- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The human oral and gut commensal microbes play vital roles in the development and maintenance of immune homeostasis, while its association with susceptibility and severity of
SARS-CoV-2 infection is barely understood. In this study, we investigated the dynamics of the oral and intestinal flora before and after the clearance of SARS-CoV-2 in 53 COVID-19 patients,
and then examined their microbiome alterations in comparison to 76 healthy individuals. A total of 140 throat swab samples and 81 fecal samples from these COVID-19 patients during
hospitalization, and 44 throat swab samples and 32 fecal samples from sex and age-matched healthy individuals were collected and then subjected to 16S rRNA sequencing and viral load
inspection. We found that SARS-CoV-2 infection was associated with alterations of the microbiome community in patients as indicated by both alpha and beta diversity indexes. Several
bacterial taxa were identified related to SARS-CoV-2 infection, wherein elevated _Granulicatella_ and _Rothia mucilaginosa_ were found in both oral and gut microbiome. The SARS-CoV-2 viral
load in those samples was also calculated to identify potential dynamics between COVID-19 and the microbiome. These findings provide a meaningful baseline for microbes in the digestive tract
of COVID-19 patients and will shed light on new dimensions for disease pathophysiology, potential microbial biomarkers, and treatment strategies for COVID-19. SIMILAR CONTENT BEING VIEWED
BY OTHERS TEMPORAL ASSOCIATION BETWEEN HUMAN UPPER RESPIRATORY AND GUT BACTERIAL MICROBIOMES DURING THE COURSE OF COVID-19 IN ADULTS Article Open access 18 February 2021 IMPACT OF SARS-COV-2
INFECTION ON RESPIRATORY AND GUT MICROBIOME STABILITY: A METAGENOMIC INVESTIGATION IN LONG-TERM-HOSPITALIZED COVID-19 PATIENTS Article Open access 13 November 2024 TEMPORAL DYNAMICS OF
OROPHARYNGEAL MICROBIOME AMONG SARS-COV-2 PATIENTS REVEALS CONTINUED DYSBIOSIS EVEN AFTER VIRAL CLEARANCE Article Open access 24 August 2022 INTRODUCTION The ongoing COVID-19 pandemic caused
by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has jeopardized global public health and safety. As of June 21, 2021, the COVID-19 pandemic has infected more than 179
million people, resulting in over 3.8 million causalities globally. Transmission of SARS-CoV-2 has commonly occurred via breathing or direct contact with virus-containing droplets and
aerosols from infected people through coughing and sneezing. When the SARS-CoV-2 particle reaches the nasal cavity of the host, it enters the host epithelial cells through the
angiotensin-converting enzyme 2 (ACE2) receptor, which is prominently presented on epithelial cells lining in the respiratory and digestive tract systems1,2. Most infections lead to a prompt
innate immune response and the virus gets eradicated or controlled quickly, manifesting none or mild symptoms. Whereas in some patients, viruses located in the upper airway replicate
further toward the lower respiratory tract to activate enhanced pro-inflammatory responses, therefore resulting in severe outcomes including acute respiratory syndrome, organ malfunctioning,
and even death3. Not limited to the respiratory tract, SARS-CoV-2 is known to target different organ systems4. For instance, around 55% of patients were observed with prolonged viral RNA
present in the feces, even weeks after the viral clearance in their respiratory tract5. It has been reported that abnormal immune response, comorbid conditions, and advanced age are risk
factors linked to COVID-19 severity. However, these factors are insufficient to offer a satisfactory explanation of all patients’ severe disease outcomes. As global mass vaccination and
proven effective therapy for COVID-19 remain unclear, explorative efforts towards new perspectives of protection and therapeutic approaches for COVID-19 are indispensable6,7. The human
microbiome is important for developing and maintaining immune homeostasis and it is known that microbiota imbalance or dysbiosis are highly associated with various diseases. The intestinal
tract and oral cavity, with the largest and second-largest microbiota in the human body, play significant roles in the pathogenesis of infectious disease. Previous studies have reported that
oral-lung microbes can influence the outcome of many infectious diseases by regulating the host mucosal immunity8,9,10. Intestinal flora can affect the occurrence and progression of viral
infection through the gut–lung axis11,12,13. Conversely, the indigenous microbiome could be disturbed by a viral infection, leading to alterations in susceptibility and disease severity
through dysbiotic community structure and function14. The unequivocal association between influenza viral and bacterial co-infection and disease severity has been proved in early studies15.
Likewise, evidence has suggested that SARS-CoV-2 infection could predispose patients to bacterial co-infections and superinfections, resulting in increased disease severity and mortality14.
Further, the dysbiosis of influenza-infected individuals progresses toward microbiota homeostasis, coinciding with viral clearance and recovery, suggesting that the health status of
microbial flora is likely a fine indication of disease recovery16. Zuo and colleagues have also suggested that fecal microbiomes from COVID-19 patients were characterized by proliferation of
opportunistic pathogens and depletion of favorable commensals compared to healthy controls with the shot-gun metagenomics approach17; however, this study was limited by its size of subjects
used. Moreover, validation of the profiles at the genus level is currently suggested to be performed through 16S rRNA gene microbial profiling to evaluate the presence of undetected
microbes in the marker gene-based profiles18. Accumulated evidence for the oral–gut axis has revealed its role in modulating the pathogenesis process in numerous diseases19,20. It is
compelling to look into the oral and intestinal microbiome combined with SARS-CoV-2 infection and the crosstalk among them, which may provide an improved understanding of the initiation of
viral infection and the path of disease deterioration. RESULTS OVERVIEW OF MICROBIAL COMPOSITION IN THE STUDY SUBJECTS A total of 53 patients diagnosed with COVID-19 and 76 healthy
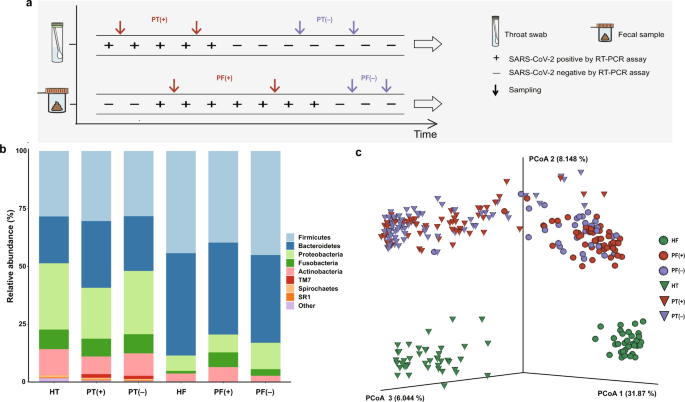
individuals were included in this study. As indicated in Fig. 1a, serial throat swab samples and fecal specimens were collected from COVID-19 patients during hospitalization, covering both
the positive viral RNA test period (P-VRTP) and the negative viral RNA test period (N-VRTP). Depending on the sample availability, a total of 140 throat swab samples, including 52 during the
P-VRTP [PT(+)] and 88 during the N-VRTP [PT(−)], and a total of 81 fecal samples, including 50 during the P-VRTP [PF(+)] and 31 during the N-VRTP [PF(−)] were collected from hospitalized
patients (P: patient). In addition, 44 throat swab samples (HT) and 32 fecal samples (HF) from sex and age-matched healthy individuals (H: healthy individual) were included in our study as
controls. The sample distribution, demographics, and relevant clinical information about the subjects recruited in the study were summarized in Supplementary Table 1. To characterize the
microbiotas in those aforementioned samples, we sequenced the V3–V4 region of the bacterial 16S rRNA gene. As expected, our data confirmed a distinct microbiota composition between oral and
gut samples21. The three most abundant phyla _Firmicutes_, _Proteobacteria_, and _Bacteroidetes_ accounted for 79.3% of the community in the oral samples. The two dominant phyla _Firmicutes_
and _Bacteroidetes_ accounted for 83.7% of the bacterial community in the gut samples (Fig. 1b). The difference was further illustrated by the principal coordinates analysis (PCoA) plot and
Permutational multivariate analysis of variance (PERMANOVA) with unweighted UniFrac distance calculated at the sequence feature level. The microbiota composition of the oral and gut samples
diverged from each other along the first axis in which 31.87% of the total variance was explained, indicating that the largest source of variation was the sample types (Fig. 1c). Similarly,
the PERMANOVA result indicated 30.80% of the total variance was explained by the sample types (oral samples vs. gut samples, PERMANOVA, _R_2 = 0.308, _p_ < 0.001). More importantly,
samples from patients and healthy individuals seemed to possess strong dissimilarities for both oral (PERMANOVA, _R_2 = 0.175, _p_ < 0.001) and gut microbiome communities (PERMANOVA, _R_2
= 0.196, _p_ < 0.001), suggesting overwhelming shifts of microbiome structure were gained after SARS-CoV-2 infection in both oral cavity and feces. Meanwhile, the microbial compositions
of COVID-19 patients collected during the P-VRTP and the N-VRTP presented no significant discrepancy in oral samples (PERMANOVA, _R_2 = 0.012, _p_ = 0.080) or in gut samples (PERMANOVA, _R_2
= 0.016, _p_ = 0.139). Due to their distinctive characteristics, the oral and gut microbiome profiles and their potential roles in SARS-CoV-2 infection were addressed in separate sections
followed. ALTERATIONS OF THE ORAL MICROBIOTA IN PATIENTS Given the observation that the SARS-CoV-2 infection may impact the structure of the microbiome (Fig. 1c), we first investigated the
effects of different disease severity conditions and treatment therapies on the oral samples from patients. The diversity metrics among oral microbiomes from the healthy controls (HT),
patients with the non-severe condition [PT(NS)], and patients with the severe condition [PT(S)] during hospitalization were compared. Alpha diversity of Faith’s phylogenetic index revealed
that the oral microbial diversity was decreased in COVID-19 patients compared to the healthy controls (ANOVA, PT(NS) vs. HT, _p_ = 0.016; PT(S) vs. HT, _p_ < 0.0001). Further, a
significant deduction of diversity was observed in the PT(S) group when comparing to the PT(NS) group (ANOVA, _p_ < 0.0001, Fig. 2a). Similarly, inter-individual beta diversity unweighted
UniFrac dissimilarity revealed that the microbiomes of the PT(NS) group (PERMANOVA, _R_2 = 0.238, _p_ < 0.001) and the PT(S) group (PERMANOVA, _R_2 = 0.233, _p_ < 0.001) clustered
apart from that of the healthy controls. We also found a significant dissimilarity between the PT(S) group and the PT(NS) group (PERMANOVA, _R_2 = 0.049, _p_ < 0.001, Fig. 2b). The
significant differences were partially attributed to the different dispersions between the PT(S) group and the PT(NS) group (PERMDISP, _p_ = 0.002). In contrast, the difference in dispersion
between the HT group and the PT(NS) group was not significant (PERMDISP, _p_ = 0.166). We also investigated the impact of clinical antibiotic usage. The PCoA based on unweighted UniFrac
dissimilarity demonstrated that bacterial communities in the HT group, the patients without antibiotic interference [PT(abx−)] group and the patients with antibiotic interference [PT(abx+)]
group were mutually separated (PERMANOVA, PT(abx−) vs. HT, _R_2 = 0.258, _p_ = 0.001; PT(abx+) vs. HT, _R_2 = 0.204, _p_ = 0.001; PT(abx−) vs. PT(abx+), _R_2 = 0.016, _p_ = 0.016; Fig. 2c).
To identify potential microbial biomarkers associated with COVID-19 patients, we employed a combined approach with LEfSe and MaAsLin2 to minimize the influence of confounding factors.
Seventeen significantly different taxa were identified in the oral microbiome between the healthy controls and COVID-19 patients (HT and PT, Fig. 2d and Supplementary Table 2). Specifically,
significant decreases of _Neisseria_, _Corynebacterium_, _Actinobacillus_, _Moryella_, _Aggregatibacter_, _Treponema,_ and _Pseudomonas_ at the genus level, as well as _P. intermedia_ and
_T. amylovorum_ were observed in the patients comparing to the controls. In contrast, _Veillonella_, _Campylobacter_, _Granulicatella_, _Kingella,_ and _Filifactor_ at the genus level, as
well as _H. parainfluenzae_, _R. mucilaginosa,_ and _N. subflava_ were enriched in the oral microbial communities of COVID-19 patients compared to those of the controls. A similar comparison
was conducted between the PT(NS) and the PT(S) groups to assess the impact of disease severity on bacteria (Supplementary Table 3). Bacterial taxa including _Treponema_, _Aggregatibacter,_
and _P. intermedia_ were identified further depleted in the PT(S) group, implying their associations with disease severity. The comparison between the controls and the PT(abx–) group was
carried out to assess the impact of infection on bacteria by excluding the antibiotics interference (Supplementary Table 3). Lastly, the potential functional consequence resulted from the
drastic taxa compositional alteration was evaluated. The global pattern from Procrustes analysis displayed a good-fit correlation between the oral microbial community composition and
microbial function (Mantel, rho = 0.403, _p_ = 0.001, Supplementary Fig. 1a). The significant difference in functional pathways of the oral microbial communities between COVID-19 patients
and the controls was shown in Fig. 2e. Notably, the top four depleted pathways in patients were involved in the TCA cycle used by all aerobic organisms to generate energy, indicating a
disturbed microbial community. ALTERATIONS OF THE GUT MICROBIOTA IN PATIENTS Next, we explored whether dysbiosis occurred in the gut microbiome. Faith’s phylogenetic diversity index showed
that its diversity was significantly lower in patients with the non-severe condition [PF(NS)] (ANOVA, _p_ < 0.0001) and patients with the severe condition [PF(S)] (ANOVA, _p_ < 0.0001)
in comparisons to the healthy controls (HF, Fig. 3a). The diversity of the PF(NS) group was slightly higher than the PF(S) group (ANOVA, _p_ = 0.73). Besides, the PCoA plot with unweighted
UniFrac distances revealed a cluster separation in the fecal microbiota between PF(NS) and PF(S) (PERMANOVA, _R_2 = 0.067, _p_ < 0.001), and between the patients and the healthy controls
(PERMANOVA, PF(NS) vs. HF, _R_2 = 0.232, _p_ < 0.001; PF(S) vs. HF, _R_2 = 0.267, _p_ < 0.001, Fig. 3b). Unlike the oral microbiome, the gut microbiota in the PF(NS) group seemed more
impacted after infection, as reflected by its greater statistical dispersion than the HF group (PERMDISP, _p_ = 0.002). Moreover, the gut microbiota among patients with antibiotic
interference [PF(abx+)], patients without antibiotic interference [PF(abx−)], and the HF group exhibited mutual clustering separation with unweighted UniFrac distances based PCoA plot
(PERMANOVA, PF(abx−) vs. HF, _R_2 = 0.262, _p_ = 0.001; PF(abx+) vs. HF, _R_2 = 0.230, _p_ = 0.001; PF(abx−) vs. PF(abx+), _R_2 = 0.030, _p_ = 0.002; Fig. 3c). In terms of potential
microbial biomarkers, we identified 17 bacterial taxa again with confounding covariates adjusted. Concretely, decreased taxa included _Blautia_, _Coprococcus_, and _Collinsella_ at the genus
level, and _B. caccae_, _B. coprophilus_, _B. obeum,_ and _C. colinum_ species; and increased taxa included _Streptococcus_, _Weissella_, _Enterococcus_, _Rothia_, _Lactobacillus_,
_Actinomyces_, and _Granulicatella_ at the genus level as well as _C. citroniae_, _B. longum,_ and _R. mucilaginosa_ species (Fig. 3d and Supplementary Table 4). More interestingly, two
normal components of the respiratory tract flora, _Granulicatella_ and _R. mucilaginosa_, were significantly increased in the patients for both oral and gut samples, and _R. mucilaginosa_
seemed further associated with the disease severity in the fecal samples (Supplementary Fig. 2). Additional analysis focusing on the bacteria changes associated with disease severity and
antibiotics usage in fecal samples were described in Supplementary Table 5. Notably, no difference in the bacterial taxa was identified between the PF(NS) group and the PF(S) group. Similar
to the oral microbiome, the global pattern from Procrustes analysis displayed a good-fit correlation between the microbial composition and predicted function profile in the gut microbiota
community (Mantel, rho = 0.268, _p_ = 0.001, Supplementary Fig. 1b). Also, the major distinct functional pathways associated with SARS-CoV-2 infection were shown in Fig. 3e, including two
depleted pyrimidine deoxyribonucleotides biosynthesis pathways involved in viral suppression through innate immunity in patients22. RELATIONSHIP BETWEEN SARS-COV-2 VIRUS AND BACTERIAL
SPECIES The viral loads in the throat swab and fecal samples spanning the full collection period were compared. The median time span of the throat swabs collection was 10 (IQR: 6–12) days
for P-VRTP and 10 (IQR: 3–18) days for N-VRTP. The median time span of the fecal samples collection was 8 (IQR: 3.75–14) days for P-VRTP and 8 (IQR: 2–12) days for N-VRTP. The viral copy
numbers derived from the E gene showed a relatively higher abundance over the numbers derived from the N gene (Fig. 4a). As E-gene and N-gene copy numbers showed a strong correlation
therefore both can accurately reflect the abundance of viral copy (throat swab samples: Spearman rho = 0.872, _p_ < 0.001; fecal samples: Spearman rho = 0.923, _p_ < 0.001). The
overall viral loads in throat swab samples seemed equally abundant with fecal specimens collected from patients during the P-VRTP, which is consistent with the previous studies23. Based on
unweighted UniFrac distance, it was found that the differences in beta diversity between the HT and PT(+) groups (PERMANOVA, _R_2 = 0.234, _p_ = 0.001), the HT and PT(−) groups (PERMANOVA,
_R_2 = 0.218, _p_ = 0.001), were significant from each other. Likewise, unweighted UniFrac dissimilarity revealed that the microbiomes of the PF(+) group (PERMANOVA, _R_2 = 0.233, _p_ =
0.001) and the PF(−) group (PERMANOVA, _R_2 = 0.250, _p_ = 0.001) were significantly different from that of the healthy controls. During the N-VRTP, we observed a microbial community
recovery towards the healthy controls, that is, unweighted UniFrac distance between PT(−) vs. HT was lower than that between PT(+) vs. HT (Fig. 4b). Compared to the oral microbiome, this
recovery trend in the microbiome in the gut was less evident, which may be attributed to a relatively shorter time span of sample collection and microbiome robustness in the fecal samples.
Additionally, we investigated whether the SARS-CoV-2 viral load was associated with any oral and gut bacterial species. In the oral microbiota, _Pelomonas_ was identified as positively
correlated with the viral load of SARS-CoV-2 (Fig. 4c). While in the gut microbiota, _P. copri_ and _E. dolichum_ were identified positively correlated with the viral load of SARS-CoV-2, and
_S. anginosus_, _Dialister_, _Alistipes_, _Ruminococcus_, _C. citroniae_, _Bifidobacterium_, _Haemophilus_, and _H. parainfluenzae_ were identified negatively correlated with the viral load
of SARS-CoV-2. DISCUSSION The oral and gut microbiome offers a range of valuable properties to the host. Several most significant contributions of these microbes are to boost metabolism,
improve digestive health and strengthen resistance against pathogens. Furthermore, the complex crosstalk between commensal microbes and different body systems is essential for the
functioning of the immune system24. Our data revealed profound alterations in both oral and gut microbiomes, which were reflected in the dramatic changes in community structure, potential
bacterial marker species, and predicted functional profile. The decline in commensal bacterial diversity has been considered as a key dysbacteriosis indicator in several diseases25.
Coherently, the oral and gut microbiome of COVID-19 patients in our study exhibited decreased diversities compared to the healthy controls. This trend of microbial imbalance in patients with
severe conditions compared to non-severe patients was also observed in both sample types, though the alpha diversity was not significant (ANOVA, _p_ = 0.73) in the fecal samples. Moreover,
the microbiomes from the severe and the non-severe patients were partitioned into two clusters in both microbial populations. Overall, diversity results of impaired microbiota suggested the
strong association between the microbiome community complexity and the disease severity in COVID-19 patients. Though experimental validation is needed, our result has highlighted the
possibility of personalized microbiota to affect the disease outcome of COVID-19 patients26. Numerous studies have reported high occurrences of bacterial co-infection in hospitalized
COVID-19 patients, and the odds of the bacterial infection get even higher for ICU patients27,28. Among the list of eight bacterial taxa with enlarged relative abundance in patients in Fig.
2d, _Veillonella_, _Campylobacter_, _R. mucilaginosa_, _Granulicatella_, _Kingella_, and _Filifactor_ belongs to a group of periodontitis-correlated taxa, adding evidence to support the
previous work denotative of a close relationship between periodontitis and SARS-CoV-2 infection29. Periodontal-associated cytokines may proliferate contacts of bacteria between the lungs and
the mouth via driving the alteration of the respiratory epithelium and thereby promote respiratory infection in COVID-19 patients14. _H. parainfluenzae_ and _Kingella_, two enriched taxa in
the oral cavity of COVID-19 patients, are opportunistic pathogens well-known for respiratory tract infections, infective endocarditis, and meningitis30,31. The abrupt loss of _Neisseria_ in
patients, which is the highly abundant genus in the normal oral cavity, could raise serious damage to the oral microbiota32,33. In addition, all increased bacteria were previously
classified as bacteremia-associated bacteria, implying a potential association between oral dysbiosis and secondary bacterial infection in COVID-19 patients34. Typically, mechanical
ventilation supports may predispose the COVID-19 patients with severe symptoms of dyspnea to pulmonary bacterial co-infection, as the penetration of the device provides an entrance for those
opportunistic infectious agents to access the lower respiratory tract from the oral cavity. Given the non-ignorable association between the oral microbiome with bacterial co-infections
suggested by our and others’ data35,36, correct and frequent oral health care measurements should be recommended by physicians to protect COVID-19 patients from secondary infections and
improve survival, especially for the ones with the severe disease condition. Elevated levels of _Streptococcus_, _Rothia_, and _Actinomyces_ were identified in COVID-19 patients’ feces,
which is consistent with previous findings37. The host immunity symbionts beneficial bacterial species _B. obeum_, whose parent genus _Blautia_ was listed as the top decreased taxon in
COVID-19 patients’ feces in our data, was identified to be depleted in another study17. Notably, two normal components of the respiratory tract flora, _Granulicatella,_ and _R.
mucilaginosa_, were identified enriched in patients’ feces, which is likely a reflection of the migration of extra-intestinal microbes into the gut or the flourishment of potentially
pathogenic bacteria. Typically, the fecal enrichment of specific oral taxa, which was suggested to be linked with the increased oral–fecal microbial transmission, has frequently been
regarded as a hallmark of disease20. As SARS-CoV-2 viral RNA was detected in stools, the route of viral transmission from the respiratory tract to the intestinal tract can be hinted by the
path of the oral flora translocation and colonization38. Moreover, _R. mucilaginosa_ seemed further associated with the disease severity in the fecal samples in our study. As some severe
patients with COVID-19 linked with cardiovascular disease comorbidities, this finding supports previous work wherein patients with cardiovascular disease comorbidities tended to have a
higher prevalence of the _Rothia_ ASV associated with SARS-CoV-239,40. In addition, increased expression of ACE2 receptor was observed in _B. longum_ treated mice; hence changes of _B.
longum_ might impact the individual’s susceptibility to SARS-CoV-2 through modulating ACE2 expression41. It is worth noticing that a decreased relative abundance of _B. caccae_ and _B.
coprophilus_ was observed in the gut flora of COVID-19 patients. A recent study by Martino et al.42, has demonstrated that _Bacteroides_ may regulate viral adhesion via modifying heparan
sulfate (HS), and loss of HS-modifying _Bacteroides_ strains could predispose individuals to SARS-CoV-2 infection. Concordance of these findings with ours provides evidence for a potential
relationship between _B. caccae_ and _B. coprophilus_ and susceptibility to SARS-CoV-2 infection. The Mantel and Procrustes analysis indicated the predicted function profiles aligned well
with the bacterial taxonomy profile in both oral and fecal samples as expected. Compared to the gut microbiome, our analysis suggested a stronger impairment in the oral microbiome after
SARS-CoV-2 infection, wherein a significant reduction of _Neisseria_, an essential oral microbial genus, was found, along with several fatal metabolic pathways involving the TCA cycle were
suppressed. The result echoes a previous study that the gut microbial community possesses higher taxa-function robustness over oral community43, since the bacterial community in the gut
generally has a higher gene and functional redundancy in comparison to communities inhabiting other body sites. Microbiome dysbiosis persists during the COVID-19 disease course, even after
the viral clearance. Moreover, previous studies have revealed that some recovered patients were re-detectable positive for SARS-CoV-2 RNA after discharge44. The cause of re-detectable
positive remains unclear. One possibility is that a prolonged detrimental effect on the microbiomes exerted by SARS-CoV-2 infection persists even after discharge, which might render
convalescent patients susceptible to the residual viremia or reinfection via a more long-lasting dysfunction of the immune system. The microbiome recovery at the taxonomy level seemed to be
slowly healed in the oral samples as the PT(−) group shifts towards the healthy controls group based on the dissimilarity distance. In contrast, in the gut samples, the PF(−) group shifts
even away from the healthy controls (Fig. 4b). The inconsistency may be due to the period span of sample collection or microbiome characteristics within the oral and gut samples. In a
previous metagenomics study, Zuo and colleagues suggested four negatively associated _Bacteroides_ species could be involved in the downregulation of ACE2 expression, and positively
regulated _E. bacterium 2_2_44A_ may promote augmenting SARS-CoV-2 infection in the gut17. However, a similar analysis method in our study did not identify the same agents to be associated
with the viral load of SARS-CoV-2 (Fig. 4c). Thus, we urge validation of the results that should be taken before recognizing the results. It is well accepted that unnecessary use of
antibiotics should be avoided in the episode of acute respiratory infections as antibiotics have no role in treatment. In reality, 67.9% of patients in our study were treated with
antibiotics to prevent potential secondary bacterial co-infections at the early phase of the pandemic. Besides, antibiotics prescribing was elevated in patients with severe conditions as
compared to non-severe patients. In a meta-analysis study, Langford has reported that 74.6% of COVID-19 patients received antimicrobial treatment28. Antibiotics treatment can not only
eliminate pathogens but commensal microorganisms indiscriminately, which may lead to microbiota dysbiosis and antimicrobial resistance. To circumvent and minimize the effect of antibiotics
on the microbiome analysis, we employed a method to count antibiotics treatment as an adjusting variable in the model or to stratify the data for only patients without antibiotics treatment.
In addition, unsupervised dimensionality reduction results demonstrated that COVID-19 patients with and without antibiotics treatment shared similar bacterial community structures with a
declined diversity in both oral and gut microbiome, shifting away from healthy individuals. It seems that the impacts derived from infection are overwhelming over that of the antibiotic
treatment that may apply to the microbiome. The taxa differential analysis also supported this finding, that the differential taxa between the controls and the antibiotics-free patients were
largely overlapped with those identified between the controls and all patients (Supplementary Tables 3 and 5). Collectively, we reported the alterations in both oral and gut microbiomes of
SARS-CoV-2 infected patients during hospitalization and made comprehensive analyses to evaluate their potential consequences and implication. The associations between microbial species with
disease severity and viral load in patients have suggested the potential of microbiome-based intervention in the prevention and treatment of COVID-19. We believe that the data provides new
knowledge with innovative perspectives for tackling and managing the ongoing COVID-19 pandemic. METHODS STUDY SUBJECT AND SAMPLE COLLECTION A total of 53 COVID-19 patients and 76 healthy
individuals were included in this study. Patients with suspected SARS-CoV-2 infection were confirmed after two sequential positive respiratory tract sample real-time RT-PCR results. Patients
were kept hospitalized and under strict observation until the virus was completely eliminated in both respirational and intestinal territories by real-time RT-PCR results. Depending on the
sample availability, serial samples were collected from a patient throughout his/her hospitalization period. More specifically, throat swab and fecal samples were collected in both the
positive viral RNA test period (P-VRTP, defined as the period of positive nucleic acid tests until the first day of continuous negative tests) and the negative viral RNA test period (N-VRTP,
defined as the interval between the first day of negative nucleic acid test until the hospital discharge) for both sample types. Fecal samples were collected from patients who were ever
detected with viral RNA in their feces. Only one sample, either throat swab or fecal specimen, was collected from healthy individuals during their physical examination. None of the COVID-19
patients was received antibiotics nor probiotics within 8 weeks before the infection, and none of the healthy individuals was either before this study recruitment. Patients were categorized
into two groups based on disease severity: the non-severe group (mild/moderate) and the severe group (severe/critical) following the instruction of the New Coronavirus Pneumonia Prevention
and Control Program (7th edition) published by the National Health Commission of China. The demographic information, underlying diseases, clinical indexes, and treatments were summarized
from official patients’ medical records. This study was reviewed and approved by the Medical Ethical Committee of the Fifth Affiliated Hospital of Sun Yat-Sen University (approval # K162-1).
Written informed consent was obtained from each enrolled subject. SAMPLE LIBRARY PREPARATION AND SEQUENCING Samples were inactivated at 56 °C for 30 min before DNA extraction. Extraction of
nucleic acids was performed with the CFDA approved nucleic acid extraction kits (QIAamp Viral RNA Mini Kit, Catalog #: 52904, QIAGEN). The concentration and the purity were measured using
the NanoDrop One (ThermoFisher Scientific, MA, USA). Sequencing libraries were generated using NEBNext® UltraTM II DNA Library Prep Kit for Illumina (New England Biolabs, MA, USA) following
manufacturer’s recommendations, and index codes were added. The universal primers 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) were used for amplification of the
V3–V4 region of the bacterial 16S rRNA gene. DNA libraries were generated from PCR amplicons targeting the hypervariable regions V3–V4 of the bacterial 16S rRNA gene. After quality
assessment with the Qubit 2.0 Fluorometer (ThermoFisher Scientific, MA, USA), the library was then sequenced on an Illumina NovaSeq 6000 platform, and a minimum of 50,000 of 250 bp
paired-end reads was generated for the samples. BIOINFORMATICS ANALYSIS Raw FASTQ files were demultiplexed using the QIIME 2 demux plugin based on their unique barcodes45. Demultiplexed
sequences from each sample were stitched, quality filtered, trimmed, de-noised, and then the chimeric sequences were identified and removed using the QIIME 2 dada2 plugin to obtain the
feature table46. The QIIME 2 feature-classifier plugin was then used to align feature sequences to a pre-trained GREENGENES 13_8 99% database to generate the taxonomy table47. The data was
rarefied prior to alpha and beta diversity analysis using a depth of 13,111 reads. Any contaminating mitochondrial and chloroplast sequences were filtered using the QIIME 2 feature-table
plugin. Diversity metrics were calculated and plotted using the core-diversity plugin and the emperor plugin within QIIME 248. The beta diversity significance among groups was examined with
PERMANOVA and PERMDISP tests using QIIME 2 plugins and Vegan package in R (version 4.0.2). The differences in the relative abundance of taxa between the patients and healthy control groups
were identified in both sample types separately using the linear discriminant analysis effect size (LEfSe)49. Given the possible confounding impact, the output from LEfSe was further
validated with Multivariate Association with Linear Models (MaAsLin2) by adjusting confounding factors (age, sex, antibiotic usage, PCR detection result, and patient ID), so only the
bacteria taxa agreed by both methods were presented50. PICRUSt2 was used to predict the microbial metabolic pathways to assess the potential functional implication51. QIIME 2 Procrustes
plugin was used to examine the fitness of the functional properties and the bacterial composition in the microbiome community with the Procrustes plot, and the correlation between functional
properties (Bray Curtis distance) and the bacterial composition (unweighted UniFrac distance) in the microbiome community was examined by two-sided Mantel test. DETECTION OF SARS-COV-2
VIRAL LOAD SARS-CoV-2 viral loads in the throat and fecal swabs were measured using a real-time RT-PCR assay. Viral RNA from throat swabs and fecal samples were extracted using QIAamp Viral
RNA Mini Kit (QIAamp Viral RNA Mini Kit, Catalog #: 52904, QIAGEN). Up to 0.1 g of stool or throat swab was suspended in a 2 mL viral transport medium (in 1:10 dilution), followed by
centrifugation at 3000 × _g_ for 30 min. The aliquot of the filtrate was used as the starting material. The real-time RT-PCR was carried out with the Novel Coronavirus (2019-nCoV) real-time
RT-PCR kit from LifeRiver Ltd. (Catalog #: RR-0479-02). Nucleocapsid gene (N), membrane gene (E), and RNA dependent RNA polymerase gene (RdRp) were the three targeted genes simultaneously
amplified and tested. According to the manufacturer’s instructions, a combined result of the three SARS-CoV-2 viral gene targets was used to yield a positive result. Samples were considered
negative if the cycle threshold values exceeded 43 cycles. Plasmids containing the full N gene and E gene were obtained to assess SARS-CoV-2 viral copy in the samples (PCDNA6B-SARS-CoV-2-N
and PCDNA6B-SARS-CoV-2-E, gifts from Peihui Wang, Cheeloo College of Medicine, Shandong University). Serial 10-fold dilutions of known copies of these plasmids were prepared separately for
generating the standard curve. The Ct values of real-time RT-PCR from patient samples were converted into viral RNA copies based on a standard curve. STATISTICAL ANALYSIS Categorical
variables were presented as numbers and percentages (n/N, %) whereas continuous variables were reported as median and interquartile ranges (IQR). According to the distribution of data sets,
the one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test or the Kruskal–Wallis test followed by Dunn’s multiple comparisons test were performed using Prism
8.3.0 (GraphPad Software). Given the nature of compositional data for the 16S microbial relative abundance information, we implemented CCLasso to define the correlation between the
longitudinal viral load and the timepoint-matched relative abundances of different taxa in the throat swab and fecal samples separately52. The Spearman rank test was used to calculate the
correlation between the viral load represented by the abundances of the N gene and the E gene, as their values did not follow the normal distribution according to the Kolmogorov–Smirnov
test. All statistical tests were two-sided, and a _p_-value, of <0.05 was considered significant. Statistical analysis was performed using SPSS version 25.0 (SPSS Inc). Additional custom
R scripts were used to making the plots. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this article. DATA
AVAILABILITY Raw FASTQ files of 16S rRNA gene sequences were archived in the Sequence Read Archive (SRA) under Bioproject accession number PRJNA684070. All supporting data are publicly
available at https://github.com/XiaominCheng/COVID19-16S. CODE AVAILABILITY The code used for analyses is publicly available at https://github.com/XiaominCheng/COVID19-16S. CHANGE HISTORY *
_ 15 DECEMBER 2021 A Correction to this paper has been published: https://doi.org/10.1038/s41522-021-00262-z _ REFERENCES * Gheblawi, M. et al. Angiotensin-converting enzyme 2: SARS-CoV-2
Receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. _Circ. Res._ 126, 1456–1474 (2020). Article CAS PubMed Google Scholar *
Saponaro, F. et al. ACE2 in the era of SARS-CoV-2: controversies and novel perspectives. _Front. Mol. Biosci._ 7, 588618 (2020). Article CAS PubMed PubMed Central Google Scholar *
Mueller, A. L., McNamara, M. S. & Sinclair, D. A. Why does COVID-19 disproportionately affect older people? _Aging_ 12, 9959–9981 (2020). Article CAS PubMed PubMed Central Google
Scholar * Ni, W. et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. _Crit. Care_ 24, 422 (2020). Article PubMed PubMed Central Google Scholar * Wu, Y. et al. Prolonged
presence of SARS-CoV-2 viral RNA in faecal samples. _Lancet Gastroenterol. Hepatol._ 5, 434–435 (2020). Article PubMed PubMed Central Google Scholar * Kumari, P. et al. Potential
diagnostics and therapeutic approaches in COVID-19. _Clin. Chim. Acta_ 510, 488–497 (2020). Article CAS PubMed PubMed Central Google Scholar * Hodgson, S. H. et al. What defines an
efficacious COVID-19 vaccine? A review of the challenges assessing the clinical efficacy of vaccines against SARS-CoV-2. _Lancet Infect. Dis._ 21, e26–e35 (2021). Article CAS PubMed
Google Scholar * Bao, L. et al. Oral Microbiome and SARS-CoV-2: Beware of Lung Co-infection. _Front. Microbiol._ 11, 1840 (2020). Article PubMed PubMed Central Google Scholar * Mammen,
M. J., Scannapieco, F. A. & Sethi, S. Oral-lung microbiome interactions in lung diseases. _Periodontology_ 83, 234–241 (2020). Article Google Scholar * Li, Y. et al. Salivary mycobiome
dysbiosis and its potential impact on bacteriome shifts and host immunity in oral lichen planus. _Int J. Oral. Sci._ 11, 13 (2019). Article CAS PubMed PubMed Central Google Scholar *
Budden, K. F. et al. Emerging pathogenic links between microbiota and the gut-lung axis. _Nat. Rev. Microbiol._ 15, 55–63 (2017). Article CAS PubMed Google Scholar * Zhang, D. et al. The
cross-talk between gut microbiota and lungs in common lung diseases. _Front. Microbiol._ 11, 301 (2020). Article CAS PubMed PubMed Central Google Scholar * Enaud, R. et al. The
Gut-lung axis in health and respiratory diseases: a place for inter-organ and inter-kingdom crosstalks. _Front. Cell Infect. Microbiol._ 10, 9 (2020). Article CAS PubMed PubMed Central
Google Scholar * Xiang, Z. et al. Potential implications of SARS-CoV-2 oral infection in the host microbiota. _J. Oral. Microbiol._ 13, 1853451 (2020). Article PubMed PubMed Central
Google Scholar * Jia, L. et al. Mechanisms of severe mortality-associated bacterial co-infections following influenza virus infection. _Front. Cell Infect. Microbiol._ 7, 338 (2017).
Article PubMed PubMed Central Google Scholar * Kaul, D. et al. Microbiome disturbance and resilience dynamics of the upper respiratory tract during influenza A virus infection. _Nat.
Commun._ 11, 2537 (2020). Article CAS PubMed PubMed Central Google Scholar * Zuo, T. et al. Alterations in gut microbiota of patients with Covid-19 during time of hospitalization.
_Gastroenterology_ https://doi.org/10.1053/j.gastro.2020.05.048 (2020). * Lugli, G. A. et al. A microbiome reality check: limitations of in silico-based metagenomic approaches to study
complex bacterial communities. _Environ. Microbiol. Rep._ 11, 840–847 (2019). PubMed Google Scholar * Kitamoto, S. et al. The intermucosal connection between the mouth and gut in commensal
pathobiont-driven colitis. _Cell_ 182, 447–462 (2020). Article CAS PubMed PubMed Central Google Scholar * Schmidt, T. S. et al. Extensive transmission of microbes along the
gastrointestinal tract. _Elife_ 8, e42693 (2019). * Human Microbiome Project, Consortium. Structure, function and diversity of the healthy human microbiome. _Nature_ 486, 207–214 (2012).
Article Google Scholar * Lucas-Hourani, M. et al. Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. _PLoS Pathog_. 9, e1003678 (2013). * Jeong,
H. W. et al. Viable SARS-CoV-2 in various specimens from COVID-19 patients. _Clin. Microbiol. Infect._ 26, 1520–1524 (2020). Article CAS PubMed PubMed Central Google Scholar * Rooks,
M. G. & Garrett, W. S. Gut microbiota, metabolites and host immunity. _Nat. Rev. Immunol._ 16, 341–352 (2016). Article CAS PubMed PubMed Central Google Scholar * Kriss, M.,
Hazleton, K. Z., Nusbacher, N. M., Martin, C. G. & Lozupone, C. A. Low diversity gut microbiota dysbiosis: drivers, functional implications and recovery. _Curr. Opin. Microbiol._ 44,
34–40 (2018). Article PubMed PubMed Central Google Scholar * Fischbach, M. A. Microbiome: focus on causation and mechanism. _Cell_ 174, 785–790 (2018). Article CAS PubMed PubMed
Central Google Scholar * Lansbury, L., Lim, B., Baskaran, V. & Lim, W. S. Co-infections in people with COVID-19: a systematic review and meta-analysis. _SSRN Electron. J_.
https://doi.org/10.2139/ssrn.3594598 (2020). * Langford, B. J. et al. Antibiotic prescribing in patients with COVID-19: rapid review and meta-analysis. _Clin. Microbiol. Infect_.
https://doi.org/10.1016/j.cmi.2020.12.018 (2021). * Sahni, V. & Gupta, S. COVID-19 & periodontitis: the cytokine connection. _Med. Hypotheses_ 144, 109908 (2020). Article CAS
PubMed PubMed Central Google Scholar * Yagupsky, P. Kingella kingae: from medical rarity to an emerging paediatric pathogen. _Lancet Infect. Dis._ 4, 358–367 (2004). Article PubMed
Google Scholar * Kosikowska, U., Rybojad, P., Stepien-Pysniak, D., Zbikowska, A. & Malm, A. Changes in the prevalence and biofilm formation of _Haemophilus influenzae_ and _Haemophilus
parainfluenzae_ from the respiratory microbiota of patients with sarcoidosis. _BMC Infect. Dis._ 16, 449 (2016). Article PubMed PubMed Central Google Scholar * Weyand, N. J. Neisseria
models of infection and persistence in the upper respiratory tract. _Pathog. Dis._ 75, 1–13 (2017). Article Google Scholar * Donati, C. et al. Uncovering oral _Neisseria tropism_ and
persistence using metagenomic sequencing. _Nat. Microbiol._ 1, 16070 (2016). Article CAS PubMed Google Scholar * Gonzalez Navarro, B., Jane Salas, E., Estrugo Devesa, A., Lopez Lopez, J.
& Vinas, M. Bacteremia associated with oral surgery: a review. _J. Evid. Based Dent. Pr._ 17, 190–204 (2017). Article Google Scholar * Sampson, V., Kamona, N. & Sampson, A. Could
there be a link between oral hygiene and the severity of SARS-CoV-2 infections? _Br. Dent. J._ 228, 971–975 (2020). Article PubMed PubMed Central Google Scholar * Patel, J. &
Sampson, V. The role of oral bacteria in COVID-19. _Lancet Microbe_ 1, e105 (2020). Article CAS PubMed PubMed Central Google Scholar * Gu, S. et al. Alterations of the gut microbiota in
patients with COVID-19 or H1N1 influenza. _Clin. Infect. Dis._ https://doi.org/10.1093/cid/ciaa709 (2020). * Prodan, A., Levin, E. & Nieuwdorp, M. Does disease start in the mouth, the
gut or both? _Elife_ 8, e45931 (2019). * Wu, L. et al. SARS-CoV-2 and cardiovascular complications: from molecular mechanisms to pharmaceutical management. _Biochem. Pharm._ 178, 114114
(2020). Article CAS PubMed Google Scholar * Marotz, C. et al. SARS-CoV-2 detection status associates with bacterial community composition in patients and the hospital environment.
_Microbiome_ 9, 1–15 (2021). Article Google Scholar * Machado, A. S. et al. Oral probiotic bifidobacterium longum supplementation improves metabolic parameters and alters the expression of
the renin-angiotensin system in obese mice liver. _Biol. Res. Nurs._ 23, 100–108 (2021). Article CAS PubMed Google Scholar * Martino, C. et al. Bacterial modification of the host
glycosaminoglycan heparan sulfate modulates SARS-CoV-2 infectivity. Preprint at https://biorxiv.org/content/10.1101/2020.08.17.238444v1 (2020). * Eng, A. & Borenstein, E. Taxa-function
robustness in microbial communities. _Microbiome_ 6, 45 (2018). Article PubMed PubMed Central Google Scholar * Tao, W. et al. Re-detectable positive SARS-CoV-2 RNA tests in patients who
recovered from COVID-19 with intestinal infection. _Protein Cell_ https://doi.org/10.1007/s13238-020-00778-8 (2020). * Caporaso, J. G. et al. QIIME allows analysis of high-throughput
community sequencing data. _Nat. Methods_ 7, 335–336 (2010). Article CAS PubMed PubMed Central Google Scholar * Callahan, B. J. et al. DADA2: high-resolution sample inference from
Illumina amplicon data. _Nat. Methods_ 13, 581–583 (2016). Article CAS PubMed PubMed Central Google Scholar * Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene
amplicon sequences with QIIME 2’s q2-feature-classifier plugin. _Microbiome_ 6, 90 (2018). Article PubMed PubMed Central Google Scholar * Vazquez-Baeza, Y., Pirrung, M., Gonzalez, A.
& Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. _Gigascience_ 2, 16 (2013). Article PubMed PubMed Central Google Scholar * Segata, N. et al.
Metagenomic biomarker discovery and explanation. _Genome Biol._ 12, R60 (2011). Article PubMed PubMed Central Google Scholar * Mallick, H. et al. Multivariable association discovery in
population-scale meta-omics studies. Preprint at https://biorxiv.org/content/10.1101/2021.01.20.427420v1 (2021). * Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions.
_Nat. Biotechnol._ 38, 685–688 (2020). Article CAS PubMed PubMed Central Google Scholar * Fang, H., Huang, C., Zhao, H. & Deng, M. CCLasso: correlation inference for compositional
data through Lasso. _Bioinformatics_ 31, 3172–3180 (2015). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank Peihui Wang from Cheeloo
College of Medicine, Shandong University for providing PCDNA6B-SARS-CoV-2-N and PCDNA6B-SARS-CoV-2-E. We also thank Dr. Zulfiqar Sahito from Zhejiang University, Dr. Muhammad Younis from the
Fifth Affiliated Hospital of Sun Yat-sen University for their help in the language editing. We are thankful to all the study participants. This work was supported by grants from the
National Natural Science Foundation of China (82072062), National Science and Technology Key Projects for Major Infectious Diseases (2017ZX10302301-002), Guangzhou Science and Technology
Planning Project (201704020226 and 201604020006), National Key Research and Development Program of China (2016YFC1200105), The Three Major Scientific Research Projects of Sun Yat-sen
University (20200326236), Guangdong Scientific and Technological Research Project for COVID-19 containment (2020A111128022, 2020B111112003), Guangdong Scientific and Technological Research
for COVID-19 (202020012612200001), Zhuhai Scientific and Technological Research Project for COVID-19 containment (ZH22036302200029PWC). AUTHOR INFORMATION Author notes * These authors
contributed equally: Yongjian Wu, Xiaomin Cheng, Guanmin Jiang. AUTHORS AND AFFILIATIONS * Center for Infection and Immunity, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai,
Guangdong, China Yongjian Wu, Guanmin Jiang, Huishu Tang, Siqi Ming, Lantian Tang & Xi Huang * School of Public Health, Sun Yat-sen University, Guangzhou, Guangdong, China Yongjian Wu,
Xiaomin Cheng & Jiahai Lu * Guangdong Provincial Engineering Research Center of Molecular Imaging, Guangdong Provincial Key Laboratory of Biomedical Imaging, and Department of
Interventional Medicine, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, Guangdong, China Yongjian Wu, Guanmin Jiang, Huishu Tang, Siqi Ming, Lantian Tang, Hong Shan & Xi
Huang * Southern Marine Science and Engineering Guangdong Laboratory, Zhuhai, Guangdong, China Yongjian Wu, Hong Shan & Xi Huang * National Clinical Research Center for Infectious
Disease, Shenzhen Third People’ s Hospital; The Second Affiliated Hospital of Southern University of Science and Technology, Shenzhen, Guangdong, China Yongjian Wu, Siqi Ming & Xi Huang
* Department of Clinical Laboratory, The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, Guangdong, China Guanmin Jiang * Center for Infection and Immunity, Mailman School of
Public Health, Columbia University, New York, NY, USA Cheng Guo Authors * Yongjian Wu View author publications You can also search for this author inPubMed Google Scholar * Xiaomin Cheng
View author publications You can also search for this author inPubMed Google Scholar * Guanmin Jiang View author publications You can also search for this author inPubMed Google Scholar *
Huishu Tang View author publications You can also search for this author inPubMed Google Scholar * Siqi Ming View author publications You can also search for this author inPubMed Google
Scholar * Lantian Tang View author publications You can also search for this author inPubMed Google Scholar * Jiahai Lu View author publications You can also search for this author inPubMed
Google Scholar * Cheng Guo View author publications You can also search for this author inPubMed Google Scholar * Hong Shan View author publications You can also search for this author
inPubMed Google Scholar * Xi Huang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Y.W., X.C. and G.J. contributed equally to this work.
Conceptualization: X.H., H.S., C.G. and J.L.; Funding acquisition: X.H. and H.S.; investigation and lab work: Y.W., G.J., H.T., S.M. and L.T.; microbiota data collection: G.J., H.T. and
Y.W.; project administration: Y.W. and X.H.; supervision: X.H., H.S., C.G. and J.L.; bioinformatics and biostatistical analysis: C.G. and X.M.C; writing—original draft: Y.W., X.C. and
G.M.J.; writing—review and editing: all. All the authors read and approved the final manuscript. CORRESPONDING AUTHORS Correspondence to Jiahai Lu, Cheng Guo, Hong Shan or Xi Huang. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Wu, Y., Cheng, X., Jiang, G. _et al._ Altered oral and gut
microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. _npj Biofilms Microbiomes_ 7, 61 (2021). https://doi.org/10.1038/s41522-021-00232-5
Download citation * Received: 23 January 2021 * Accepted: 01 July 2021 * Published: 22 July 2021 * DOI: https://doi.org/10.1038/s41522-021-00232-5 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative