- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Several mutations that cause Parkinson’s disease (PD) have been identified over the past decade. These account for 15–25% of PD cases; the rest of the cases are considered sporadic.
Currently, it is accepted that PD is not a single monolithic disease but rather a constellation of diseases with some common phenotypes. While rodent models exist for some of the PD-causing
mutations, research on the sporadic forms of PD is lagging due to a lack of cellular models. In our study, we differentiated PD patient-derived dopaminergic (DA) neurons from the induced
pluripotent stem cells (iPSCs) of several PD-causing mutations as well as from sporadic PD patients. Strikingly, we observed a common neurophysiological phenotype: neurons derived from PD
patients had a severe reduction in the rate of synaptic currents compared to those derived from healthy controls. While the relationship between mutations in genes such as the _SNCA_ and
_LRRK2_ and a reduction in synaptic transmission has been investigated before, here we show evidence that the pathogenesis of the synapses in neurons is a general phenotype in PD. Analysis
of RNA sequencing results displayed changes in gene expression in different synaptic mechanisms as well as other affected pathways such as extracellular matrix-related pathways. Some of
these dysregulated pathways are common to all PD patients (monogenic or idiopathic). Our data, therefore, show changes that are central and convergent to PD and suggest a strong involvement
of the tetra-partite synapse in PD pathophysiology. SIMILAR CONTENT BEING VIEWED BY OTHERS SYNAPTIC DYSFUNCTION AND EXTRACELLULAR MATRIX DYSREGULATION IN DOPAMINERGIC NEURONS FROM SPORADIC
AND E326K-_GBA1_ PARKINSON’S DISEASE PATIENTS Article Open access 19 February 2024 UPREGULATED ECM GENES AND INCREASED SYNAPTIC ACTIVITY IN PARKINSON’S HUMAN DA NEURONS WITH _PINK1_/ _PRKN_
MUTATIONS Article Open access 18 May 2024 SINGLE-CELL TRANSCRIPTOMICS OF HUMAN IPSC DIFFERENTIATION DYNAMICS REVEAL A CORE MOLECULAR NETWORK OF PARKINSON’S DISEASE Article Open access 13
January 2022 INTRODUCTION Parkinson’s disease (PD) was first described in 1817 by James Parkinson1, who wrote about patients with shaking palsy, i.e., involuntary tremulous motion with
lessened muscular power. PD occurs in approximately two of 1000 people and is highly correlated with aging, affecting about 1% of the older population above 60 years of age2,3 The main
neuropathological symptoms are α-synuclein-containing Lewy bodies and loss of dopaminergic (DA) neurons in the substantia nigra pars compacta. PD patients experience movement difficulties
with three cardinal signs: tremor, rigidity, and bradykinesia. Some of the main non-motor symptoms of PD include loss of smell, depression, sleep disorders, and dementia, but a wide range of
other symptoms such as excessive saliva4 and susceptibility to melanoma5 may also be present. With the progression of the disease, Lewy body pathology spreads to many areas of the brain,
and Lewy bodies containing α-synuclein aggregates are seen throughout many brain areas such as the hippocampus, hypothalamus, neocortex, and cortex6,7,8. Approximately 15% of PD cases report
familial inheritance of the disease. This percentage may vary significantly in different populations9. Hundreds of variants have been observed in several PD genes that showed a clear
association, including _α-synuclein_ (_SNCA, PARK4_), _parkin_ (_PARK2_), _UCH-L1_ (_PARK5_), _PINK1_ (_PARK6_), _DJ-1_ (_PARK7_), _LRRK2_ (_PARK8_), _ATP13A2_ (_PARK9_), _GBA, VPS35
(PARK17)_, _EIF4G1_, and _PARK16_10,11. Thus, PD is not a single disease but a constellation of phenotypes that are displayed variably by patients with different co-morbidities but some
commonalities12. Some of the genetically characterized forms of the disease are rapidly evolving, with early-onset ages of the mid-30s or mid-40s13,14,15, but some genetic forms have a
similar age-onset to idiopathic PD16. α-Synuclein immunohistochemistry is currently considered one of the gold standards in the neuropathological evaluation of PD. Aggregates of misfolded
α-synuclein in Lewy bodies in DA neurons are now considered a hallmark of PD, but it is not clear whether these Lewy bodies are the cause of neuronal atrophy or a byproduct of the
disease17,18,19. In fact, injection of synthetic α-synuclein fibrils into the dorsal striatum of wild-type mice was enough to elicit and transmit disease pathology and neurodegeneration20.
Similarly, mice carrying mutations that disrupt physiological tetramers of the α-synuclein protein develop brain pathology and neurodegeneration typical of PD21. In neurons, α-synuclein
physiologically localizes mainly to presynaptic terminals22,23,24. The aggregates of α-synuclein have been observed in cell soma but also in neurites25, and they are widespread in various
brain areas in PD patients. However, reports show that micro-aggregates are present in the neurites close to presynaptic terminals, causing synaptic impairment sometimes long before the
large aggregates that eventually make up the Lewy bodies form17,26,27,28. Mutations as well as copy number variations in the _SNCA_ (_PARK1_) gene, which codes for the α-synuclein protein,
have been shown to have a causative effect in PD13,14,15,29,30,31,32,33,34,35. α-Synuclein is localized mainly in presynaptic terminals18. It helps to maintain the size of the presynaptic
vesicular pool as well as vesicle recycling36,37, and it functions to help neurotransmitter release, especially dopamine38,39,40. Animal models for mutations and copy number variations in
the SNCA gene recapitulate the human motor symptoms and neuronal loss41,42,43. Surprisingly, there is an almost immediate neurophysiological phenotype of a reduction in synaptic activity
after the introduction of different SNCA mutations in rodent and human models37,44,45,46,47,48. A reduction in synaptic activity has been shown on its own to cause neuronal atrophy in
different types of neurons37,48,49, suggesting a positive feedback mechanism that further increases neuronal cell death. Leucine-rich repeat kinase 2 (_LRRK2, PARK8_) is another gene with a
causative association with PD10. It is the most common form of genetic PD, and mutations are highly prevalent in certain populations50. The precise physiological function of _LRRK2_ is not
completely understood, but recent studies have shown that _LRRK2_ is involved in cellular functions such as neurite outgrowth, cytoskeletal maintenance, vesicle trafficking, autophagic
protein degradation, and the immune system51. Drosophila models with overexpression of _LRRK2_ recapitulate PD phenotypes of motor dysfunction and DA cell death52,53 and so do C. Elegans
overexpression models54. However, flies and nematodes do not express a-synuclein and are therefore not a great model for studying PD. Rodent models with _LRRK2_ mutations show minimal
evidence of neurodegeneration55,56 but do show locomotor impairments56 and reductions in stimulated dopamine neurotransmission and D2 receptor function. Interestingly, _LRRK2_ was found to
regulate synaptic vesicle endocytosis and recycling and neurotransmitter release57,58,59. RNA-mediated silencing of _LRRK2_ affected postsynaptic currents as well as presynaptic vesicle
trafficking and recycling60. Parkin (encoded by _PARK2_) is a ubiquitin-ligase enzyme expressed in the CNS and peripheral tissues61. It is a multifunctional protein that is involved in many
intracellular processes, and several substrates for it have been identified. Parkin is localized on synaptic vesicles and displays a distribution pattern similar to that of synapsin I, a
protein that associates with the cytoplasmic surface of synaptic vesicles62,63. Differential fractioning of rat brain lysates revealed that parkin was enriched in the fraction containing
PSD-95, a postsynaptic marker64. Homozygous and heterozygous mutations are considered risk factors for early-onset PD, though the contribution of the heterozygous form to the onset of PD is
still considered controversial65,66,67. Loss of function mutations in the _PARK2_ gene cause early and severe degeneration of DA neurons of the substantia nigra pars compacta68. Parkin has
been shown to negatively regulate the number and strength of excitatory synapses in rats69 and to ubiquitinate synaptic proteins70. Pathogenic parkin mutations were shown to disrupt
glutamatergic synaptic transmission and plasticity by impeding NMDA and AMPA receptor trafficking71,72. Mutations in Parkin may therefore cause dysregulation of glutamatergic and DA synapses
that eventually culminates in DA neuron cell death. Overall, quite a few animal model studies suggest that several PD-associated mutations result in a synaptic pathology that occurs before
neuronal cell death and may even be the cause of neuronal degeneration27. These studies are further supported by other neuroanatomical studies of post-mortem patient brain samples from
familial PD cases73,74,75. Here, we used PD patient-derived neurons to study the neuropathology in PD using the induced pluripotent stem cells (iPSCs) technique that has recently been used
to study mechanisms of brain disorders76,77,78,79,80. Reprogramming adult cells into stem cells is known to erase aging signatures81,82; therefore, the neurons derived in this method can be
considered young, and even pre-natal, neurons. We found that in all the PD lines [both mutations-derived and sporadic PD (sPD)] there is a significant reduction in the rate of synaptic
events in these “young” neurons. The use of iPSCs allowed us to also measure neurons derived from PD patients with no known genetic mutations, who are therefore considered sporadic cases.
Our cohort consisted of a few patients with sPD. A few of the patients had mutations in the SNCA gene (one patient with a duplication, one patient with a triplication, and a genetically
engineered line with the A53T mutation). One patient had a mutation in the LRRK2 gene, and two patients had a mutation in their Parkin gene. Our results suggest a biological predisposition
that exists in PD patients, even without other epigenetic or environmental influences. Furthermore, our methods allowed us to recognize gene expression patterns and gene ontology (GO) terms
that are common to neurons derived from patients with different PD mutations as well as to sPD. RESULTS A DRASTIC REDUCTION IN SYNAPTIC ACTIVITY IS OBSERVED IN NEURONS DERIVED FROM PATIENTS
WITH DUPLICATION AND TRIPLICATION OF THE SNCA GENE Our first cohort consisted of a control line (healthy individual, the 40102 line), a PD patient with a duplication of the α- synuclein gene
(denoted as 2X), and a PD patient with a triplication of the α-synuclein gene (denoted as 3X). The patient with the α-synuclein triplication had early-onset, autosomal dominant PD at age 38
and was previously described83. We differentiated these lines into DA neurons (see “Methods”) and used whole-cell patch-clamp to assess intrinsic properties as well as synaptic activity in
the neurons. An immunostaining image for DAPI to mark cell bodies, MAP2 to mark neurons, and tyrosine hydroxylase (TH) to specifically mark dopaminergic neurons is shown in Supplementary
Fig. 12P. A total of 15 control neurons, 15 2X neurons, and 15 3X neurons were patch-clamped approximately 50 days after the start of differentiation. The total number of evoked potentials
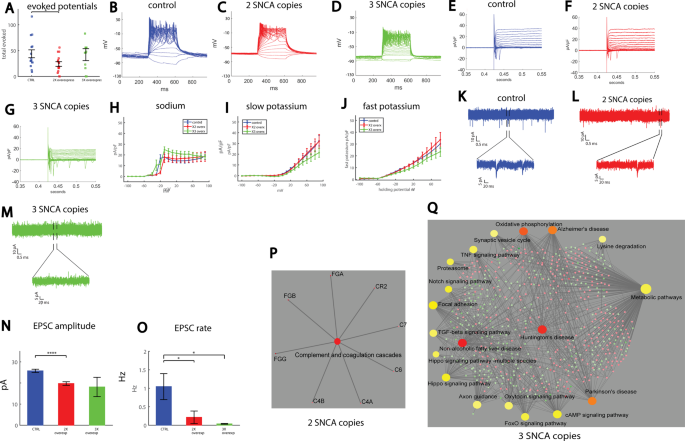
in the 17 first depolarization steps was decreased in the 2X neurons count (see “Methods”, Total evoked action potentials) (_p_ = 0.033, Fig. 1A). Representative traces of evoked action
potentials are shown in Fig. 1B–D. Next, we used voltage-clamp mode to measure the sodium and potassium currents in the control and PD neurons; representative traces of the currents are
shown in Fig. 1E–G. The average sodium currents are presented in Fig. 1H. At −20 mV, control neurons displayed a sodium current of 16.8 ± 3.3 pA/pF, whereas 2X neurons displayed a sodium
current of 3.7 ± 1.3 pA/pF (_p_ = 0.002 compared to controls), indicating later opening of the sodium channels pf the 2X PD neurons. The slow potassium currents are presented in Fig. 1I. An
ANOVA test indicated no significant differences. The fast potassium currents are shown in Fig. 1J. Similarly, running an ANOVA did not indicate significant differences. Analysis of the shape
of the action potential is presented in Supplementary Fig. 1A–D. Recording of Excitatory postsynaptic currents (EPSCs) revealed an altered network activity in both the 2X and 3X neurons.
Figure 1K displays a representative recording example of EPSCs in a control neuron, Fig. 1L displays an example recording of EPSCs in a 2X neuron, and Fig. 1M displays an example recording
of a 3X neuron. The cumulative distribution of the amplitudes of EPSCs was left-shifted in 2X neurons and further left-shifted in 3X neurons, indicating lower amplitudes of EPSCs
(Supplementary Fig. 1E). The average EPSC amplitude was decreased in the 2X neurons (_p_ = 0.0006 between the control and 2X neurons, Fig. 1N). The rate of synaptic events per recorded cell
is shown in Fig. 1O. The average frequency of EPSCs was 1.04 ± 0.35 Hz for control neurons, 0.21 ± 0.17 Hz for 2X neurons (_p_ = 0.04), and 0.037 ± 0.012 Hz for 3X neurons (_p_ = 0.01). The
cell capacitance was not significantly different between the three groups (Supplementary Fig. 1G). The input conductance was increased in 3X neurons (_p_ = 0.0016 between 3X and control
neurons, and _p_ = 0.0019 between 2X and 3X neurons, Supplementary Fig. 1H). To summarize, a reduction in synaptic activity was observed and exaggerated with α-synuclein gene dosage.
Analyzing the RNA sequencing data and looking at gene ontology and affected KEGG and MsigDB pathways, we found quite a few dysregulated pathways. Four control lines were pooled together for
this analysis: 40102, UKERf1JF-X-001, UKERf33Q-X-001, and UKERfO3H-X-001. We plotted the 15 most significant pathways in Supplementary Fig. 13 (up and downregulated). The full lists of GO
terms, KEGGS, and MSigDB pathways are presented in Supplementary Tables 3–5. Figure 1P presents the KEGG signaling pathway for RNA extracted from the 2X neurons compared to the control
neurons. Similarly, Fig. 1Q presents the top enriched KEGG signaling pathways for RNA extracted from the 3X neurons compared to the control neurons. Interestingly, in the patient with the
SNCA triplication, there were many synapse-related dysregulated pathways, with dozens of affected genes (Supplementary Fig. 13D and Supplementary Tables 3, 5), such as synapse part, synaptic
vesicle cycle, synaptic transmission, and many more. In the neurons derived from this patient, we also saw a very severe reduction in the rate of synaptic currents, further confirming that
synapses were severely disrupted. The full list of the genes involved in the affected pathways is presented in Supplementary Table 6 and consists of both presynaptic and postsynaptic genes.
We hypothesize that PD neurons are unable to form or maintain effective synapses, which we detect as a reduction in the rate of synaptic activity with electrophysiology, and this failure, in
turn, triggers compensation mechanisms that cause further dysregulation of synapse-related genes. Additionally, in the upregulated pathways, we found “Parkinson’s disease,” Alzheimer’s
disease” and “Huntington’s disease” showing commonalities between these neurodegenerative diseases. Several oxidative phosphorylation-related pathways also significantly overlapped with
upregulated genes, supporting the known role of oxidative stress in PD, as well as mitochondrial genes that were dysregulated. In the pathways that significantly overlapped with
downregulated genes, we found several metabolic pathways and pathways related to protein folding, as well as a few pathways that were not known before to relate to PD and are common in many
of our mutation lines as well as sPD (as will be presented in the subsequent figures), such as extracellular matrix pathways (probably the most dysregulated pathways), and PI3K-Akt signaling
pathways, lysosome membrane degradation, and focal adhesion. When performing immunocytochemistry (ICC) for two extracellular matrix (ECM) proteins fibronectin and collagen IV, we have
observed reduced puncta size in the SNCA triplication neurons compared to the control neurons (see Supplementary Fig. 11). In the neurons derived from the patient with the SNCA duplication,
the “ion transport” pathway and other transporter pathways were dysregulated (See Supplementary Tables 3, 5), which may be related to the changes that we observed in neuronal excitability
and ionic currents. In the electrophysiology data, the reduction in synaptic activity was less pronounced than in the patient with the SNCA triplication, further emphasizing the need for
electrophysiology for targeting smaller changes that are related to functionality. Several extracellular matrix-related pathways were also dysregulated in the patient with the SNCA
duplication, suggesting that there might be changes to the extracellular matrix structure that complement or even precede a possible synaptic degradation. Overall dysregulation of
extracellular matrix pathways is common to almost all the PD lines that we worked with. A DRASTIC REDUCTION IN SYNAPTIC ACTIVITY IS OBSERVED IN DA NEURONS DERIVED FROM PATIENTS WITH LRRK2
AND PARKIN MUTATIONS Our second cohort consisted of two control (two healthy subjects, 40102 and UKERfO3H-X-001 lines) individuals, one patient with a mutation in the LRRK2 and two patients
with mutations in the Parkin gene. We differentiated these lines using a DA human neuronal protocol84 (see “Methods”) and used whole-cell patch-clamp to assess intrinsic properties as well
as synaptic activity in the neurons (_n_ = 20 control neurons, _n_ = 31 from the patient with the LRRK2 mutation, _n_ = 12 from one patient with a Parkin mutation and _n_ = 15 neurons from
the second patient with a Parkin mutation, for a total of 27 neurons recorded for the Parkin mutation). The total number of evoked potentials in the 17 first depolarization steps was
decreased in the LRRK2 DA neurons (see “Methods”, Total evoked action potentials) and in the neurons with the Parkin mutation (_p_ = 0.0005 between control and LRRK2 mutation, _p_ = 0.059
between control and the two Parkin mutations, pooling the data of the two mutations together, Fig. 2A). Representative traces of evoked action potentials are shown in Fig. 2B–E. Next, we
measured the sodium and potassium currents in voltage-clamp mode in the control, LRRK2, and Parkin DA neurons. The average sodium currents are presented in Fig. 2F. The average slow
potassium currents are presented in Fig. 2G and the average fast potassium currents are presented in Fig. 2H. The sodium, slow, and fast potassium currents were reduced in one of the Parkin
lines (PET) (using an ANOVA, _p_ = 0.0002 for sodium currents, _p_ = 0.02 for slow potassium currents, and _p_ = 0.01 for fast potassium currents). To assess network activity, we next
measured EPSCs. Representative traces of measured EPSCs are shown in Fig. 2I (control), 2J (LRRK2 mutation), 2K (first patient with a Parkin mutation termed Parkin1), and 2L (the second
patient with a Parkin mutation, termed Parkin2). There was no significant change in the average amplitude of the synaptic currents (Fig. 2M). There was a significant reduction in the
frequency of synaptic currents in all three PD-causing mutations. Control neurons had 0.95 ± 0.25 Hz, LRRK2 0.27 ± 0.1 Hz, Parkin1 0.16 ± 0.07 and Parkin2 0.21 ± 0.08 Hz (LRRK2 compared to
control _p_ = 0.009, Parkin1 compared to control _p_ = 0.027, Parkin2 compared to control _p_ = 0.019, Fig. 2N). When we performed imaging of calcium transients (see Supplementary Fig. 10),
we observed smaller correlations between the spontaneously active neurons of the DA neurons with the Parkin mutation compared to the control neurons, suggesting that neurons with the Parkin
mutation are less connected, further supporting the data of the patch-clamp showing a reduction in the EPSC frequency. We also analyzed several features of the shape of the spikes. There was
a significant decrease in the spike amplitude in the LRRK2 mutation (LRRK2 vs. control, _p_ = 0.01; Parkin pooled together vs. control, _p_ = 0.07, Supplementary Fig. 2A). There were no
significant changes in the spike threshold (Supplementary Fig. 2B), the spike width (Supplementary Fig. 2C), or the fast afterhyperpolarization (AHP) amplitude (Supplementary Fig. 2D). The
cumulative distribution of the amplitude of EPSCs was similar between the control and the LRRK2 and Parkin lines (Supplementary Fig. 2E). There were no significant differences in the
capacitance of the neurons (Supplementary Fig. 2F), and the input conductance was significantly larger in the first line with the Parkin mutation compared to the control (_p_ = 0.03,
Supplementary Fig. 2G). The top 10 GO terms and KEGG and MSigDB over-representation analyses for the DA neurons with the LRRK2 and Parkin mutations are presented in Supplementary Fig. 14,
and the full list is shown in Supplementary Tables 3, 5. The signaling network analysis with the top enriched KEGG pathways is presented in Fig. 2O for the LRRK2 mutant neurons compared to
the control neurons. The differentially expressed gene list is presented in Supplementary Table 4. Interestingly, several synaptic transmission pathways were significantly overlapped with
upregulated genes in the LRRK2 mutation, in agreement with our electrophysiological data, with approximately 10–20 affected genes in each of these pathways; these included “regulation of
synaptic transmission”, “excitatory synapse” and more (these are shown in Supplementary Tables 3, 5). Similar to the neurons derived from patients with the SNCA copy number variation,
Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease, oxidative phosphorylation, and oxidation reduction-related pathways were significantly overrepresented among upregulated
genes. In addition, mitochondrial pathways were overrepresented among upregulated genes, presenting more evidence that mitochondrial defects are present in PD. Among the terms significantly
overlapping with downregulated genes we found many affected pathways that were related to the extracellular matrix, PI3K-Akt signaling pathway, lysine degradation, focal adhesion, and FoxO
signaling pathways, as well as protein folding-related pathways. When performing ICC for two ECM proteins fibronectin and collagen IV, we have observed reduced puncta size in the LRRK2
neurons compared to the control neurons (see Supplementary Fig. 11). Some of these pathways were not previously known to be associated with PD, but they were dysregulated in most of our PD
lines, showing new possible cellular defects that may be targeted for treatment. For the DA neurons derived from the PD patient with the Parkin mutation, there were not many significantly
dysregulated pathways. These did include “transport vesicle membrane.” A REDUCTION IN SYNAPTIC ACTIVITY IS OBSERVED IN NEURONS DERIVED FROM SPD PATIENTS We continued the study by
differentiating neurons from a PD patient with no PD-causing mutations (UKERfAY6-X-001). We recorded _n_ = 20 control neurons (two patients: 40102 and UKERfO3H-X-001) and _n_ = 33 neurons
from the sPD patient. There was no significant change in the total evoked action potentials (Fig. 3A for averages and 3B, 3C for representative traces). The average of the sodium currents is
presented in Fig. 3D. The average over the slow potassium currents is presented in Fig. 3E and the average of the fast potassium currents is presented in Fig. 3F. There was no significant
difference between the sPD and the control neurons in any of the sodium, slow potassium, and fast potassium currents. Representative EPSC traces are presented in Fig. 3G, H. There were no
significant differences in the mean amplitude (Fig. 3I) or the mean rate (Fig. 3J) of EPSCs. However, when counting the number of neurons that had synaptic activity (see “Methods”), we did
see a significant reduction in this number in the sPD neurons (_p_ = 0.0013, Fig. 3K). Furthermore, analysis of the decay time constant of the synaptic events revealed a faster decay in sPD
neurons, with an average of 2.3 ± 0.1 ms for control neurons and 1.5 ± 0.07 ms for sPD neurons (_p_ = 9e−12, the distribution is presented in Supplementary Fig. 3). Spike shape analysis did
not reveal any significant changes in the spike height (Supplementary Fig. 4A). The spike was significantly narrower in the sPD neurons (control 6 ± 0.8 ms, sPD 3.6 ± 0.2 ms, _p_ = 0.0035,
Supplementary Fig. 4B). The spike threshold was significantly more negative in the sPD neurons (control −18.4 ± 1.3 mV, sPD −24.6 ± 1 mV, _p_ = 0.0006, Supplementary Fig. 4C). There was no
significant change in the fast AHP (Supplementary Fig. 4D) or the capacitance (Supplementary Fig. 4F), and the input conductance was larger in sPD neurons (control 0.44 ± 0.12 nS, sPD 0.89 ±
0.16 nS, _p_ = 0.05, Supplementary Fig. 4G). The cumulative distribution of amplitudes of EPSCs was similar between the control and sPD neurons (Supplementary Fig. 4E). Three sPD lines
(UKERfRJO-X-001, UKERfAY6-X-001, and UKERfM89-X-001) were pooled together for the analysis of gene expression, plus four control (lines 40102, UKERf1JF-X-001, UKERf33Q-X-001, and
UKERfO3H-X-001). The top 10 GO terms, KEGG, and MSigDB dysregulated pathways for the DA neurons derived from sPD patients are presented in Supplementary Fig. 15, and the full list is shown
in Supplementary Tables 3–5. The signaling network analysis with the top enriched KEGG pathways for the sPD neurons compared to the control neurons is presented in Fig. 3L. There were a few
dysregulated pathways that were synapse-related such as regulation of synaptic structure, regulation of synaptic organization, regulation of synaptic plasticity, and more. The most
dysregulated pathways were related to nasopharyngeal carcinoma, with almost 250 affected genes and a false discovery rate (FDR) of 1.6e–105. Other highly dysregulated pathways were related
to the cilium. Like the other genetic PD lines, the extracellular matrix was highly affected, and more dysregulated pathways that were repeatedly affected in our PD lines were focal
adhesion, collagen processes, PI3K-Akt, protein digestion and absorption, and pathways related to reactive oxygen species and metabolic processes. When performing ICC for two ECM-proteins
fibronectin and collagen IV, we have observed reduced puncta size in the sPD neurons compared to the control neurons (see Supplementary Fig. 11). Several hypoxia-related pathways were
affected as well. A REDUCTION IN SYNAPTIC ACTIVITY IS OBSERVED IN NEURONS DERIVED FROM MORE SPD PATIENTS USING A DIFFERENT PROTOCOL AND SELECTING A SUBSET OF THE NEURONS We next analyzed
data that were acquired using a different differentiation protocol (see “Methods”, second DA differentiation); in addition, only neurons that were defined as type 5 neurons (see definition
in “Methods”) were used for the analysis. Strikingly, despite the different methods used both in differentiating the neurons and in analyzing the data, the main phenotype of a reduction in
synaptic activity was present in the sPD neurons as well. We recorded from control individual (line 40102) and two sPD patients (UKERfRJO-X-001, UKERfR66-X-001). It should be noted that one
of the patients had a heterozygous missense mutation in the EIF4G1 gene (UKERfRJO-X-001). The number of total evoked action potentials was not different between control and sPD neurons
(averages presented in Fig. 4A and representative traces are presented in Fig. 4B, C). The averages of the sodium currents are presented in Fig. 4D, the averages of the slow potassium
currents are presented in Fig. 4E, and the averages of the fast potassium currents are presented in Fig. 4F. Representative traces of synaptic activity are presented in Fig. 4G (control) and
4H (sPD). The average amplitude of the synaptic currents was increased in sPD neurons, but not significantly (Fig. 4I). The average rate of synaptic currents was significantly reduced in
the sPD neurons (control 3 ± 0.6 Hz, sPD 1.4 ± 0.2 Hz, _p_ = 0.0037, Fig. 4J). The cumulative distribution of the amplitude of synaptic currents was slightly right-shifted in the sPD
neurons. The capacitance was reduced in the sPD neurons, indicating smaller cells (control 64 ± 38 pF, sPD 45 ± 33 pF, _p_ = 0.02). Performing action potential shape analysis, we did not
observe any significant changes in the spike height (Supplementary Fig. 5A) or the spike width (Supplementary Fig. 5B). The amplitude of the fast AHP was larger in the sPD neurons (control
−5.7 ± 0.8 mV, sPD −10.9 ± 0.6 mV, _p_ = 0.0000035, Supplementary Fig. 5C), and the threshold for evoking an action potential was more depolarized (control −43 ± 0.8 mV, sPD −39.9 ± 0.6 mV,
_p_ = 0.0016, Supplementary Fig. 5D). A REDUCTION IN SYNAPTIC ACTIVITY IS OBSERVED IN NEURONS DERIVED FROM AN EDITED IPSC LINE WITH AN INSERTED A53T MUTATION IN THE SNCA GENE We next
performed experiments on neurons derived from a healthy human subject whose fibroblasts were reprogrammed into iPSCs and on an engineered line, in which the A53T mutation was edited into the
SNCA gene (isogenic lines). The healthy line and the edited mutated line were differentiated into DA neurons by a differentiation technique that is proprietary to Fujifilm Cellular Dynamics
Inc (CDI) (iCell DopaNeurons85). Having an edited healthy line allowed us to observe a similar genetic background, thereby measuring the neuronal changes that occurred specifically due to
the A53T mutation. iCell DopaNeurons (CDI) were previously shown to have a protein expression pattern that supported a midbrain lineage DA phenotype85. Using whole-cell patch-clamp, we
measured the functional features of A53T neurons compared to the control neurons. Fourteen A53T and 17 control neurons were recorded. The total number of evoked potentials in 17
depolarization steps (see “Methods”) was not significantly different between A53T neurons (39 ± 5) and control neurons (43 ± 6) (Fig. 5A and representative traces in Fig. 5B, C). Using
voltage clamping we measured the sodium/potassium currents in the neurons (representative traces are shown in Fig. 5D, E). Sodium currents were not significantly different, except for the
opening of the sodium channels; at −30 mV, control neurons displayed a sodium current of 1.6 ± 0.7 pA/pF, whereas A53T neurons displayed a sodium current of 10.4 ± 2.5 pA/pF (_p_ = 0.0012,
Fig. 5F). Slow and fast potassium currents were not significantly different between A53T neurons and controls (Fig. 5G, H). Spike parameters were not altered in the A53T neurons
(Supplementary Fig. 6A–D). The cumulative distribution curve of the amplitude of synaptic currents was left-shifted in A53T compared to controls, indicating lower amplitudes of synaptic
currents (Fig. 5I). Representative example recordings of EPSCs for control neurons (Fig. 5J) and an A53T neuron (Fig. 5K) are shown. The mean amplitude of synaptic currents was significantly
reduced in A53T neurons (16.7 ± 1.2 pA) compared to control neurons (20.6 ± 0.3 pA, _p_ = 2e−6, Fig. 5L). The mean rate of synaptic events was significantly reduced in A53T neurons (0.2 ±
0.06 Hz) compared to control neurons (1 ± 0.4 Hz, Fig. 5M, _p_ = 0.05). Cell capacitance was smaller, but not significantly, in A53T neurons (20.2 ± 1.6 pF) compared to control neurons (23 ±
1.4 pF, Supplementary Fig. 6E). The input conductance was decreased, but not significantly, in A53T neurons (Supplementary Fig. 6F). Further imaging of neurons stained for TH showed that
the A53T neurons were smaller and had fewer, less arborized neurites (Supplementary Fig. 7A–H). We similarly analyzed immunostaining images for the other PD lines and did not find changes in
neurites’ lengths (see Supplementary Fig. 8). The mean percentage of neurons with neurite beading was 3.7 ± 0.6% in control cultures and 28 ± 1.8% in A53T cultures (_p_ = 2e−10,
Supplementary Fig. 7I–K). Increased expression of the SNCA gene was observed in the A53T mutant neurons, and also in the LRRK2 mutant neurons, and the neurons derived from the patient with
the triplication in the SNCA gene (Supplementary Fig. 9). The top 10 affected KEGG and MsigDB pathways and GO terms are presented in Supplementary Fig. 16, and the full list is given in
Supplementary Tables 3–5. The signaling network analysis with the top enriched KEGG pathways of the A53T mutant neurons compared to the controls is presented in Fig. 5N. There were many
dysregulated synapse-related pathways such as dopaminergic synapse, postsynaptic membrane, presynaptic membrane, chemical synaptic transmission, and more. Similar to the other PD lines
presented in this study, there were many extracellular matrix-related affected pathways as well as pathways related to focal and cell adhesion, collagen processes, protein digestion and
absorption, and PI3K-Akt signaling pathways. It is interesting to note that similar findings of synaptic defects, both at the electrophysiological and morphological level, as well as
dysregulation of pre- and post-synaptic factors and transsynaptic adhesion molecules identified by transcriptome analysis, were previously reported44. PROTEIN AGGREGATES AND ALTERED
MORPHOLOGY IN THE A53T SNCA MUTATED IDOPANEURONS COMPARED TO CONTROL NEURONS We next delved deeper to look for additional morphological and cellular alterations that occurred in the
engineered A53T iDopaNeurons neurons compared to controls. The neurons were stained for neuronal markers TUJ1 and MAP2, and both A53T and control neurons expressed these markers (Fig. 6a–d).
Misfolded protein aggregates were observed in A53T neurons but rarely seen in control neurons (Fig. 6e–j). The number of aggregates was quantified using the aggresomal kit and imaging in
ultra-high resolution, 60.8 ± 7.1% of the A53T neurons displayed protein aggregates (_n_ = 99), compared to 2.2 ± 0.6% of the control neurons (_n_ = 96, _p_ < 0.0001, Fig. 6q). To assess
the number of synapses, we co-stained for synapsin1 and the post-synaptic marker PSD95. The number of puncta pairs Syn1/PSD95 density was significantly reduced in A53T cultures; 2.3 ± 0.2
pairs in 10 µm were observed in control cultures, whereas only 1.5 ± 0.16 (_n_ = 15 neurites) pairs in 10 µm were observed in A53T cultures (_p_ = 0.009, Fig. 6k–p, r). To assess how many of
the misfolded protein aggregates were α-synuclein positive, we co-stained α-synuclein/aggresomes in the A53T neurons. Some of the aggregates contained α-synuclein protein (Fig. 6s–u).
SEEKING COMMONLY AFFECTED PATHWAYS IN PD NEURONS PD patients share similar symptoms despite completely different genes causing the disease, or even when there is no defined genetic cause.
Therefore, we sought to find common pathways between neurons derived from PD patients with different mutations. We started by pooling the RNA sequencing results from all our lines with
PD-causing mutations—SNCA duplication, SNCA triplication, LRRK2, and Parkin—and we looked for differential expression compared to the four control lines. The 10 top terms enriched among up
and down-regulated genes are presented in Fig. 7a–d (GO and KEGG pathways). The full list, as well as MsigDB dysregulated pathways, are given in Supplementary Tables 3–5. A clear picture
emerges of pathways that were strongly affected in neurons derived from all PD lines with mutations; some of them were not previously known to be associated with PD. Collagen-related
pathways and the extracellular matrix were very strongly affected in the PD lines, as was confirmed by ICC experiments at the protein level for Fibronectin and collagen IV (Supplementary
Fig. 11). Focal adhesion was another pathway that repeated throughout the monogenic PD lines, as well as the PI3K-Akt signaling pathway, pathways related to cancer and oxidoreductase
activity, and protein digestion and absorption. Importantly, synapse-related pathways such as synapse, synaptic membrane, postsynapse, and more were commonly dysregulated in our monogenic PD
lines. In the up-regulated pathways, we found cell adhesion molecules (CAM), which are molecules that interact with the extracellular matrix and may be a compensation mechanism for the
reduced collagen and other extracellular matrix-related genes. We also looked for overrepresented GO terms for genes that were common in the monogenic PD lines and the sPD lines and these
are presented in Fig. 7e, f. The extracellular matrix is commonly implicated in both monogenic and sPD. Other dysregulated pathways were collagen pathways, focal and cell adhesion, PI3K-Akt
signaling pathway, protein digestion and absorption, and a few pathways related to hypoxia. Pathways related to oxidative stress such as reactive oxygen species and oxidative stress were
also dysregulated. Age-related pathways such as brain up, Alzheimer’s disease up, and cellular senescence were also commonly dysregulated in both monogenic and our sPD lines. Pathways that
might be related to synapse function that were dysregulated included cell-cell junction, axon guidance, cell junction assembly, vasculature development, and regulation of neuron development
projection. SEEKING PATHWAYS RELATED DIRECTLY TO A REDUCTION IN SYNAPTIC TRANSMISSION RATE The common electrophysiological phenotype that we observed in all PD lines was a drastic and
significant reduction in the rate of synaptic current events. Therefore, we were interested to determine the affected pathways when the differential expression was taken relative to the EPSC
rate of the investigated cell line (see “Methods”). The 10 top upregulated pathways for neurons with reduced EPSC rates are presented in Fig. 7g–j and the entire list of dysregulated GO
terms and KEGG and MsigDB pathways are presented in Supplementary Tables 3–5. As expected, many synaptic pathways were dysregulated when comparing neurons that had a high-frequency rate of
synaptic activity vs. neurons with a low rate of synaptic events. These included, for example, dopaminergic synapse, glutamatergic synapse, GABAergic synapse, serotonergic synapse, aminergic
neurotransmitter loading into synaptic vesicles, filopodium, axon guidance, and many more. Other dysregulated pathways were similar to those that were dysregulated in PD, probably since PD
lines exhibited a reduced synaptic event rate. These included many extracellular matrix pathways, focal adhesion, CAMs, melanosome, lysosome, cilium, mitochondrial pathways, endoplasmic
reticulum-related pathways, and several cancer-related pathways. DISCUSSION PD affects the lives of nearly one million people in the US and is the most prevalent movement disorder. The
hallmarks of PD are aggregates of the α-synuclein protein that are more specific to areas in the brain with a high density of DA neurons. In some PD cases, Lewy bodies appear in those
DA-dense areas. These Lewy bodies are composed of protein aggregates whose main component is the α-synuclein protein. However, it is not clear if the Lewy bodies are causing the neuronal
cell death that is observed in high-density DA neuron areas or is a side effect of other processes that occur and are the actual triggers for neurodegeneration. Recent studies suggest that
degeneration starts as micro aggregates of the α-synuclein protein form, long before Lewy bodies appear. Strikingly, there are several different PD-causing mutations, but most patients do
not have a known genetic origin and are considered idiopathic or sporadic. Finding phenotypes that are common to PD early in the lifetime of neurons (and the patients) will help to
understand disease mechanisms and develop an effective treatment. Furthermore, the use of patient-derived neurons helps to mitigate the problem of the lack of animal models for sporadic PD.
Here we report a neurophysiological phenotype that is common to DA neurons derived from PD patients for the following mutations: the A53T SNCA mutation, SNCA copy number variations
(duplication as well as triplication), LRRK2, Parkin, and importantly, sPD patients. Using whole-cell patch-clamp, we found that neurons derived from PD patients all exhibited a significant
reduction in the rate of synaptic activity. It is known that neurons that are plated sparsely have a low survival rate86,87; neurons need interactions with their neighboring neurons for
survival. Therefore, the low connectivity that we observed in the PD neurons may exacerbate neuronal death caused by different mechanisms. Several labs have shown that reprogramming of adult
cells into iPSCs erases aging signatures and epigenetic modifications81,82, and therefore the neurons in our cultures are considered to be young, and even pre-natal neurons. Consequently,
the observed phenotype of a reduction in synaptic activity in these very young neurons indicates an early process and a biological predisposition that is present in the patients’ DA neurons,
probably long before patients exhibit any motor deficits. Similar findings were described in SNCA mutations in mice42,43,45,47,48. In these mice, the earliest phenotype that was observed
was a reduction in synaptic activity, followed by protein aggregates that, after weeks and sometimes months, evolved into the loss and death of DA neurons and motor dysfunction. Overall, our
results imply that neuronal cultures derived from human patients using iPSC techniques are a good model for studying PD progression, starting with this prodromal phenotype in the neurons.
Moreover, this phenotype may have important implications for the early diagnosis and prediction of disease onset. The α-synuclein gene plays a major role in PD. The SNCA gene was only
dysregulated in the neurons derived from the patients with the LRRK2, A53T, and SNCA triplication mutations, with a very large increase in the neurons derived from the patient with the SNCA
triplication (Supplementary Fig. 9). It is interesting to note that the neurons derived from the patient with the SNCA duplication do not significantly overexpress SNCA, perhaps due to
compensation mechanisms. However, we revealed shared dysregulated pathways analyzing gene expression of neurons derived from PD patients with different genetic mutations as well as the
sporadic form of the disease and this was often consistent with, and supportive of the electrophysiology. Among these dysregulated pathways and genes, we found the CAMs. Several CAM families
have been shown to localize at the synapses88,89 and to influence the assembly and function of synapses in the CNS. The PI3K-Akt signaling pathways were also overrepresented among
downregulated genes in both monogenic and sPD, and they have been shown to recruit PSD-95 to synapses90. The FoxO pathway, another commonly overrepresented pathway among downregulated genes
in our PD lines, has also been shown to play an important role in synaptic growth, synaptic vesicle exocytosis91, and the promotion of synaptic plasticity92. The latter two pathways have
been shown to interact93. Proteoglycans and collagen fibers also exhibited dysregulation in both our monogenic and sPD lines. These make up the extracellular matrix, which more and more
evidence suggests is a part of the tetrapartite synapse. The extracellular matrix-related pathways were strongly dysregulated in almost all our PD lines. Overall, we saw the dysregulation of
many genes and pathways that affected synaptic formation. We hypothesize that this gene expression dosage dysregulation prevents neurons derived from PD patients from establishing effective
synapses, measured in our electrophysiological experiments as a reduced rate of synaptic events. We hypothesize that PD neurons have a reduced ability to form operative synapses, which
reduces the rate of synaptic events and triggers homeostatic mechanisms that take place; a vast dysregulation of genes associated with synaptic transmission-related pathways then occurs, as
we observed when analyzing gene expression in our PD lines. It is interesting to note that, in those PD lines where there was a stronger decrease in the synaptic event’s rate, we also
observed more abundant synaptic-related pathways that were overrepresented among upregulated genes. In the LRRK2 and the SNCA duplication, where the reduction in the synaptic rate was not as
severe, we detected fewer synaptic pathways that were significantly enriched among upregulated genes, further stressing the importance of electrophysiology for the detection of subtler
changes. Overall, our results strongly suggest that PD pathology starts at the synapse (as our in vitro neurons are young, and therefore reflect early brain events), in agreement with
previous reports and hypotheses regarding the role of disrupted synapses in PD27,94,95. Studies have shown that loss of synaptic terminals exceeds the loss of DA cell bodies74 and that
α-synuclein aggregates at the presynaptic terminals before forming Lewy bodies18,28; these results are supported by our findings of young and rejuvenated DA neurons that already exhibit
synaptic deficits. To summarize, our work demonstrates an early phenotype that is common to neurons from several PD mutations as well as sPD patients. It also demonstrates that there are
common pathways that are affected in and common to PD patients (mutation-driven or sporadic). Importantly, these findings reveal that PD can be studied using iPSC-derived neuron technology,
allowing us to trace the disease progression step by step. The affected pathways that we have identified through the analysis of gene expression should now be considered important targets
for further research. METHODS A written informed consent was provided by all the participants in the study to take part in the study. ETHICS The study was approved by the Salk institute with
the following approvals: IRB 09-0003 and ESCRO 07-009. HUMAN PATIENTS Supplementary Tables 1 and 2 (see Supplementary) present the clinical features of the human patients who participated
in this study. The first cohort, listed in Supplementary Table 1, was diagnosed by Dr. Juergen Winkler, and the second cohort (Supplementary Table 2) was diagnosed by Dr. Alexis Brice. A
written informed consent was provided by all the participants in the study to take part in the study. DOPAMINERGIC (DA) NEURONS DA neurons were generated from iPSCs based on a previously
described protocol84 with modifications96. This protocol robustly produces over 80% DA neurons within the neuronal population in the culture. We performed an assessment of the neuronal types
in our different PD lines and controls, and the protocols yields a high percentage of DA neurons with almost no glutamatergic neurons and approximately 5–10% GABAergic neurons (see
Supplementary Fig. 12). In brief, iPSCs were dissociated into a single cell suspension with TrypLE and replated on Matrigel-coated plates at a density of 40,000 cells/cm2 in mTesR medium
(Stem Cell Technologies). Cells were allowed to propagate with a daily change of medium for two days. Two days after the propagation of the cells, the differentiation process was started by
switching into KSR medium (DMEM F-12 with Glutamax, 15% KO-SR, 1% NEAA, 1% Antibiotic-Antimycotic, 0.1 mM b-mercaptoethanol); this day was regarded as day 0. The medium was gradually changed
to N2 medium (DMEM F-12 with Glutamax, 1% N2 supplement, 1% Antibiotic-Antimycotic) from day 5 to day 10 (day 5 and 6: 75% KSR : 25% N2; day 7 and 8: 50% KSR : 50% N2; day 9 and 10: 25% KSR
: 75% N2). From Day 11 onward, the medium was switched to B27 medium (Neurobasal medium, 2% B27 supplement, 1% glutamax, 1% Antibiotic-Antimycotic, 10 ng/mL BDNF, 10 ng/mL GDNF, 1 ng/mL
TGF3, 0.2 mM ascorbic acid and 0.1 mM cAMP). Cocktails of small molecules were added to the culture throughout the differentiation process (10 µM SB431542 on day 0–4; 100 nM LDN-193189 on
day 0–12; 2 µM purmorphamine, 0.25 µM SAG, 100 ng/mL FGF8 on day 1–6; 3 µM CHIR99021 on day 3–12). Cells were replated onto Matrigel-coated coverslips on day 20 and further matured in B27
medium until day 30. On day 30, the base medium was switched to BrainPhys medium97 until, on day 45–50, whole-cell patch-clamp recording was performed. All neurons that had a good quality
patch-clamp seal were included in the analysis. The neurons in one of the experiments (results presented in Fig. 4) were grown and differentiated at the Bardy lab, using a published
protocol97,98. This protocol produces a smaller proportion of tyrosine hydroxylase (TH)-positive neurons. Furthermore, the analysis was performed only on neurons that were classified as
“type 5” neurons by this protocol. PLATING NEURONS FROM A COMMERCIAL CONTROL LINE AND AN AFFECTED LINE We utilized commercial neurons from Fujifilm Cellular Dynamics International (CDI),
which are a more homogeneous culture of neurons and produce more than 80% pure midbrain DA neurons The detailed nomenclatures/catalog numbers for the cells used in this paper are iCell
DopaNeurons, 01279, catalog number: C1028; and MyCelliDopaNeurons, C1279, catalog number: C1113, genotype: SNCA (A53T alfa synuclein mutation). The cells were defrosted according to the
protocol on the User’s Guide from CDI and counted. The medium used was Brainphys (Stem Cell Technologies), with the addition of supplements, according to the manufacturer’s instructions:
iCell Neural Supplement B (catalog number: M1029) and iCell Nervous System Supplement (catalog number: M1031), N2 supplement, Laminin, and antibiotics (penicillin/streptomycin). For patch
clamp electrophysiology experiments, we used Poly-L-ornithine- and Laminin-coated, 24-well plates with coverslips in each well, and we plated 4–6 × 105 cells per well. Recordings started
after a week in culture. For the RNA sequencing experiments, we plated 1 × 106 cells per well on six-well plates. For immunostaining, we plated about 1 × 105 cells per well of 8 chamber
ibidi slides. ICELL DOPANEURON CULTURES ON MULTI-ELECTRODE ARRAYS (MEAS) Control iCell DopaNeurons (Fujifilm Cellular Dynamics Inc.) and MyCell _SNCA_ A53T DopaNeurons were thawed and dotted
(~80k cells) onto a 48-well MEA plate (Axion Biosystems) using the iCell DopaNeuron MEA application protocol. Cultures were treated with BrainPhys medium (Stem Cell Technologies), with 50%
medium changes performed every two to three days for 20 days. Five-minute recordings were made on DIV20 post-plating (_N_ = 24). Raw voltage recordings were processed via a Butterworth
(200–4 kHz) filter; action potentials were then detected by a 5.5 standard deviation detection threshold, and network-level bursting behaviors were analyzed off-line using the Axion’s
NeuralMetric toolbox via the ‘Envelope’ algorithm (threshold factor 3, minimum inter-burst-interval 10 s, burst inclusion percentage 75, minimum number of electrodes percentage 25) and
synchrony index (20 ms)48. The activity and network-level bursting behaviors assessed included mean firing rate, network bursting rate (bursts per minute: BPM), intensity (Hz), and duration
(seconds). A two-tailed student t-test was used to assess statistical differences on each measure independently between the two groups of neurons. ELECTROPHYSIOLOGY—WHOLE-CELL PATCH-CLAMP
Neurons on glass coverslips were transferred to a recording chamber in a standard recording medium containing (in mM) 10 HEPES, 4 KCl, 2 CaCl2, 1 MgCl2, 139 NaCl, 10 D-glucose (310 mOsm, pH
7.4). Whole-cell patch-clamp recordings were performed on days 9–12 days after plating for A53T vs. iDOPA CDI lines and for 40 days after plating for the other DA protocols. Patch electrodes
were filled with internal solutions containing (in mM) 130 K-gluconate, 6 KCl, 4 NaCl, 10 Na-HEPES, 0.2 K-EGTA, 0.3 GTP, 2 Mg-ATP, 0.2 cAMP, 10 D-glucose, 0.15% biocytin and 0.06%
rhodamine. The pH and osmolarity of the internal solution were brought close to physiological conditions (pH 7.3, 290–300 mOsmol). Signals were amplified with a Multiclamp700B amplifier and
recorded with Clampex 10.2 software (Axon Instruments). Data were acquired at a sampling rate of 20 kHz and analyzed using Clampfit-10 and the software package Matlab (2018b, The MathWorks
Inc., Natick, MA, 2000). All measurements were conducted at room temperature. For post-synaptic current measurements, 40 µM of bicuculline was applied and cells were held at −60 mV when
currents were recorded (−70 mV after correction of junction liquid potential). ANALYSIS ELECTROPHYSIOLOGY TOTAL EVOKED ACTION POTENTIALS Cells were typically held in current-clamp mode near
−60 mV with a steady holding current, and current injections were given starting 12 pA below the steady holding current in 3 pA steps 400 ms in duration. A total of 35 depolarization steps
were injected in current-clamp mode. Neurons that needed more than 50 pA to be held at −60 mV were discarded from the analysis. The total number of action potentials was counted in the first
17 depolarization steps. ACTION POTENTIAL SHAPE ANALYSIS The first evoked action potential was used for spike shape analysis (with the lowest injected current needed for eliciting an action
potential). Spike threshold was the membrane potential at which the slope of the depolarizing membrane potential increased drastically, resulting in an action potential (the first maximum
in the second derivative of the voltage vs. time). The fast (5 ms) AHP amplitude was calculated as the difference between the threshold of the action potential and the value of the membrane
potential 5 ms after the potential returned to cross the threshold value at the end of the action potential. The spike amplitude was calculated as the difference between the maximum membrane
potential during a spike and the threshold. Action potential width was calculated as the time it took for the membrane potential to reach half the spike amplitude in the rising part of the
spike to the descending part of the spike (Full Width at Half Maximum). INPUT CONDUCTANCE The input conductance was calculated around the resting membrane potential by measuring the current
with the cell held in voltage-clamp mode first at −70 mV and then at −50 mV. The difference in currents divided by the difference in membrane potential (of 20 mV) is the calculated input
conductance. SODIUM AND POTASSIUM CURRENTS The sodium and potassium currents were acquired in voltage-clamp mode. Cells were held at −60 mV, and voltage steps of 400 ms were made in the
range of −90 mV to 80 mV. Currents were normalized by the cell capacitance. FAST AND SLOW POTASSIUM CURRENTS We measured the fast potassium current by the maximum current immediately
following a depolarization step, typically within a time window of a few milliseconds. The slow potassium currents were obtained at the end of the 400 ms depolarization step. CAPACITANCE The
capacitance was measured by the membrane test of the Clampex SW. SYNAPTIC ACTIVITY Excitatory synaptic activity was measured in voltage-clamp mode with 40 μM bicuculline applied in the
recording medium. The neurons were held at −60 mV and currents were measured in the patched neuron. We measured both the amplitude of these currents and their rates. A neuron was defined as
having synaptic activity if it had more than 20 events in a 60 s recording and as not having synaptic activity if it had fewer than 20 events in 60 s. RNA-SEQUENCING ANALYSIS Sequenced reads
were quality-tested using FASTQC99 v0.11.5 and aligned to the hg19100 human genome using the STAR aligner101 version 2.5.3a. Mapping was carried out using default parameters, filtering
non-canonical introns and allowing up to 10 mismatches per read, and only keeping uniquely mapped reads. The genome index was constructed using the gene annotation supplied with the hg19
Illumina iGenomes collection102 and sjdbOverhang value of 100. Raw or TPM (Transcripts per million) gene expression was quantified across all gene exons with HOMER103 using the top-expressed
isoform as a proxy for gene expression, and differential gene expression was carried out on the raw counts using the edgeR104 package version 3.28.1. For each disease type, differentially
expressed genes were defined as having a false discovery rate (FDR) <0.05 when comparing two experimental conditions. We also separated monogenic subjects from sPD and compared each group
separately to controls using the exact test function in edgeR. We then combined all subjects (monogenic, sPD, and control) and treated the EPSC rate as a continuous variable; we conducted
differential expression analysis using the GLM model in edgeR. In both cases, differentially expressed genes were defined as having an FDR < 0.05. A GO enrichment test and KEGG pathway
analysis were performed using the program DAVID Bioinformatics Resources 6.8105. Overrepresentation of GO terms and KEGG pathway was determined by FDR < 0.05. MsigDB106 overrepresentation
analysis was carried out using HOMER findGO.pl using the corrected Benjamini & Yakutieli method for multiple testing correction107. IMMUNOCYTOCHEMISTRY AND CELL IMAGING Neuronal
cultures were fixed with 4% paraformaldehyde for 15 min at room temperature and then treated with PBS containing 0.1% Triton X-100. After a 15 min PBS wash, cells were blocked with 5% BSA in
PBS for 1 h, then incubated with the primary antibody in PBS at 4 °C overnight and the next day after a few PBS washes with secondary antibodies for 1 hour at ambient temperature. This
process was followed by a 10 min incubation with DAPI and a final set of PBS washes. The coverslips were mounted on glass slides using PVA-DABCO. Primary antibodies used were rabbit
anti-Tuj1, (1:500, Covance), chicken anti-MAP2 (1:400, Abcam), rabbit anti-αSynuclein (1:500, Invitrogen), rabbit anti-TH (1:500, Pel-Freez), mouse anti-PSD95 (1:500, Life Tech), rabbit
anti-Synapsin I (1:500, Calbiochem), anti-Fibronectin (1:200, Sigma). Corresponding Alexa FluorTM secondary antibodies were then used (1:1000). For detecting protein aggregates, the
PROTEOSTAT® Aggresome Detection kit was used according to the manufacturer’s instructions. Confocal z-stacks were acquired with a Zeiss LSM 880 Airy scan microscope (Carl Zeiss, Microimaging
Inc.) using 405 nm Diode laser, 488 nm Argon, and 543 nm HeNe lasers with a Plan NeoFluar 40×/1.3 oil DIC or a Plan-Apochromat 63×/1.4 oil DIC objective. IMAGING OF CELL MORPHOLOGY Neurons
stained for TH were imaged and analyzed with Neurolucida SW (MBF Bioscience), where neurites and soma were manually traced. CALCIUM IMAGING To image calcium transient neuronal cultures were
incubated for 60 min in the recording solution (128 mM NaCl, 4 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 45 mM sucrose, 10 mM glucose, and 10 mM HEPES; pH is titrated to 7.4) in the presence of 4
μg/ml cell-permeant Fluo5-AM (Abcam). Cultures were then placed in fresh recording solution and imaged. The calcium transients were imaged at 10 Hz similar to previous experiments
described108. STATISTICAL ANALYSIS Unless otherwise stated, _p_ values were calculated using a two-sample t-test (two-tailed). DATA AVAILABILITY The datasets generated and analyzed during
the current study are available in the figshare repository in the following links: https://figshare.com/articles/dataset/ephys_data_7z/14635452,
https://figshare.com/articles/dataset/MsigDB_xlsx/19665885, https://figshare.com/articles/dataset/DEGs_xlsx/19665897. The patients information is provided in
https://github.com/Precision-Disease-Modeling-Lab/NPJ-Parkinson-Disease-NPJPARKD-00785R1. The RNA sequencing raw data is available in GEO Series record GSE207533. CODE AVAILABILITY The
electrophysiological and Statistical analysis were performed using Matlab (2018b, The MathWorks Inc., Natick, MA, 2000). The Matlab scripts used for the analysis are available from the
corresponding author upon reasonable request. REFERENCES * Parkinson, J. An essay on the shaking palsy. 1817. _J. Neuropsychiatry Clin. Neurosci._ 14, 223–236 (2002). Article PubMed Google
Scholar * Tysnes, O. B. & Storstein, A. Epidemiology of Parkinson's disease. _J. Neural Transm._ 124, 901–905 (2017). Article PubMed Google Scholar * von Campenhausen, S. et
al. Prevalence and incidence of Parkinson's disease in Europe. _Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol._ 15, 473–490 (2005). Article CAS Google Scholar *
Bagheri, H. et al. A study of salivary secretion in Parkinson's disease. _Clin. Neuropharmacol._ 22, 213–215 (1999). CAS PubMed Google Scholar * Huang, P., Yang, X. D., Chen, S. D.
& Xiao, Q. The association between Parkinson's disease and melanoma: A systematic review and meta-analysis. _Transl. Neurodegeneration_ 4, 21 (2015). Article Google Scholar *
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson's disease. _Neurobiol. Aging_ 24, 197–211 (2003). Article PubMed Google Scholar * Burke, R. E., Dauer, W. T.
& Vonsattel, J. P. A critical evaluation of the Braak staging scheme for Parkinson's disease. _Ann. Neurol._ 64, 485–491 (2008). Article PubMed PubMed Central Google Scholar *
Jellinger, K. A. Critical evaluation of the Braak staging scheme for Parkinson's disease. _Ann. Neurol._ 67, 550 (2010). Article PubMed Google Scholar * Tran, J., Anastacio, H. &
Bardy, C. Genetic predispositions of Parkinson's disease revealed in patient-derived brain cells. _NPJ Parkinsons Dis._ 6, 8 (2020). Article PubMed PubMed Central Google Scholar *
Li, J. Q., Tan, L. & Yu, J. T. The role of the LRRK2 gene in Parkinsonism. _Mol. Neurodegener._ 9, 47 (2014). Article CAS PubMed PubMed Central Google Scholar * Nuytemans, K.,
Theuns, J., Cruts, M. & Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. _Hum.
Mutat._ 31, 763–780 (2010). Article CAS PubMed PubMed Central Google Scholar * Cummings, J. L. The dementias of Parkinson's disease: Prevalence, characteristics, neurobiology, and
comparison with dementia of the Alzheimer type. _Eur. Neurol._ 28(Suppl 1), 15–23 (1988). PubMed Google Scholar * Chartier-Harlin, M. C. et al. Alpha-synuclein locus duplication as a cause
of familial Parkinson's disease. _Lancet_ 364, 1167–1169 (2004). Article CAS PubMed Google Scholar * Golbe, L. I., Di Iorio, G., Bonavita, V., Miller, D. C. & Duvoisin, R. C. A
large kindred with autosomal dominant Parkinson's disease. _Ann. Neurol._ 27, 276–282 (1990). Article CAS PubMed Google Scholar * Singleton, A. B. et al. alpha-Synuclein locus
triplication causes Parkinson's disease. _Science_ 302, 841 (2003). Article CAS PubMed Google Scholar * Domingo, A. & Klein, C. Genetics of Parkinson disease. _Handb. Clin.
Neurol._ 147, 211–227 (2018). Article PubMed Google Scholar * Kramer, M. L. & Schulz-Schaeffer, W. J. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration
in dementia with Lewy bodies. _J. Neurosci. : Off. J. Soc. Neurosci._ 27, 1405–1410 (2007). Article CAS Google Scholar * Schulz-Schaeffer, W. J. Is cell death primary or secondary in the
pathophysiology of idiopathic Parkinson's disease? _Biomolecules_ 5, 1467–1479 (2015). Article CAS PubMed PubMed Central Google Scholar * Tanaka, M. et al. Aggresomes formed by
alpha-synuclein and synphilin-1 are cytoprotective. _J. Biol. Chem._ 279, 4625–4631 (2004). Article CAS PubMed Google Scholar * Luk, K. C. et al. Pathological alpha-synuclein
transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. _Science_ 338, 949–953 (2012). Article CAS PubMed PubMed Central Google Scholar * Nuber, S. et al.
Abrogating native alpha-synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson's disease. _Neuron_ 100, 75–90.e75 (2018). Article CAS
PubMed PubMed Central Google Scholar * Burre, J. The synaptic function of alpha-synuclein. _J. Parkinsons Dis._ 5, 699–713 (2015). Article CAS PubMed PubMed Central Google Scholar *
George, J. M. The synucleins. _Genome Biol._ 3, REVIEWS3002 (2002). PubMed Google Scholar * Withers, G. S., George, J. M., Banker, G. A. & Clayton, D. F. Delayed localization of
synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. _Brain Res. Dev. Brain Res._ 99, 87–94 (1997). Article CAS PubMed Google Scholar * Stefanis, L.
alpha-Synuclein in Parkinson's disease. _Cold Spring Harb. Perspect. Med._ 2, a009399 (2012). Article PubMed PubMed Central CAS Google Scholar * Bridi, J. C. & Hirth, F.
Mechanisms of alpha-synuclein induced synaptopathy in Parkinson's disease. _Front. Neurosci._ 12, 80 (2018). Article PubMed PubMed Central Google Scholar * Soukup, S. F.,
Vanhauwaert, R. & Verstreken, P. Parkinson’s disease: Convergence on synaptic homeostasis. _EMBO J_ 37, e98960 (2018). Article PubMed PubMed Central CAS Google Scholar * Spinelli,
K. J. et al. Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson's disease. _J. Neurosci. : Off. J. Soc. Neurosci._ 34, 2037–2050 (2014). Article CAS Google Scholar
* Fuchs, J. et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. _Neurology_ 68, 916–922 (2007). Article CAS PubMed Google Scholar * Ki, C.
S. et al. The Ala53Thr mutation in the alpha-synuclein gene in a Korean family with Parkinson disease. _Clin. Genet._ 71, 471–473 (2007). Article PubMed Google Scholar * Konno, T., Ross,
O. A., Puschmann, A., Dickson, D. W. & Wszolek, Z. K. Autosomal dominant Parkinson's disease caused by SNCA duplications. _Parkinsonism Relat. Disord._ 22(Suppl 1), S1–S6 (2016).
Article PubMed Google Scholar * Kruger, R. et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. _Nat. Genet._ 18, 106–108 (1998). Article CAS
PubMed Google Scholar * Polymeropoulos, M. H. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. _Science_ 276, 2045–2047 (1997). Article
CAS PubMed Google Scholar * Puschmann, A. et al. A Swedish family with de novo alpha-synuclein A53T mutation: evidence for early cortical dysfunction. _Parkinsonism Relat. Disord._ 15,
627–632 (2009). Article PubMed PubMed Central Google Scholar * Zarranz, J. J. et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. _Ann. Neurol._
55, 164–173 (2004). Article CAS PubMed Google Scholar * Murphy, D. D., Rueter, S. M., Trojanowski, J. Q. & Lee, V. M. Synucleins are developmentally expressed, and alpha-synuclein
regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. _J. Neurosci. : Off. J. Soc. Neurosci._ 20, 3214–3220 (2000). Article CAS Google Scholar * Scott, D.
& Roy, S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. _J. Neurosci. : Off. J. Soc. Neurosci._ 32, 10129–10135 (2012). Article CAS
Google Scholar * Liu, S. et al. alpha-Synuclein produces a long-lasting increase in neurotransmitter release. _EMBO J._ 23, 4506–4516 (2004). Article CAS PubMed PubMed Central Google
Scholar * Lotharius, J. & Brundin, P. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson's disease. _Hum. Mol.
Genet._ 11, 2395–2407 (2002). Article CAS PubMed Google Scholar * Yavich, L., Tanila, H., Vepsalainen, S. & Jakala, P. Role of alpha-synuclein in presynaptic dopamine recruitment.
_J. Neurosci. : Off. J. Soc. Neurosci._ 24, 11165–11170 (2004). Article CAS Google Scholar * Eslamboli, A. et al. Long-term consequences of human alpha-synuclein overexpression in the
primate ventral midbrain. _Brain : J. Neurol._ 130, 799–815 (2007). Article Google Scholar * Ip, C. W. et al. AAV1/2-induced overexpression of A53T-alpha-synuclein in the substantia nigra
results in degeneration of the nigrostriatal system with Lewy-like pathology and motor impairment: A new mouse model for Parkinson's disease. _Acta Neuropathologica Commun._ 5, 11
(2017). Article CAS Google Scholar * Kirik, D. et al. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. _J. Neurosci. :
Off. J. Soc. Neurosci._ 22, 2780–2791 (2002). Article CAS Google Scholar * Kouroupi, G. et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of
familial Parkinson's disease. _Proc. Natl Acad. Sci. USA_ 114, E3679–E3688 (2017). Article CAS PubMed PubMed Central Google Scholar * Lundblad, M., Decressac, M., Mattsson, B.
& Bjorklund, A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. _Proc. Natl Acad. Sci. USA_ 109, 3213–3219 (2012). Article CAS PubMed
PubMed Central Google Scholar * Nemani, V. M. et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after
endocytosis. _Neuron_ 65, 66–79 (2010). Article CAS PubMed PubMed Central Google Scholar * Paumier, K. L. et al. Behavioral characterization of A53T mice reveals early and late stage
deficits related to Parkinson's disease. _PLoS One_ 8, e70274 (2013). Article CAS PubMed PubMed Central Google Scholar * Wu, N., Joshi, P. R., Cepeda, C., Masliah, E. & Levine,
M. S. Alpha-synuclein overexpression in mice alters synaptic communication in the corticostriatal pathway. _J. Neurosci. Res._ 88, 1764–1776 (2010). CAS PubMed PubMed Central Google
Scholar * Fishbein, I. & Segal, M. Miniature synaptic currents become neurotoxic to chronically silenced neurons. _Cereb. Cortex_ 17, 1292–1306 (2007). Article PubMed Google Scholar
* Bardien, S., Lesage, S., Brice, A. & Carr, J. Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson's disease. _Parkinsonism Relat. Disord._ 17,
501–508 (2011). Article PubMed Google Scholar * Rideout, H. J. & Stefanis, L. The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson's disease. _Neurochem. Res._
39, 576–592 (2014). Article CAS PubMed Google Scholar * Venderova, K. et al. Leucine-rich repeat kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of
Parkinson's disease. _Hum. Mol. Genet._ 18, 4390–4404 (2009). Article CAS PubMed Google Scholar * Liu, Z. et al. A Drosophila model for LRRK2-linked parkinsonism. _Proc. Natl Acad.
Sci. USA_ 105, 2693–2698 (2008). Article CAS PubMed PubMed Central Google Scholar * Saha, S. et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans.
_J. Neurosci. : Off. J. Soc. Neurosci._ 29, 9210–9218 (2009). Article CAS Google Scholar * Li, Y. et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of
Parkinson's disease. _Nat. Neurosci._ 12, 826–828 (2009). Article CAS PubMed PubMed Central Google Scholar * Tong, Y. et al. R1441C mutation in LRRK2 impairs dopaminergic
neurotransmission in mice. _Proc. Natl Acad. Sci. USA_ 106, 14622–14627 (2009). Article CAS PubMed PubMed Central Google Scholar * Cirnaru, M. D. et al. LRRK2 kinase activity regulates
synaptic vesicle trafficking and neurotransmitter release through modulation of LRRK2 macro-molecular complex. _Front. Mol. Neurosci._ 7, 49 (2014). Article CAS PubMed PubMed Central
Google Scholar * Inoshita, T. et al. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. _Hum. Mol. Genet._ 26, 2933–2948
(2017). Article CAS PubMed Google Scholar * Shin, N. et al. LRRK2 regulates synaptic vesicle endocytosis. _Exp. Cell Res._ 314, 2055–2065 (2008). Article CAS PubMed Google Scholar *
Piccoli, G. et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. _J. Neurosci. : Off. J. Soc. Neurosci._ 31, 2225–2237 (2011). Article CAS Google
Scholar * Sassone, J. et al. The synaptic function of parkin. _Brain : J. Neurol._ 140, 2265–2272 (2017). Article Google Scholar * Huttner, W. B., Schiebler, W., Greengard, P. & De
Camilli, P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. _J. Cell
Biol._ 96, 1374–1388 (1983). Article CAS PubMed Google Scholar * Kubo, S. I. et al. Parkin is associated with cellular vesicles. _J. Neurochem._ 78, 42–54 (2001). Article CAS PubMed
Google Scholar * Fallon, L. et al. Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. _J. Biol. Chem._
277, 486–491 (2002). Article CAS PubMed Google Scholar * Lucking, C. B. et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. _N. Engl. J.
Med._ 342, 1560–1567 (2000). Article CAS PubMed Google Scholar * Pankratz, N. et al. Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations.
_Neurology_ 73, 279–286 (2009). Article CAS PubMed PubMed Central Google Scholar * Pankratz, N. et al. Genomewide association study for susceptibility genes contributing to familial
Parkinson disease. _Hum. Genet._ 124, 593–605 (2009). Article CAS PubMed Google Scholar * Poulopoulos, M., Levy, O. A. & Alcalay, R. N. The neuropathology of genetic Parkinson's
disease. _Mov. Disord. : Off. J. Mov. Disord. Soc._ 27, 831–842 (2012). Article CAS Google Scholar * Helton, T. D., Otsuka, T., Lee, M. C., Mu, Y. & Ehlers, M. D. Pruning and loss of
excitatory synapses by the parkin ubiquitin ligase. _Proc. Natl Acad. Sci. USA_ 105, 19492–19497 (2008). Article CAS PubMed PubMed Central Google Scholar * Trempe, J. F. et al. SH3
domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. _Mol. Cell_ 36, 1034–1047 (2009). Article CAS PubMed Google Scholar *
Coetzee, S. G. et al. Enrichment of risk SNPs in regulatory regions implicate diverse tissues in Parkinson's disease etiology. _Sci. Rep._ 6, 30509 (2016). Article CAS PubMed PubMed
Central Google Scholar * Zhu, M., Cortese, G. P. & Waites, C. L. Parkinson's disease-linked Parkin mutations impair glutamatergic signaling in hippocampal neurons. _BMC Biol._ 16,
100 (2018). Article PubMed PubMed Central CAS Google Scholar * Burke, R. E. & O'Malley, K. Axon degeneration in Parkinson's disease. _Exp. Neurol._ 246, 72–83 (2013).
Article CAS PubMed Google Scholar * Cheng, H. C., Ulane, C. M. & Burke, R. E. Clinical progression in Parkinson disease and the neurobiology of axons. _Ann. Neurol._ 67, 715–725
(2010). Article PubMed PubMed Central Google Scholar * Hornykiewicz, O. Biochemical aspects of Parkinson's disease. _Neurology_ 51, S2–S9 (1998). Article CAS PubMed Google
Scholar * Brennand, K. J. et al. Modelling schizophrenia using human induced pluripotent stem cells. _Nature_ 473, 221–225 (2011). Article CAS PubMed PubMed Central Google Scholar *
Mertens, J. et al. Age-dependent instability of mature neuronal fate in induced neurons from Alzheimer's patients. _Cell Stem Cell_ 28, 1533–1548.e1536 (2021). Article CAS PubMed
PubMed Central Google Scholar * Quraishi, I. H. et al. An epilepsy-associated KCNT1 mutation enhances excitability of human iPSC-derived neurons by increasing slack KNa currents. _J.
Neurosci._ 39, 7438–7449 (2019). Article CAS PubMed PubMed Central Google Scholar * Schafer, S. T. et al. Pathological priming causes developmental gene network heterochronicity in
autistic subject-derived neurons. _Nat. Neurosci._ 22, 243–255 (2019). Article CAS PubMed PubMed Central Google Scholar * Stern, S. et al. A physiological instability displayed in
hippocampal neurons derived from lithium-nonresponsive bipolar disorder patients. _Biol. Psychiatry_ 88, 150–158 (2020). Article CAS PubMed Google Scholar * Mertens, J. et al. Directly
reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. _Cell Stem Cell_ 17, 705–718 (2015). Article CAS PubMed
PubMed Central Google Scholar * Rando, T. A. & Chang, H. Y. Aging, rejuvenation, and epigenetic reprogramming: Resetting the aging clock. _Cell_ 148, 46–57 (2012). Article CAS PubMed
PubMed Central Google Scholar * Byers, B. et al. SNCA triplication Parkinson's patient's iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative
stress. _PLoS One_ 6, e26159 (2011). Article PubMed PubMed Central CAS Google Scholar * Kriks, S. et al. Dopamine neurons derived from human ES cells efficiently engraft in animal
models of Parkinson's disease. _Nature_ 480, 547–551 (2011). Article CAS PubMed PubMed Central Google Scholar * Wakeman, D. R. et al. Cryopreservation maintains functionality of
human iPSC dopamine neurons and rescues Parkinsonian phenotypes in vivo. _Stem Cell Rep._ 9, 149–161 (2017). Article CAS Google Scholar * Banker, G. A. Trophic interactions between
astroglial cells and hippocampal neurons in culture. _Science_ 209, 809–810 (1980). Article CAS PubMed Google Scholar * Banker, G. A. & Cowan, W. M. Rat hippocampal neurons in
dispersed cell culture. _Brain Res._ 126, 397–342 (1977). Article CAS PubMed Google Scholar * Benson, D. L. & Huntley, G. W. Building and remodeling synapses. _Hippocampus_ 22,
954–968 (2012). Article PubMed Google Scholar * Sudhof, T. C. Towards an understanding of synapse formation. _Neuron_ 100, 276–293 (2018). Article CAS PubMed PubMed Central Google
Scholar * Yoshii, A. & Constantine-Paton, M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. _Nat. Neurosci._ 10, 702–711
(2007). Article CAS PubMed Google Scholar * Mahoney, R. E., Azpurua, J. & Eaton, B. A. Insulin signaling controls neurotransmission via the 4eBP-dependent modification of the
exocytotic machinery. _Elife_ 5, e16807 (2016). Article PubMed PubMed Central Google Scholar * McLaughlin, C. N. & Broihier, H. T. Keeping neurons young and foxy: FoxOs promote
neuronal plasticity. _Trends Genet._ 34, 65–78 (2018). Article CAS PubMed PubMed Central Google Scholar * Al-Mubarak, B., Soriano, F. X. & Hardingham, G. E. Synaptic NMDAR activity
suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. _Channels_ 3, 233–238 (2009). Article CAS PubMed Google Scholar * Bagetta, V., Ghiglieri, V.,
Sgobio, C., Calabresi, P. & Picconi, B. Synaptic dysfunction in Parkinson's disease. _Biochem. Soc. Trans._ 38, 493–497 (2010). Article CAS PubMed Google Scholar * Picconi, B.,
Piccoli, G. & Calabresi, P. Synaptic dysfunction in Parkinson's disease. _Adv. Exp. Med. Biol._ 970, 553–572 (2012). Article CAS PubMed Google Scholar * Zhang, P., Xia, N.
& Reijo Pera, R. A. Directed dopaminergic neuron differentiation from human pluripotent stem cells. _J. Vis. Exp.: JoVE_ 51737 (2014).. * Bardy, C. et al. Neuronal medium that supports
basic synaptic functions and activity of human neurons in vitro. _Proc. Natl Acad. Sci. USA_ 112, E2725–E2734 (2015). Article CAS PubMed PubMed Central Google Scholar * Bardy, C. et al.
Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. _Mol. Psychiatry_ 21, 1573–1588 (2016). Article CAS PubMed PubMed Central
Google Scholar * Andrews, S. A quality control tool for high throughput sequence data. _Babraham Bioinform_. https://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010). * Lander, E.
S. et al. Initial sequencing and analysis of the human genome. _Nature_ 409, 860–921 (2001). Article CAS PubMed Google Scholar * Dobin, A. et al. STAR: Ultrafast universal RNA-seq
aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS PubMed Google Scholar * Illumina. _iGenomes online_. https://support.illumina.com/sequencing/sequencing_software/igenome.html
(2015). * Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. _Mol. cell_ 38,
576–589 (2010). Article CAS PubMed PubMed Central Google Scholar * Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. _Bioinformatics_ 26, 139–140 (2010). Article CAS PubMed Google Scholar * Da Wei Huang, B. T. S. R. A. L. Systematic and integrative analysis of large
gene lists using DAVID bioinformatics resources. _Nat. Protoc._ 4, 44–57 (2008). Article CAS Google Scholar * Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based
approach for interpreting genome-wide expression profiles. _Proc. Natl Acad. Sci. USA_ 102, 15545–15550 (2005). Article CAS PubMed PubMed Central Google Scholar * Benjamini Yoav, Y. D.
The control of the false discovery rate in multiple testing under dependency. _Ann. Stat._ 29, 1165–1188 (2001). Google Scholar * Stern, S., Rotem, A., Burnishev, Y., Weinreb, E. &
Moses, E. External excitation of neurons using electric and magnetic fields in one- and two-dimensional cultures. _J. Vis. Exp._ https://doi.org/10.3791/54357 (2017). Download references
ACKNOWLEDGEMENTS The authors would like to thank K.E. Diffenderfer for technical assistance and M.L. Gage for editorial comments. They would also like to acknowledge the Salk Institute Stem
Cell Core, Waitt Biophotonics Core, and Next Generation Sequencing Core for technical support. Funding to the cores provided in part by NIH-NCI CCSG: P30 014195. This work was supported by
the JPB Foundation, American Heart Association/Paul G. Allen Frontiers Group Brain Health & Cognitive Impairment Initiative (19PABHI34610000), The Milky Way Research Foundation, Annette
C. Merle-Smith and the G. Harold and Leila Y. Mathers Charitable Foundation, NIH AG056306 to F.H.G., NIH R01AG056411-02 to C.K.G., NIH DP5OD023071-03 to J.R.D., Zuckerman STEM leadership
program and ISF 1994/21 to S.S. Alzheimer's Association Research Fellowship (AARF) Program (A.M.), Bavarian Ministry of Science and the Arts in the framework of the ForInter network
(J.W. and B.W.), Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) WI 3567/2-1 (B.W.), The Michael J Fox Foundation, The Shake It Up Foundation Australia, The Hospital
Research Foundation: Parkinson’s South Australia, The Grosset Gaia Foundation, Michael and Angelique Boileau Corporate Philanthropy to C.B., EMBO Postdoctoral Long-term Fellowship (ALTF
1214-2014) and Human Frontiers Science Program (HFSP Long-Term Fellowship- LT001074/2015) for A.A.M. AUTHOR INFORMATION Author notes * These authors contributed equally: Maria C. Marchetto,
Fred H. Gage. AUTHORS AND AFFILIATIONS * Laboratory of Genetics, Salk Institute for Biological Studies, La Jolla, CA, USA Shani Stern, Shong Lau, Andreea Manole, Fan Qiu, Simon Schafer, Abed
AlFatah Mansour, Tchelet Stern, Polina Ofer, Yam Stern, Ana Paula Diniz Mendes, Lynne Randolph Moore, Aidan Aicher, Amanda Rhee, Thomas L. Wong, Thao Nguyen, Sara B. Linker & Fred H.
Gage * Sagol Department of Neurobiology, Faculty of Natural Sciences, University of Haifa, Haifa, Israel Shani Stern, Idan Rosh, Menachem Mendel Percia, Ran Ben Ezer, Tchelet Stern, Polina
Ofer, Yam Stern, Jose Djamus & Ritu Nayak * Razavi Newman Integrative Genomics and Bioinformatics Core, Salk Institute for Biological Studies, La Jolla, CA, USA Maxim N. Shokhirev *
Department of Psychiatry, School of Medicine, Technical University of Munich, Munich, Germany Simon Schafer * Department of Medical Neurobiology, Institute for Medical Research
Israel-Canada, Faculty of Medicine, Hebrew University of Jerusalem, Jerusalem, Israel Abed AlFatah Mansour * Fujifilm Cellular Dynamics, In, Madison, WI, 53711, USA Kile P. Mangan, Beatriz
C. Freitas & Eugenia Jones * Department of Immunology and Regenerative Biology, Weizmann Institute of Science, Rehovot, Israel Sapir Havusha Laufer & Irit Sagi * Department of Stem
Cell Biology, University Hospital Erlangen, Friedrich-Alexander-University Erlangen-Nuernberg, Erlangen, Germany Beate Winner * South Australian Health and Medical Research Institute
(SAHMRI), Laboratory for Human Neurophysiology and Genetics, Adelaide, SA, Australia Cedric Bardy * Flinders University, Flinders Health and Medical Research Institute (FHMRI), Adelaide, SA,
Australia Cedric Bardy * Sorbonne Université, Institut du Cerveau - Paris Brain Institute - ICM, Inserm, CNRS, APHP, F-75013, Paris, France Alexis Brice * Department of Molecular Neurology,
University Hospital Erlangen, Friedrich-Alexander-University Erlangen- Nürnberg, Nürnberg, Germany Juergen Winkler * Department of Anthropology, University of California San Diego, 9500
Gilman Drive, La Jolla, CA, 92093, USA Maria C. Marchetto Authors * Shani Stern View author publications You can also search for this author inPubMed Google Scholar * Shong Lau View author
publications You can also search for this author inPubMed Google Scholar * Andreea Manole View author publications You can also search for this author inPubMed Google Scholar * Idan Rosh
View author publications You can also search for this author inPubMed Google Scholar * Menachem Mendel Percia View author publications You can also search for this author inPubMed Google
Scholar * Ran Ben Ezer View author publications You can also search for this author inPubMed Google Scholar * Maxim N. Shokhirev View author publications You can also search for this author
inPubMed Google Scholar * Fan Qiu View author publications You can also search for this author inPubMed Google Scholar * Simon Schafer View author publications You can also search for this
author inPubMed Google Scholar * Abed AlFatah Mansour View author publications You can also search for this author inPubMed Google Scholar * Kile P. Mangan View author publications You can
also search for this author inPubMed Google Scholar * Tchelet Stern View author publications You can also search for this author inPubMed Google Scholar * Polina Ofer View author
publications You can also search for this author inPubMed Google Scholar * Yam Stern View author publications You can also search for this author inPubMed Google Scholar * Ana Paula Diniz
Mendes View author publications You can also search for this author inPubMed Google Scholar * Jose Djamus View author publications You can also search for this author inPubMed Google Scholar
* Lynne Randolph Moore View author publications You can also search for this author inPubMed Google Scholar * Ritu Nayak View author publications You can also search for this author
inPubMed Google Scholar * Sapir Havusha Laufer View author publications You can also search for this author inPubMed Google Scholar * Aidan Aicher View author publications You can also
search for this author inPubMed Google Scholar * Amanda Rhee View author publications You can also search for this author inPubMed Google Scholar * Thomas L. Wong View author publications
You can also search for this author inPubMed Google Scholar * Thao Nguyen View author publications You can also search for this author inPubMed Google Scholar * Sara B. Linker View author
publications You can also search for this author inPubMed Google Scholar * Beate Winner View author publications You can also search for this author inPubMed Google Scholar * Beatriz C.
Freitas View author publications You can also search for this author inPubMed Google Scholar * Eugenia Jones View author publications You can also search for this author inPubMed Google
Scholar * Irit Sagi View author publications You can also search for this author inPubMed Google Scholar * Cedric Bardy View author publications You can also search for this author inPubMed
Google Scholar * Alexis Brice View author publications You can also search for this author inPubMed Google Scholar * Juergen Winkler View author publications You can also search for this
author inPubMed Google Scholar * Maria C. Marchetto View author publications You can also search for this author inPubMed Google Scholar * Fred H. Gage View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS S.S. differentiated neurons, did patch clamp experiments, performed data analysis, design of experiments, and manuscript writing,
S.L. differentiated neurons and prepared RNA, A.M. performed immunohistochemistry (IHC), imaging of aggregated proteins, M.P. differentiated neurons and performed IHC, I.R. differentiated
neurons and performed IHC, M.P. performed ICC experiments, R.B.R., M.N.S., R.N.A., F.Q., S.S., and S.B.L. performed RNA sequencing analysis, A.A.M. performed IHC, K.M. performed MEA
experiments, TS performed confocal imaging and quantification, P.O. differentiated neurons, Y.S. performed confocal imaging, A.M.D. grew cells, J.D. performed electrophysiological recordings
analysis, L.R.M. prepared lenti-virus, R.N. differentiated neurons, S.K. helped with the ECM experiments, A.A. performed quantification of images, A.R. and T.L.W. performed quantification
of aggresome images, T.N. differentiated neurons, B.W. reprogrammed fibroblasts into iPSCs, B.C.F. differentiated neurons, E.J. helped with the design of experiments, I.S. helped with the
ECM experiments, C.B. performed path clamp experiments, A.B. and J.W. diagnosed patients, M.C.M. differentiated neurons and helped with design of experiments, F.H.G. designed research and
manuscript writing. CORRESPONDING AUTHORS Correspondence to Shani Stern or Fred H. Gage. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
SUPPLEMENTARY TABLE 3 SUPPLEMENTARY TABLE 4 SUPPLEMENTARY TABLE 5 SUPPLEMENTARY TABLE 6 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Stern, S., Lau, S., Manole, A. _et al._ Reduced synaptic activity and dysregulated
extracellular matrix pathways in midbrain neurons from Parkinson’s disease patients. _npj Parkinsons Dis._ 8, 103 (2022). https://doi.org/10.1038/s41531-022-00366-z Download citation *
Received: 12 May 2021 * Accepted: 11 July 2022 * Published: 10 August 2022 * DOI: https://doi.org/10.1038/s41531-022-00366-z SHARE THIS ARTICLE Anyone you share the following link with will
be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative