiments were performed in a minimum of three independent biological replicates. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting
Summary linked to this article. DATA AVAILABILITY All data generated during this study are included in this article and its supplementary information files. Genotype data are not readily
available because the data are part of a large population-based study (CHRIS study), and the informed consent provided by the study participants does not allow the upload of individual-level
genetic data to public repositories. REFERENCES * Bloem, B. R., Okun, M. S. & Klein, C. Parkinson’s disease. _Lancet_. https://doi.org/10.1016/S0140-6736(21)00218-X (2021). * Kouli, A.,
Torsney, K. M. & Kuan, W. L. In _Parkinson’s Disease: Pathogenesis and Clinical Aspects_ (eds Stoker, T. B. & Greenland, J. C.) Ch. 1, (Codon Publications, 2018). * Forno, L. S.
Neuropathology of Parkinson’s disease. _J. Neuropathol. Exp. Neurol._ 55, 259–272 (1996). Article CAS PubMed Google Scholar * Moustafa, A. A. et al. Motor symptoms in Parkinson’s
disease: a unified framework. _Neurosci. Biobehav. Rev._ 68, 727–740 (2016). Article PubMed Google Scholar * Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture
of Parkinson’s disease. _Lancet Neurol._ 19, 170–178 (2020). Article CAS PubMed Google Scholar * Park, J. S., Davis, R. L. & Sue, C. M. Mitochondrial dysfunction in Parkinson’s
disease: new mechanistic isights and therapeutic perspectives. _Curr. Neurol. Neurosci. Rep._ 18, 21 (2018). Article PubMed PubMed Central Google Scholar * Domingo, A. & Klein, C.
Genetics of Parkinson disease. _Handb. Clin. Neurol._ 147, 211–227 (2018). Article PubMed Google Scholar * Kasten, M. et al. Genotype-phenotype relations for the Parkinson’s disease genes
Parkin, PINK1, DJ1: MDSGene systematic review. _Mov. Disord._ 33, 730–741 (2018). Article PubMed Google Scholar * Lesage, S. et al. Characterization of recessive Parkinson’s disease in a
large multicenter study. _Ann. Neurol_. https://doi.org/10.1002/ana.25787 (2020). * Pramstaller, P. P. et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation
carriers. _Ann. Neurol._ 58, 411–422 (2005). Article CAS PubMed Google Scholar * Castelo Rueda, M. P. et al. Frequency of heterozygous Parkin (PRKN) variants and penetrance of
Parkinson’s disease risk markers in the population-based CHRIS cohort. _Front. Neurol._ 12, 706145 (2021). Article PubMed PubMed Central Google Scholar * Klein, C., Lohmann-Hedrich, K.,
Rogaeva, E., Schlossmacher, M. G. & Lang, A. E. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. _Lancet Neurol_. 6, 652–662 (2007). Article CAS
PubMed Google Scholar * Huttenlocher, J. et al. Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. _Hum. Mol. Genet._ 24, 5637–5643 (2015). Article CAS
PubMed Google Scholar * Lubbe, S. J. et al. Assessing the relationship between monoallelic PRKN mutations and Parkinson’s risk. _Hum. Mol. Genet._ 30, 78–86 (2021). Article CAS PubMed
PubMed Central Google Scholar * Zhu, W. et al. Heterozygous PRKN mutations are common but do not increase the risk of Parkinson’s disease. _Brain_. https://doi.org/10.1093/brain/awab456
(2022). * Yu, E. et al. Analysis of heterozygous PRKN variants and copy-number variations in Parkinson’s disease. _Mov. Disord._ 36, 178–187 (2021). Article CAS PubMed Google Scholar *
Weissbach, A. et al. Influence of L-dopa on subtle motor signs in heterozygous Parkin- and PINK1 mutation carriers. _Parkinsonism Relat. Disord._ 42, 95–99 (2017). Article PubMed Google
Scholar * Prasuhn, J. et al. Task matters - challenging the motor system allows distinguishing unaffected Parkin mutation carriers from mutation-free controls. _Parkinsonism Relat. Disord._
86, 101–104 (2021). Article PubMed Google Scholar * Binkofski, F. et al. Morphometric fingerprint of asymptomatic Parkin and PINK1 mutation carriers in the basal ganglia. _Neurology_ 69,
842–850 (2007). Article CAS PubMed Google Scholar * Hilker, R. et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated
with mutations in the parkin gene. _Ann. Neurol._ 49, 367–376 (2001). Article CAS PubMed Google Scholar * Guo, J. F. et al. Clinical features and [11C]-CFT PET analysis of PARK2, PARK6,
PARK7-linked autosomal recessive early onset Parkinsonism. _Neurol. Sci._ 32, 35–40 (2011). Article PubMed Google Scholar * Pavese, N. et al. Nigrostriatal dysfunction in homozygous and
heterozygous parkin gene carriers: an 18F-dopa PET progression study. _Mov. Disord._ 24, 2260–2266 (2009). Article PubMed Google Scholar * Santos, M., Morais, S., Pereira, C., Sequeiros,
J. & Alonso, I. Parkin truncating variants result in a loss-of-function phenotype. _Sci. Rep._ 9, 16150 (2019). Article PubMed PubMed Central Google Scholar * Mouton-Liger, F.,
Jacoupy, M., Corvol, J. C. & Corti, O. PINK1/Parkin-dependent mitochondrial surveillance: from pleiotropy to Parkinson’s disease. _Front. Mol. Neurosci._ 10, 120 (2017). Article PubMed
PubMed Central Google Scholar * Ge, P., Dawson, V. L. & Dawson, T. M. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. _Mol.
Neurodegener._ 15, 20 (2020). Article PubMed PubMed Central Google Scholar * Shimura, H. et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. _Nat.
Genet._ 25, 302–305 (2000). Article CAS PubMed Google Scholar * Youle, R. J. & Narendra, D. P. Mechanisms of mitophagy. _Nat. Rev. Mol. Cell Biol._ 12, 9–14 (2011). Article CAS
PubMed PubMed Central Google Scholar * Trempe, J. F. et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. _Science_ 340, 1451–1455 (2013). Article CAS PubMed
Google Scholar * Trinh, D., Israwi, A. R., Arathoon, L. R., Gleave, J. A. & Nash, J. E. The multi-faceted role of mitochondria in the pathology of Parkinson’s disease. _J. Neurochem._
156, 715–752 (2021). Article CAS PubMed Google Scholar * Gegg, M. E. et al. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy.
_Hum. Mol. Genet._ 19, 4861–4870 (2010). Article CAS PubMed PubMed Central Google Scholar * Narendra, D., Tanaka, A., Suen, D. F. & Youle, R. J. Parkin is recruited selectively to
impaired mitochondria and promotes their autophagy. _J. Cell Biol._ 183, 795–803 (2008). Article CAS PubMed PubMed Central Google Scholar * Vives-Bauza, C. et al. PINK1-dependent
recruitment of Parkin to mitochondria in mitophagy. _Proc. Natl Acad. Sci. USA_ 107, 378–383 (2010). Article CAS PubMed Google Scholar * Shin, J. H. et al. PARIS (ZNF746) repression of
PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. _Cell_ 144, 689–702 (2011). Article CAS PubMed PubMed Central Google Scholar * Wasner, K. et al. Parkin deficiency
impairs mitochondrial DNA dynamics and propagates inflammation. _Mov. Disord._ https://doi.org/10.1002/mds.29025 (2022). * Pickrell, A. M. et al. Endogenous Parkin preserves dopaminergic
substantia nigral neurons following mitochondrial DNA mutagenic stress. _Neuron_ 87, 371–381 (2015). Article CAS PubMed PubMed Central Google Scholar * Bruggemann, N. et al. Frequency
of heterozygous Parkin mutations in healthy subjects: need for careful prospective follow-up examination of mutation carriers. _Parkinsonism Relat. Disord._ 15, 425–429 (2009). Article
PubMed Google Scholar * Qadri, R. et al. Alterations in mitochondrial membrane potential in peripheral blood mononuclear cells in Parkinson’s disease: potential for a novel biomarker.
_Restor. Neurol. Neurosci_. 36, 719–727 (2018). CAS PubMed Google Scholar * Agostini, M. et al. Metabolic reprogramming during neuronal differentiation. _Cell Death Differ_. 23, 1502–1514
(2016). Article CAS PubMed PubMed Central Google Scholar * Prigione, A., Fauler, B., Lurz, R., Lehrach, H. & Adjaye, J. The senescence-related mitochondrial/oxidative stress
pathway is repressed in human induced pluripotent stem cells. _Stem Cells_ 28, 721–733 (2010). Article CAS PubMed Google Scholar * Maffezzini, C., Calvo-Garrido, J., Wredenberg, A. &
Freyer, C. Metabolic regulation of neurodifferentiation in the adult brain. _Cell Mol. Life Sci._ 77, 2483–2496 (2020). Article CAS PubMed PubMed Central Google Scholar * Castelo
Rueda, M. P. et al. Generation and characterization of induced pluripotent stem cell (iPSC) lines of two asymptomatic individuals carrying a heterozygous exon 7 deletion in Parkin (PRKN) and
two non-carriers from the same family. _Stem Cell Res._ 60, 102692 (2022). Article CAS PubMed Google Scholar * Brand, M. D. et al. Mitochondrial superoxide: production, biological
effects, and activation of uncoupling proteins. _Free Radic. Biol. Med._ 37, 755–767 (2004). Article CAS PubMed Google Scholar * Dias, V., Junn, E. & Mouradian, M. M. The role of
oxidative stress in Parkinson’s disease. _J. Parkinsons Dis._ 3, 461–491 (2013). Article CAS PubMed PubMed Central Google Scholar * Tanaka, A. et al. Proteasome and p97 mediate
mitophagy and degradation of mitofusins induced by Parkin. _J. Cell Biol._ 191, 1367–1380 (2010). Article CAS PubMed PubMed Central Google Scholar * Rakovic, A. et al. Mutations in
PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. _PLoS ONE_ 6, e16746 (2011). Article CAS PubMed PubMed Central Google Scholar * Ziviani, E., Tao, R. N. &
Whitworth, A. J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. _Proc. Natl Acad. Sci. USA_ 107, 5018–5023 (2010). Article CAS PubMed PubMed
Central Google Scholar * Ordureau, A. et al. Global landscape and dynamics of Parkin and USP30-dependent ubiquitylomes in iNeurons during mitophagic signaling. _Mol. Cell_ 77, 1124–1142
e1110 (2020). Article CAS PubMed PubMed Central Google Scholar * Nissen, S. K. et al. Alterations in blood monocyte functions in Parkinson’s disease. _Mov. Disord._ 34, 1711–1721
(2019). Article CAS PubMed Google Scholar * Scherzer, C. R. et al. Molecular markers of early Parkinson’s disease based on gene expression in blood. _Proc. Natl Acad. Sci. USA_ 104,
955–960 (2007). Article CAS PubMed PubMed Central Google Scholar * Smith, A. M. et al. Mitochondrial dysfunction and increased glycolysis in prodromal and early Parkinson’s blood cells.
_Mov. Disord._ 33, 1580–1590 (2018). Article CAS PubMed PubMed Central Google Scholar * Vida, C. et al. Lymphoproliferation impairment and oxidative stress in blood cells from early
Parkinson’s disease patients. _Int. J. Mol. Sci._ 20, 771 (2019). Article CAS PubMed PubMed Central Google Scholar * Grunewald, A. et al. Mutant Parkin impairs mitochondrial function
and morphology in human fibroblasts. _PLoS ONE_ 5, e12962 (2010). Article PubMed PubMed Central Google Scholar * Mortiboys, H. et al. Mitochondrial function and morphology are impaired
in parkin-mutant fibroblasts. _Ann. Neurol._ 64, 555–565 (2008). Article CAS PubMed PubMed Central Google Scholar * Zilocchi, M. et al. Exploring the impact of PARK2 mutations on the
total and mitochondrial proteome of human skin fibroblasts. _Front. Cell Dev. Biol._ 8, 423 (2020). Article PubMed PubMed Central Google Scholar * Muftuoglu, M. et al. Mitochondrial
complex I and IV activities in leukocytes from patients with parkin mutations. _Mov. Disord._ 19, 544–548 (2004). Article PubMed Google Scholar * Annesley, S. J. et al. Immortalized
Parkinson’s disease lymphocytes have enhanced mitochondrial respiratory activity. _Dis. Model. Mech._ 9, 1295–1305 (2016). CAS PubMed PubMed Central Google Scholar * Annesley, S. J.,
Allan, C. Y., Sanislav, O., Evans, A. & Fisher, P. R. Dysregulated Gene Expression in Lymphoblasts from Parkinson’s Disease. _Proteomes_ 10, 20 (2022). Article CAS PubMed PubMed
Central Google Scholar * Haylett, W. et al. Altered mitochondrial respiration and other features of mitochondrial function in Parkin-mutant fibroblasts from Parkinson’s disease patients.
_Parkinsons Dis._ 2016, 1819209 (2016). PubMed PubMed Central Google Scholar * Gonzalez-Casacuberta, I. et al. Mitochondrial and autophagic alterations in skin fibroblasts from Parkinson
disease patients with Parkin mutations. _Aging_ 11, 3750–3767 (2019). Article CAS PubMed PubMed Central Google Scholar * Ming, F. et al. The PARK2 mutation associated with Parkinson’s
disease enhances the vulnerability of peripheral blood lymphocytes to paraquat. _Biomed. Res. Int._ 2020, 4658109 (2020). Article PubMed PubMed Central Google Scholar * Trinh, J. et al.
Mitochondrial DNA heteroplasmy distinguishes disease manifestation in PINK1/PRKN-linked Parkinson’s disease. _Brain_ awac464 (2022). * Zheng, X. et al. Metabolic reprogramming during
neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. _Elife_ 5, e13374 (2016). Article PubMed PubMed Central Google Scholar * O’Brien, L. C., Keeney,
P. M. & Bennett, J. P. Jr Differentiation of human neural stem cells into motor neurons stimulates mitochondrial biogenesis and decreases glycolytic flux. _Stem Cells Dev._ 24, 1984–1994
(2015). Article PubMed PubMed Central Google Scholar * Schwartzentruber, A. et al. Oxidative switch drives mitophagy defects in dopaminergic parkin mutant patient neurons. _Sci. Rep._
10, 15485 (2020). Article CAS PubMed PubMed Central Google Scholar * Kumar, M. et al. Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-deficient human
dopamine neurons. _Stem Cell Rep._ 15, 629–645 (2020). Article CAS Google Scholar * Zanon, A. et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in
Parkin-deficient human iPSC-derived neurons and Drosophila. _Hum. Mol. Genet._ 26, 2412–2425 (2017). Article CAS PubMed PubMed Central Google Scholar * Bogetofte, H. et al. PARK2
mutation causes metabolic disturbances and impaired survival of human iPSC-derived neurons. _Front. Cell Neurosci._ 13, 297 (2019). Article CAS PubMed PubMed Central Google Scholar *
Okarmus, J. et al. Identification of bioactive metabolites in human iPSC-derived dopaminergic neurons with PARK2 mutation: Altered mitochondrial and energy metabolism. _Stem Cell Rep._ 16,
1510–1526 (2021). Article CAS Google Scholar * Yamaguchi, A. et al. Identifying therapeutic agents for amelioration of mitochondrial clearance disorder in neurons of familial Parkinson
disease. _Stem Cell Rep._ 14, 1060–1075 (2020). Article CAS Google Scholar * Chung, S. Y. et al. Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial
dysfunction and alpha-synuclein accumulation. _Stem Cell Rep._ 7, 664–677 (2016). Article CAS Google Scholar * Filograna, R., Beltramini, M., Bubacco, L. & Bisaglia, M. Anti-oxidants
in Parkinson’s disease therapy: a critical point of view. _Curr. Neuropharmacol._ 14, 260–271 (2016). Article CAS PubMed PubMed Central Google Scholar * Pacelli, C. et al. Mitochondrial
defect and PGC-1alpha dysfunction in parkin-associated familial Parkinson’s disease. _Biochim. Biophys. Acta_ 1812, 1041–1053 (2011). Article CAS PubMed Google Scholar * Imaizumi, Y. et
al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. _Mol. Brain_ 5, 35
(2012). Article CAS PubMed PubMed Central Google Scholar * Shaltouki, A. et al. Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2
knockout isogenic iPSC lines. _Stem Cell Rep._ 4, 847–859 (2015). Article CAS Google Scholar * Yokota, M. et al. Establishment of an in vitro model for analyzing mitochondrial
ultrastructure in PRKN-mutated patient iPSC-derived dopaminergic neurons. _Mol. Brain_ 14, 58 (2021). Article CAS PubMed PubMed Central Google Scholar * Suzuki, S. et al. Efficient
induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. _Biochem. Biophys. Res. Commun._ 483, 88–93 (2017). Article
CAS PubMed Google Scholar * Burton, E. M. & Gewurz, B. E. Epstein-Barr virus oncoprotein-driven B cell metabolism remodeling. _PLoS Pathog._ 18, e1010254 (2022). Article CAS
PubMed PubMed Central Google Scholar * Darekar, S. et al. Epstein-Barr virus immortalization of human B-cells leads to stabilization of hypoxia-induced factor 1 alpha, congruent with the
Warburg effect. _PLoS ONE_ 7, e42072 (2012). Article CAS PubMed PubMed Central Google Scholar * Fraenkel, J. R., Hyun, H.H., & Wallen, N.E. _How to Design and Evaluate Research in
Education_ 8th edn (McGraw Hill, 2012). * Gall, M. D., Gall, J. P., & Borg, W. R. _Educational Research: An Introduction_ 7th edn (Pearson Education Inc., 2003). * Prasuhn, J. et al. An
omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: concept evaluation in a double-blind randomized placebo-controlled parallel group trial. _Neurol. Res. Pract._
1, 31 (2019). Article PubMed PubMed Central Google Scholar * Prasuhn, J. et al. The use of vitamin K2 in patients with Parkinson’s disease and mitochondrial dysfunction (PD-K2): a
theranostic pilot study in a placebo-controlled parallel group design. _Front. Neurol._ 11, 592104 (2020). Article PubMed Google Scholar * Pattaro, C. et al. The Cooperative Health
Research in South Tyrol (CHRIS) study: rationale, objectives, and preliminary results. _J. Transl. Med._ 13, 348 (2015). Article PubMed PubMed Central Google Scholar * Pramstaller, P.
P., Falk, M., Schoenhuber, R. & Poewe, W. Validation of a mail questionnaire for parkinsonism in two languages (German and Italian). _J. Neurol._ 246, 79–86 (1999). Article CAS PubMed
Google Scholar * Zanon, A. et al. Generation of an induced pluripotent stem cell line (EURACi005-A) from a Parkinson’s disease patient carrying a homozygous exon 3 deletion in the
PRKNgene. _Stem Cell Res._ 41, 101624 (2019). Article CAS PubMed Google Scholar * Penno, M. B., Pedrotti-Krueger, M. & Ray, T. Cryopreservation of whole blood and isolated
lymphocytes for B-cell immortalization. _J. Tissue Culture Methods_ 15, 43–47 (1993). Article Google Scholar * Sie, L., Loong, S. & Tan, E. K. Utility of lymphoblastoid cell lines. _J.
Neurosci. Res._ 87, 1953–1959 (2009). Article CAS PubMed Google Scholar * Coore, H. G., Denton, R. M., Martin, B. R. & Randle, P. J. Regulation of adipose tissue pyruvate
dehydrogenase by insulin and other hormones. _Biochem. J._ 125, 115–127 (1971). Article CAS PubMed PubMed Central Google Scholar * Kriks, S. et al. Dopamine neurons derived from human
ES cells efficiently engraft in animal models of Parkinson’s disease. _Nature_ 480, 547–551 (2011). Article CAS PubMed PubMed Central Google Scholar * Mazzulli, J. R., Zunke, F.,
Isacson, O., Studer, L. & Krainc, D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. _Proc. Natl
Acad. Sci. USA_ 113, 1931–1936 (2016). Article CAS PubMed PubMed Central Google Scholar * Fazzini, F. et al. Association of mitochondrial DNA copy number with metabolic syndrome and
type 2 diabetes in 14 176 individuals. _J. Intern. Med._ 290, 190–202 (2021). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS The CHRIS study is a
collaborative effort between the Eurac Research Institute for Biomedicine and the Healthcare System of the Autonomous Province of Bozen/Bolzano (Südtiroler Sanitätsbetrieb/Azienda Sanitaria
dell’Alto Adige). We thank all study participants from the middle and upper Vinschgau/Val Venosta, the general practitioners, and the personnel of the Hospital of Schlanders/Silandro for
their support and collaboration. Furthermore, we thank the study assistants and the biobank of the Institute for Biomedicine for data and sample collection and elaboration. The CHRIS biobank
was assigned the “Bioresource Research Impact Factor” (BRIF) code BRIF6107. The CHRIS study is funded by the Department of Innovation, Research and the University of the Autonomous Province
of Bozen/Bolzano, Italy. This research was funded by the Department of Innovation, Research and University of the Autonomous Province of Bozen/Bolzano, Italy, through a core funding
initiative to the Institute for Biomedicine and by the Deutsche Forschungsgemeinschaft to A.A.H., A.G., C.K., and P.P.P. (FOR2488). Moreover, A.G. was supported by the Luxembourg National
Research Fund within the framework of the INTER (ProtectMove, INTER/DFG/19/14429377) and ATTRACT (Model-IPD, FNR9631103) programs. The authors thank the Department of Innovation, Research
and University of the Autonomous Province of Bozen/Bolzano for covering the Open Access publication costs. AUTHOR INFORMATION Author notes * These authors contributed equally: Andrew A.
Hicks, Irene Pichler. AUTHORS AND AFFILIATIONS * Institute for Biomedicine, Eurac Research, Affiliated Institute of the University of Lübeck, Bolzano, Italy Maria Paulina Castelo Rueda,
Alessandra Zanon, Valentina Gilmozzi, Alexandros A. Lavdas, Athina Raftopoulou, Fabiola Del Greco M, Peter P. Pramstaller, Andrew A. Hicks & Irene Pichler * Department of Economics,
University of Patras, Patras, Greece Athina Raftopoulou * Luxembourg Centre for Systems Biomedicine, University of Luxembourg, Esche-sur-Alzette, Luxembourg Sylvie Delcambre & Anne
Grünewald * Institute of Neurogenetics, University of Lübeck, Lübeck, Germany Christine Klein & Anne Grünewald * Department of Neurology, University Medical Center Schleswig-Holstein,
Campus Lübeck, Lübeck, Germany Peter P. Pramstaller Authors * Maria Paulina Castelo Rueda View author publications You can also search for this author inPubMed Google Scholar * Alessandra
Zanon View author publications You can also search for this author inPubMed Google Scholar * Valentina Gilmozzi View author publications You can also search for this author inPubMed Google
Scholar * Alexandros A. Lavdas View author publications You can also search for this author inPubMed Google Scholar * Athina Raftopoulou View author publications You can also search for this
author inPubMed Google Scholar * Sylvie Delcambre View author publications You can also search for this author inPubMed Google Scholar * Fabiola Del Greco M View author publications You can
also search for this author inPubMed Google Scholar * Christine Klein View author publications You can also search for this author inPubMed Google Scholar * Anne Grünewald View author
publications You can also search for this author inPubMed Google Scholar * Peter P. Pramstaller View author publications You can also search for this author inPubMed Google Scholar * Andrew
A. Hicks View author publications You can also search for this author inPubMed Google Scholar * Irene Pichler View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS M.P.C.R.: generated the cell lines, designed and performed all experiments, analyzed the data, and wrote the manuscript; A.Z.: designed the experiments, participated in
the interpretation of results, and helped revise the manuscript; V.G.: participated in cell lines generation and maintenance; A.A.L.: performed confocal imaging analyses; A.R.: provided
statistical advice on LCL data; S.D.: performed mtDNA experiments; F.D.G.M.: performed statistical analyses on mtDNA data; C.K.: provided critical review of the manuscript; A.G.: provided
critical review of the manuscript; P.P.P.: provided critical review of the manuscript; A.A.H.: conceived and designed the project, participated in the interpretation of results, and revised
the manuscript; I.P.: designed and supervised the project, participated in the interpretation of results, wrote and revised the manuscript. All authors read and approved the final
manuscript. CORRESPONDING AUTHORS Correspondence to Maria Paulina Castelo Rueda or Andrew A. Hicks. ETHICS DECLARATIONS COMPETING INTERESTS C.K. serves as a medical advisor to Centogene for
genetic testing reports in the field of movement disorders, except Parkinson’s disease, and as a member of the Scientific Advisory Committee of Retromer Therapeutics. The remaining authors
declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International
License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons
license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Castelo Rueda, M.P., Zanon, A., Gilmozzi, V. _et al._ Molecular phenotypes of
mitochondrial dysfunction in clinically non-manifesting heterozygous _PRKN_ variant carriers. _npj Parkinsons Dis._ 9, 65 (2023). https://doi.org/10.1038/s41531-023-00499-9 Download citation
* Received: 14 August 2022 * Accepted: 23 March 2023 * Published: 18 April 2023 * DOI: https://doi.org/10.1038/s41531-023-00499-9 SHARE THIS ARTICLE Anyone you share the following link with
will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative









