- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Little is known about the origin of germ cells in humans. We previously leveraged post-zygotic mutations to reconstruct zygote-rooted cell lineage ancestry trees in a phenotypically
normal woman, termed NC0. Here, by sequencing the genome of her children and their father, we analyze the transmission of early pre-gastrulation lineages and corresponding mutations across
human generations. We find that the germline in NC0 is polyclonal and is founded by at least two cells likely descending from the two blastomeres arising from the first zygotic cleavage.
Analyzes of public data from several multi-children families and from 1934 familial quads confirm this finding in larger cohorts, revealing that known imbalances of up to 90:10 in early
lineages allocation in somatic tissues are not reflected in mutation transmission to offspring, establishing a fundamental difference in lineage allocation between the soma and the germline.
Analyzes of all the data consistently suggest that the germline has a balanced 50:50 lineage allocation from the first two blastomeres. SIMILAR CONTENT BEING VIEWED BY OTHERS EXTENSIVE
PHYLOGENIES OF HUMAN DEVELOPMENT INFERRED FROM SOMATIC MUTATIONS Article 25 August 2021 CLONAL DYNAMICS IN EARLY HUMAN EMBRYOGENESIS INFERRED FROM SOMATIC MUTATION Article 25 August 2021 THE
MUTATIONAL LANDSCAPE OF HUMAN SOMATIC AND GERMLINE CELLS Article 25 August 2021 INTRODUCTION Little is known about the origin of germ cells in humans. Human primordial germ cells (PGCs) are
thought to be specified around 2 weeks post-fertilization1,2. While modeling germ cells kinetics suggested that there are 2–3 founder germ cells (FGC)3, there is no direct evidence on the
number of FGC and their ancestry, i.e., whether they are monoclonal or polyclonal. The major limitation to studying early lineages, including the germline, is ethical restrictions when
working with human embryos. One way to overcome this limitation is to study cell lineages in parents using naturally occurring post-zygotic developmental mutations and then analyze their
transmission to children. Post-zygotic mutations in the cells of a developing embryo start to accumulate from the very first cleavage and are acquired in almost every cell
division4,5,6,7,8,9. As such, they represent records of development that are archived in the genome of every cell of a living person allowing to examine cell type and cell lineage
allocations to all tissues including germline. De novo variation in a child is defined as a mutation that happened in the germline of either parent. However, it was estimated that up to 6%
of seemingly de novo variants in children can be detected at low frequency in parental blood, indicating that these “de novo” mutations are, in fact, early post-zygotic mutations of
developmental origin in parents, arising prior to germline specification4,10,11,12,13. The standard trio approach commonly used to discover de novo variants by comparing blood-derived
genomes of children to the blood-derived genomes of their parents is ineffective in finding early developmental mutations transmitted to the germline and then to children, since such
mutations can be relatively frequent in parental blood and would be filtered out by this approach4,10,11,12,13. Using pedigrees with three generations (i.e., including the genome of
grandparents in the analysis) allows discovering developmental mutations in parents11, yet often cannot distinguish true de novo from early post-zygotic mutations. This challenge can be
addressed by the joint analysis of the genomes of large sibships (~10 or more siblings), which are rare, where “de novo” mutations shared by multiple children represent early parental mosaic
mutations transmitted to multiple cells in the germline. Such an analysis is inevitably indirect10. When applied to large Icelandic families, this approach suggested the existence of 2 (or
sometimes 3) FGCs10. We previously leveraged post-zygotic mutations detected in clonal induced pluripotent stem cell (iPSC) lines to reconstruct zygote-rooted cell lineage ancestry trees in
two living individuals5. Briefly, we sequenced dozens of clonal lines derived from skin fibroblasts of each individual, discovered mutations present in the genome of founder cells for each
line, analyzed shared mutations across lines, genotyped the mutation in tissues derived from the three germ layers, and reconstructed early developmental cell lineages starting from the
likely first zygotic cleavage (Fig. 1). Lineages were assigned to the first zygotic division from the observations that descendants of the putative two blastomeres accounted for about 100%
of cells in the tissues of the studied individuals, as measured by the frequency of corresponding mutations in blood, saliva, and urine. Similar results have also been obtained in
post-mortem individuals6,7,8,9. A common finding emerging from these studies was that the two blastomeres resulting from the likely first cleavage of the zygote contribute asymmetrically to
the soma. Such an asymmetry was as high as 90:10 in blood for both living individuals in our study, leading to our definition of dominant lineage (i.e., accounting for the majority of
somatic cells) and recessive lineage (i.e., accounting for the minority of somatic cells)5. Here, by acquiring and sequencing the whole genome of the children of NC0, one of the two living
individuals in our previous lineage study, and their biological father, we analyzed the transmission of early developmental mutations from a phenotypically normal woman to her children and
elucidated the contribution of early developmental lineages to the germline. We next expanded our analyses to analyze transmission in a cohort of almost two thousand familial quads from the
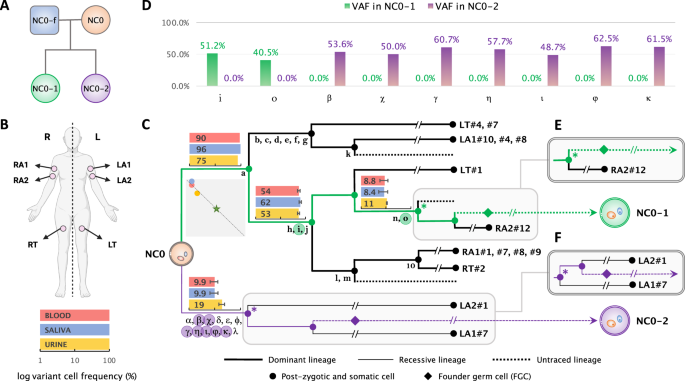
Simons Simplex Collection. RESULTS MUTATION TRANSMISSION IN THE FAMILY OF NC0 We found that 2 and 7 early post-zygotic developmental mutations traceable in the NC0 lineage tree originating
from the dominant and recessive lineages, respectively, were transmitted to each child (NC0-1 and NC0-2) and were present in 100% of children’s blood cells, corresponding to a variant allele
frequency (VAF) of 50% (Figs. 1C, D; Supplementary Data 1; Methods). In each lineage (dominant or recessive) mutations randomly originate on one of the two haplotypes. However, again
randomly, only one haplotype will be inherited by the next generation, accounting for roughly 50% transmission of mutations in a lineage to the next generation. Consistent with that, each
child had about half of the mutations transmitted from the inherited cell lineage (2 out of 5 for NC0-1 and 7 out of 12 for NC0-2) (Fig. 1C). We next applied a common trio comparison
approach to discover de novo mutations in each child by comparing their blood-derived genomes to those from the blood of the parents. Such a comparison discovered 84 and 94 (79 and 82 with
more stringent filtering) candidate de novo variants in NC0-1 and NC0-2, respectively (Methods). Counts of de novo variants were consistent with the reported age of around 40 years old for
both parents at the time of conception14. Importantly, only 1 to 4 (depending on the stringency of filtering) out of the 9 transmitted mutations could be identified as de novo using the trio
strategy (Methods). This is because the trio comparison excludes from the “de novo” catalog all mutations detectable in parental blood, often missing variants (such as early developmental
mutations) that are present in a significant proportion of cells in the blood of a parent (10%–90%; Fig. 1C). Consequently, the standard trio comparison4,10,12,14,15 mostly reveals de novo
mutations that develop later within the parental germlines (and therefore absent in parental blood), and underestimates the number of mutations occurring prior to the specification of PGCs
and transmitted to offspring; such that a child may have several (up to 6 in NC0-2) transmitted mutations not detected14,15. This observation also highlights the unique power of direct
lineage reconstruction and of the analysis of multi-children families (described below) for understanding mutation transmission across generations. Each of the children of NC0 inherited
early post-zygotic mutation arising from either the dominant or the recessive lineages in the mother. These observations proved that progenies from both dominant and recessive lineages in
the soma are also FGCs in the germline of NC0. Therefore, the germline in NC0 consists of at least two clones (i.e., is polyclonal), each descending from one of the likely two blastomeres
from the first zygotic cleavage. MUTATION TRANSMISSION IN LARGE FAMILIES We sought to confirm the observations in the family of NC0 using results from the Jonsson et al. study of transmitted
mutations in families with multiple children10. In that study, personal haplotypes in children and parents were reconstructed using co-occurance of germline variants in the parental and
children genomes. Transmitted mutations were inferred when a locus from the same haplotype was present in multiple children, carried a mutation in at last one child and did not carry the
variant in at least one other child (Fig. 2A and Supplementary Fig. 1). Such transmitted mutations were often observed in parental blood and, thus, represent early post-zygotic mutations in
parents. Because of relying on variant comparison across children and parents this approach is powered in revealing transmitted mutations when analyzing large families, with the power
approaching saturation when there are 10 children in a family10. The analysis by Jonsson et al. of six large families with 17, 10, 9, 9, 9 and 9 children pointed to the existence of two (in
a few cases of three) lineages in the germline of parents and their counterparts in the blood. For each parent in those families, we used transmitted mutation(s) with the highest variant
allele frequency (VAF) in the blood to define the dominant lineage in the soma and estimate its frequency (Methods). In seven parents, we unambiguously identified the dominant lineage
(contributing to > 70% of cells) in the soma (Table 1) (see also Fig. 2, S2, and S3 in Jonsson et al.10), similarly to what we observed in NC0. Strikingly, different from the soma,
dominant and recessive lineages in those parents were roughly equally probable to be transmitted to children, and indeed, for all parents the frequency of transmitting the dominant lineage
to children was always smaller than its frequency in the blood. Thus, the difference in the frequency of each lineage in the parental blood and its transmission to children was significant
in four parents and, when combining analyzes, across all parents and NC0 (combined _p_-value = 3.6 × 10−17). Jonsson et al. analyzed 251 families with multiple children and given the
observed _p_-values (Table 1), random sampling of just one parent with such a difference is improbable. Thus, this analysis suggests that lineage allocation to germline and soma are
drastically different, and the contributions of progenies from the first two blastomeres is asymmetric to soma but is symmetric to germline. MUTATION TRANSMISSION IN FAMILIES FROM SIMONS
SIMPLEX COLLECTION The Simons Simplex Collection contains whole-genome sequencing (WGS) data for 7736 blood samples from 1934 quartet families, each composed of a mother, a father, a child
affected by autism spectrum disorder (ASD), and an unaffected sibling control16. We leveraged that data to discover post-zygotic developmental mutations marking the dominant lineage in
parents and analyzed the mutations’ transmission to children. Developmental mutations were discovered by finding loci with evidence for three haplotypes. Namely, for a given locus every cell
has two germline haplotypes A and B. When a locus has a developmental mutation (e.g., on haplotype A) then mutated cells will have a mutated haplotype A (Am) and an unaffected haplotype B.
Bulk blood will consist of mutated and non-mutated cells and will, thus, have three haplotypes: A, Am, and B. We developed an algorithm that considered pairs of nearby (within 100 bps)
single nucleotide variants detected in the blood such that overlapping reads can phase the somatic with germline variants and indicate the existence of three haplotypes (Fig. 2A). In such a
pair of variants, one is an inhered heterozygous variant that serves as an anchor to phase the second one—an early developmental mutation—to haplotypes. We only considered developmental
mutations at high frequency to ensure that they mark the dominant lineage. To calibrate this approach and access its accuracy, we first applied it to mutation discovery in children in the
quads (Fig. 2B). A true early post-zygotic mutation is likely to be unique and is unlikely to match variants in the corresponding parents. Thus, we considered a phased early mutation as a
false positive if it had a matching variant call in one of the corresponding parents. We required VAF between 35% and 45% (corresponding to a cell frequency of 70–90%, since the dominant
lineage should be in the majority of blood cells), and defined criteria on the number of reads supporting the minor (out of three) haplotype to eliminate most of the false positives. We
arrived at a set of 35 early post-zygotic mutations in children with just 6 (17%) false positives (Fig. 2C; Supplementary Data 3). Of the remaining 29 early mutations in children, 25 (86%)
have been previously identified as de novo using the standard approach by An et al.16 indicating that early mutations we discover are hardly distinguishable from de novo mutations. We then
analyzed mutations in parents, assuming the same false positive rate found in children, as WGS data for parents and children had similar coverage and other sequencing characteristics. We
discovered a comparable number of 40 post-zygotic mutations in 40 parents of 40 families (Fig. 2D; Supplementary Data 3). Since every family in SSC has just 2 children, analysis of mutation
transmission in each family lacks statistical power. We, therefore, analyzed the distribution of the 40 families by the outcomes of mutation transmission to their children, which were:
transmission to none of the children, transmission to only one child, and transmission to both children (Fig. 2E). In this analysis, we considered four scenarios for the contribution of the
dominant lineage to gonads and therefore to children: (i) frequency is the same as in blood (i.e., gonads and soma have similar lineage composition); (ii) frequency is 50% (i.e., equal
contribution of dominant and recessive lineages to gonads); (iii) frequency is 100% (i.e., only dominant lineage is present in the gonads and passed to the next generation); (iv) frequency
is random (Supplementary Fig. 2). For each scenario we derived the expected distribution of mutation transmission to none, one, or both children and compared it to the observed distribution
(Fig. 2E & Supplementary Fig. 3). We could confidently reject the scenario of the same frequency of the dominant lineage in gonads and blood (_p_-value = 0.03) supporting such an
inference made above. We could also confidently reject the scenario that only dominant lineage is present in the gonads (_p_-value = 10−4) (Supplementary Fig. 3B), supporting the inference
made above for NC0 that the gonads are derived from at least two founder cells each descending from one blastomere likely arising from the first zygotic cleavage. At the same time, the
observed distribution was consistent with the equal contribution of dominant and recessive lineages to the gonads (_p_-value = 0.62) supporting the same observation made above for NC0 and
large families (Table 1). Тhe scenario of random distribution of lineages in the gonads is also consistent with the observations (_p_-value = 0.41). Yet note, that even in this scenario
almost always the germline would consist of at least two cells—one from the dominant lineage and one from the recessive. DISCUSSION In the described study we leveraged three different
approaches to study transmission of early mutations from parents to the next generation. The approaches included (i) direct reconstruction of developmental lineages in a parent and tracing
their transmission to the progeny, (ii) identification of early mutations in parents in a collection of 1934 familial quads, and (iii) analysis of early mutations in parents discovered as
shared de novo mutations across multiple children. All approaches consistently revealed that both dominant and recessive developmental lineages in parental soma contribute to the germline,
which consequently has at least two FGCs. Similarly, all the approaches consistently revealed differential frequencies of the dominant lineage in the germline and blood, suggesting a
fundamental difference in lineage allocation between the soma and the germline. Moreover, all the approaches consistently suggest a balanced (i.e., 50:50) allocation of dominant and
recessive lineage to the germline, and we hypothesize that such an allocation is a common phenomenon in humans. This hypothesis is supported by direct measurement of frequencies of dominant
lineage in the testis of individual PD28690 in a previous study7. We and others previously hypothesized that asymmetric contribution of early lineages to soma could be the result of one of
the first two blastomeres committing mostly to the extraembryonic (trophectoderm) lineages, with the other blastomere mostly committing to inner cell mass (ICM), becoming the dominant
lineage in the embryo5. A recent imaging study of the developing human embryo supported the asymmetric contribution of the first two blastomeres to the ICM but suggested that the asymmetry
arises from stochasticity in internalizing just a few, 1 to 4, cells to the ICM at the 8-to-16-cell stage17. If allocation of PGCs (that develop from the ICM) is also stochastic, then one
could expect roughly the same bias in lineage contribution for the germline as for the soma. However, the results presented above (i.e., different frequencies of early lineages in germline
and blood) contradict this expectation. This may imply that allocation of PGCs may not be stochastic and may depend on yet unknown factors, e.g., relative position in the ICM of the cells
from dominant and recessive lineages, which eventually results in a different frequency of allocation of the lineages to soma and germline. In such scenario, similar contribution of dominant
and recessive lineages to the germline may imply that PGCs are initiated at the border between one “dominant” and one “recessive” cell assemblies. Such a border may have roughly the same
number of surrounding cells form dominant and recessive lineage committing to PGCs. This scenario could be consistent with the proposed dual origin of PGCs from amnion and epiblast in
humans18,19,20,21. It is intriguing to speculate that there could be a molecular mechanism for maintaining such a border interface from the first zygotic cleavage until PGC specification
perhaps through asymetric division of the blastomeres (Fig. 3). Larger studies of mutation transmission in multi-children families combined with analysis of somatic and germline lineages in
parents will likely shed light on this issue. More broadly, leveraging naturally occurring somatic mutations is a powerful approach to study human development, which will inform not only
about the formation of the germline but also about other developmental steps prior and after gastrulation. Finally, these findings highlight the unique nature of early post-zygotic
mutations, which is their ability to be transmitted to progeny and therefore contribute to the evolution of the human genome. METHODS ETHICS STATEMENT Human subjects were recruited through
several research projects at the Yale Child Study Center. The participants did not receive compensation for taking part in the study. Written informed consent was obtained from each
participant enrolled in the study, and all research was approved by the Yale University Institutional Review Board (HIC# 1104008337) and Yale Center for Clinical Investigation at Yale
University. Analysis of SSC data was conducted under IRB 23-012592 approved by Mayo Clinic IRB. SOMATIC MOSAICISM AND RECONSTRUCTION OF A CELL LINEAGE TREE IN THE MOTHER In our previous
study5, we reconstructed a cell lineage tree in early development by leveraging early post-zygotic mosaic mutations that we identified in the mother (NC0). In brief, we obtained whole genome
sequencing data for 25 clonal induced pluripotent stem cell (iPSC) lines derived from skin fibroblasts (average coverage of 35x), as well as for bulk blood, saliva, and urine samples
(average coverage of 238x) from NC0. Using the all-2-all exhaustive comparison22 of all sequenced iPSC lines, we discovered mosaic mutations that arose in development and accumulated in cell
lineages. Based on the sharing of discovered early mutations and their estimated cell frequencies in bulk tissues, we reconstructed an early developmental cell ancestry tree starting from
the likely first zygotic cleavage. COLLECTION OF SAMPLES AND WHOLE GENOME SEQUENCING Informed consent was obtained from each participant enrolled in the study according to the regulations of
the Institutional Review Board and Yale Center for Clinical Investigation at Yale University. The participant agreed to data sharing of genomic de-identified data using controlled data
access. About 14 mL of whole blood was collected for each individual using BD Vacutainer ACD tubes. DNA was extracted using the Maxwell® RSC Blood DNA Kit (Promega) in accordance with the
manufacturer’s instructions. Whole Genome Sequencing was conducted to 30x coverage at BGI Americas Corporation for NC0-1 and NC0-2 and at the Yale Center for Genome Analysis for NC0-f.
Measurements of DNA quantity by Qubit, and quality, by Agarose Gel Electrophoresis, were performed before and after the PCR-free library preparations. CALLING CONSTITUTIONAL/INHERITED
GERMLINE VARIATIONS IN CHILDREN AND THE FATHER OF NC0 FAMILY The blood of two children of NC0 (NC0-1 and NC0-2) and father (NC0-f) was sequenced to 36x and 48x coverage on average,
respectively. We aligned the raw sequences against the human reference genome hg19. The duplicated reads were marked and removed by Picard (http://broadinstitute.github.io/picard), and local
realignment and base quality score recalibration were performed with GATK23. We used GATK HaplotypeCaller version 4.2.6 to call constitutional/inherited germline variants, resulting in
4,057,075 variants passing GATK filters with VQSR (Variant Quality Score Recalibration) in the father, and 4,231,719 and 4,167,927 in the two daughters. IDENTIFICATION OF TRANSMITTED EARLY
MUTATIONS IN NC0 Out of all discovered post-zygotic mosaic mutations in the mother (18,419; see above), we retained only the mutations matching constitutional/inherited germline variations
found in each daughter (see above) as candidates for transmitted early post-zygotic mutations. This resulted in 27 candidates, of which 18 were excluded as they were present in almost 100%
(i.e., at 50% VAF) of the father’s blood cells, thereby indicating they were inherited from the father (Supplementary Data 2). Moreover, those excluded mutations were not shared by any other
sampled lineage and lacked supporting reads in bulk tissues (blood, saliva, and urine) in the mother, suggesting they occurred very late in development, are private to the specific somatic
lineages where they were found and not relevant to the maternal germline lineage. Consequently, we identified 2 and 7 early post-zygotic mutations in the mother transmitted to each of the
offspring. The constitutional nature of these mutations in the daughters was evidenced by their VAFs of around 50% in the blood (Fig. 1D and Supplementary Data 1), which indicated that they
were also present in germline lineages in the mother, thus had occurred prior to the differentiation and proliferation of primordial germ cells (PGCs) (Fig. 1C). EXTRACTION OF DE NOVO
CANDIDATES THROUGH THE WHOLE GENOME COMPARISON WITHIN THE PARENTS-OFFSPRING TRIO We searched de novo mutations within the trio by comparing the genomes of two children to the maternal and
paternal genomes. To mitigate the potential effects of coverage bias, we down-sampled the maternal genome from its original average coverage of 238x to 48x, which is equivalent to the
average coverage of the paternal genome. We used Mutect223 and Strelka224 to call mutations in each child against their parental genomes and considered only consensus “PASS” calls made by
both callers. Calls were required to have a VAF of between 30% and 70% in the child and a minimum depth of 20 reads in the child and both parents. We also excluded sites within known
segmental duplication and simple tandem repeat regions, as defined by the UCSC. For each child, we defined mutations as de novo if they satisfied such criteria and were called relative to
both parental genomes. This yielded 84 de novo mutation candidates in one child and 94 in the other. We observed that only 4 out of 9 early post-zygotic mutations in the mother, which were
transmitted to the offspring, were also identified as de novo mutations in children using this trio whole genome comparison strategy (Supplementary Data 1). There was only one shared de novo
candidate variant between daughters. Given that lineage reconstruction in mother suggest that transmitted mutations to daughters can’t be shared (transmitted lineages as originating from
the likely first zygotic cleave share not common mutations), this variant likely originated in father. Alternatively, it could be a systematic false positive. Furthermore, we additionally
applied the parental presence filter as in other trio studies4,14,15, where de novo mutations were required to have a maximum depth of 1 read and a maximum allele frequency of 5% for
alternative alleles in both parents. Only 1 out of the 9 early post-zygotic mutations in the mother transmitted to children was retained as de novo, with a total of 79 in one child and 82 in
the other. This demonstrates that the number of early developmental mutations transmitted to offspring, which likely occurred before the specification of PGCs (PGCS), may have been
underestimated with the standard trio approach4,14,15. This is due to the exclusion of variations with evidence of presence in parents, which removes a large proportion of transmitted
mutations that arose in pre-PGCS lineages. Some of these de novo mutations may be recovered with the haplotype sharing by siblings’ approach10,11, but the approach still misses de novo
mutations in the germline that are carried by all or none of siblings who share the same parental haplotype at the locus10. ANALYZING LARGE FAMILIES FROM ICELAND PEDIGREE STUDY We utilized
data presented in Jonsson et al.10 in Fig. 2 and Supplementary Figs. 1 and 2. For each parent we defined lineages by branches from the ancestral states defined by Jonsson et al. There were 2
to 3 of such lineages/branches for parents in the family. Since no lineage tree were reconstructed in each parent, no clear mutation marker(s) for the lineages were defined. Therefore, we
estimated frequencies of lineages in the blood from the frequencies of transmitted mutations in the following ways. Mutations with higher frequencies mark earlier originating lineages, so,
for each lineage, we first identified a primary marker mutation as a mutation with the highest VAF in the blood. We then defined a set of lineage markers as mutations with frequencies
different by less than 10% VAF from the frequency of the primary marker. We then averaged VAFs of the defined marker set and estimated cell frequencies of the corresponding lineages as a
double of the average VAF. We used 7 parents with an obvious dominant lineage in the soma (i.e., at least 70% of cells in the blood). Frequency of the recessive lineage was defined as a
complement to the frequency of the dominant lineage. Accordingly, counts of children derived from the transmitted recessive lineage were calculated as the number of children without
transmitted dominant lineage. When testing for consistency with the count of children with the transmitted lineage and the frequency of the lineage in the blood, we used a one-tailed
binomial test for a less or equal count of children with the dominant lineage. ANALYZING TRANSMISSION OF EARLY MUTATIONS ACROSS GENERATIONS IN THE SIMONS SIMPLEX COLLECTION We downloaded
CRAM files (aligned to the human reference genome GRCh38) and VCF files from SFARI Base (https://www.sfari.org/resource/sfari-base). To select inherited SNPs for phasing with mutation
candidates (Fig. 2A), we searched heterozygous single nucleotide variant calls from VCF files. We retained only calls that passed GATK filters, were in accessible genomic regions according
to the 1000 Genomes Project’s mappablility mask (bases marked as “P”) and had a population allele frequency of more than 10−3 in gnomAD database (https://gnomad.broadinstitute.org). Mutation
candidates were selected from single nucleotide variant calls within 100 bps upstream and downstream of the selected inherited SNPs. To retain the most likely early mutations we required
the calls to be in genomic regions with the “P” mask, pass GATK filters, have a population allele frequency of less than 10−5, and be outside the known repeat regions defined by the UCSC
table browser. We then used a phasing tool in MosaicForecast package25 to phase two nearby SNVs (inherited SNP and candidate mutation). We retained candidate mutations that were phased to 3
haplotypes, had at least 3 supporting reads for the minor haplotype, and had a VAF of more than 35% for further consideration. For the final sets of phased early post-zygotic mutations we
required that the minor haplotype had 4 to 6 supporting reads for VAF [35%,40%] and 5 or 6 supporting reads for VAF [40%,45%]. This was based on the analysis for children, for which we
considered a phased early mutation as a false positive if there was a matching variant call in parents, i.e., the mutation was an inherited SNP. We additionally removed phased clustered
within 100 bps mutations. For the final set of 35 early developmental mutations in children the false positive rate was estimated to be 17% (Fig. 2C). Except for these false positives, none
other variants in the set had any supporting reads in the parents, suggesting the calls are genuine early developmental mutations. Because WGS data for parents and children had similar
coverage and other sequencing characteristics, we assume the same false positive rate for the final set of phased early mutations in parents (Fig. 2D). We calculated transmission
probabilities for early mutations in a parent to none, one, or two children for four possible scenarios based on their expected frequencies in germ cells of the parent, i.e., based on the
contribution of the dominant lineage to germline development (Supplementary Fig. 2B). For the scenario where the frequency of the dominant lineage in gonads is same as in blood, we first
calculated transmission probabilities for each mutation and then averaged the probabilities across the entire set of mutations. To derive the expected distribution of mutation transmission,
we applied the 17% false positive rate for inherited variants (Fig. 2E and Supplementary Fig. 3), as estimated in the analysis for children. We considered a candidate early mutation in
parents as a transmitted one if it had a matching variant call that passes GATK filters in one or two corresponding children. To test the statistical difference between expected and observed
transmission distributions, we used two-tailed _×_2 test with 2 degrees of freedom. STATISTICS & REPRODUCIBILITY For large families from the Iceland pedigree study, we conducted a
statistical test for consistency between the count of children with the transmitted lineage and the frequency of the lineage in the blood, using one-tailed binomial test for less or equal
count of children with the dominant lineage. We also performed an _×_2 test (two-tailed) with 2 degrees of freedom to test the statistical difference between expected and observed
transmission distribution of early mutations across generations in the Simons Simplex Collection. Regarding the family of NC0, no statistical method was used to predetermine sample size, as
this is an observational study for a specific family. We have excluded 18 mosaic mutations found in the mother (NC0) because they were not relevant to maternal germline lineages as described
above. The experiments were not randomized, as it was not applicable since we analyzed a specific family. There was no blinding in this study, as it was not relevant for genomic data
analyzes. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Sequencing data for NC0
was previously analyzed5 and is available at the NIMH Data Archive (NDA) under collection #2961 [https://nda.nih.gov/edit_collection.html?id=2961]. Aligned sequencing reads from children and
father are available at the dbGaP for General Research Use (GRU) under accession phs003781.v1.p1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003781.v1.p1]. For
the reason of participants’ privacy, as specified in the consent they signed, data access is controlled. Qualified researchers should submit a Data Access Request to dbGaP. The exact
requirements for data access and data request process are determined by the National Institute of Health (https://sharing.nih.gov). WGS and phenotype data for the SSC collection were
obtained from the Simons Foundation for Autism Research Initiative (SFARI) Base (https://base.sfari.org/). All processed data necessary for a full evaluation of this work is included in the
text, Supplementary Information, and Supplementary Data files. The coordinates of mutations in Supplementary Data 1 and 2 were de-identified. Full information on all post-zygotic mutations
discovered in NC0 was part of the previous study5 and is available in the NDA study #1057 [https://nda.nih.gov/study.html?id=1057]. CHANGE HISTORY * _ 10 MARCH 2025 A Correction to this
paper has been published: https://doi.org/10.1038/s41467-025-56705-0 _ REFERENCES * Wen, L. & Tang, F. Human germline cell development: from the perspective of single-cell sequencing.
_Mol. Cell_ 76, 320–328 (2019). Article CAS PubMed MATH Google Scholar * Samuels, M. E. & Friedman, J. M. Genetic mosaics and the germ line lineage. _Genes (Basel)_ 6, 216–237
(2015). Article CAS PubMed MATH Google Scholar * Zheng, C. J., Luebeck, E. G., Byers, B. & Moolgavkar, S. H. On the number of founding germ cells in humans. _Theor. Biol. Med Model_
2, 32 (2005). Article PubMed PubMed Central MATH Google Scholar * Rahbari, R. et al. Timing, rates and spectra of human germline mutation. _Nat. Genet_ 48, 126–133 (2016). Article CAS
PubMed MATH Google Scholar * Fasching, L. et al. Early developmental asymmetries in cell lineage trees in living individuals. _Science_ 371, 1245–1248 (2021). Article ADS CAS PubMed
PubMed Central MATH Google Scholar * Bae, T. et al. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. _Science_ 359, 550–555 (2018). Article
ADS CAS PubMed MATH Google Scholar * Coorens, T. H. H. et al. Extensive phylogenies of human development inferred from somatic mutations. _Nature_ 597, 387–392 (2021). Article ADS
CAS PubMed MATH Google Scholar * Park, S. et al. Clonal dynamics in early human embryogenesis inferred from somatic mutation. _Nature_ 597, 393–397 (2021). Article ADS CAS PubMed
MATH Google Scholar * Spencer Chapman, M. et al. Lineage tracing of human development through somatic mutations. _Nature_ 595, 85–90 (2021). Article ADS CAS PubMed MATH Google Scholar
* Jonsson, H. et al. Multiple transmissions of de novo mutations in families. _Nat. Genet_ 50, 1674–1680 (2018). Article CAS PubMed MATH Google Scholar * Sasani, T. A. et al. Large,
three-generation human families reveal post-zygotic mosaicism and variability in germline mutation accumulation. _Elife_ 8, e46922 (2019). Article PubMed PubMed Central Google Scholar *
Acuna-Hidalgo, R. et al. Post-zygotic point mutations are an underrecognized source of De Novo genomic variation. _Am. J. Hum. Genet_ 97, 67–74 (2015). Article CAS PubMed PubMed Central
MATH Google Scholar * Campbell, I. M. et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. _Am. J. Hum. Genet_ 95, 173–182 (2014).
Article CAS PubMed PubMed Central MATH Google Scholar * Kaplanis, J. et al. Genetic and chemotherapeutic influences on germline hypermutation. _Nature_ 605, 503–508 (2022). Article
ADS CAS PubMed PubMed Central MATH Google Scholar * Jonsson, H. et al. Parental influence on human germline de novo mutations in 1548 trios from Iceland. _Nature_ 549, 519–522 (2017).
Article ADS PubMed Google Scholar * An, J. Y. et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. _Science_ 362, eaat6576 (2018). Article
ADS PubMed PubMed Central Google Scholar * Junyent, S. et al. The first two blastomeres contribute unequally to the human embryo. _Cell_ 187, 2838–2854.e17 (2024). Article CAS PubMed
MATH Google Scholar * Chen, D. et al. Human primordial germ cells are specified from lineage-primed progenitors. _Cell Rep._ 29, 4568–4582.e4565 (2019). Article CAS PubMed PubMed
Central MATH Google Scholar * Kobayashi, T. & Surani, M. A. On the origin of the human germline. _Development_ 145, dev150433 (2018). Article PubMed MATH Google Scholar * Saitou,
M. & Hayashi, K. Mammalian in vitro gametogenesis. _Science_ 374, eaaz6830 (2021). Article CAS PubMed Google Scholar * De Felici, M. in _Oogenesis_ (eds G. Coticchio, D. F.
Albertini, & L. De Santis) 19-37 (Springer London, 2013). * Sarangi, V. et al. All2: a tool for selecting mosaic mutations from comprehensive multi-cell comparisons. _PLoS Comput Biol._
18, e1009487 (2022). Article CAS PubMed PubMed Central MATH Google Scholar * McKenna, A. et al. The Genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA
sequencing data. _Genome Res_ 20, 1297–1303 (2010). Article CAS PubMed PubMed Central MATH Google Scholar * Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic
variants. _Nat. Methods_ 15, 591–594 (2018). Article CAS PubMed MATH Google Scholar * Dou, Y. et al. Accurate detection of mosaic variants in sequencing data without matched controls.
_Nat. Biotechnol._ 38, 314–319 (2020). Article CAS PubMed PubMed Central MATH Google Scholar Download references ACKNOWLEDGEMENTS We are grateful to members of NC0 family that
participated in this study by donating tissue and/or blood samples. We are grateful to members of families in SSC that donated blood samples for WGS and to the Simons Foundation that
provided access to the data (approved project 2343.4). This work was funded by the NIH Common Fund SMaHT program (grants UG3 NS132128 and UG3 NS132146) and by the Simons Foundation (grant
399558). Y.J. was also supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant number
2022R1A6A3A03055692). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Quantitative Health Sciences, Center for Individualized Medicine, Mayo Clinic, Rochester, MN, USA Yeongjun
Jang, Taejeong Bae & Alexej Abyzov * Child Study Center, Yale University, New Haven, CT, USA Livia Tomasini & Flora M. Vaccarino * Department of Neurology, Yale University, New
Haven, CT, USA Anna Szekely * Department of Neuroscience, Yale University, New Haven, CT, USA Flora M. Vaccarino * Yale Kavli Institute for Neuroscience, New Haven, CT, USA Flora M.
Vaccarino Authors * Yeongjun Jang View author publications You can also search for this author inPubMed Google Scholar * Livia Tomasini View author publications You can also search for this
author inPubMed Google Scholar * Taejeong Bae View author publications You can also search for this author inPubMed Google Scholar * Anna Szekely View author publications You can also search
for this author inPubMed Google Scholar * Flora M. Vaccarino View author publications You can also search for this author inPubMed Google Scholar * Alexej Abyzov View author publications
You can also search for this author inPubMed Google Scholar CONTRIBUTIONS F.M.V. and A.A. conceived the study. A.A. and F.M.V. supervised the study. L.T. and A.S. collected samples and
performed experiments. Y.J., T.B., and A.A. performed computational data analyses. Y.J. and A.A. prepared display items. F.M.V., A.A., and Y.J. drafted the initial text of the manuscript.
CORRESPONDING AUTHORS Correspondence to Flora M. Vaccarino or Alexej Abyzov. ETHICS DECLARATIONS COMPETING INTERESTS Alexej Abyzov is a paid consultant at OmniTier Inc. Other authors declare
no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Magdalena Zernicka-Goetz, and the other, anonymous, reviewers for their contribution to the peer
review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2
SUPPLEMENTARY DATA 3 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License,
which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this
article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the
material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to
obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Jang, Y., Tomasini, L., Bae, T. _et al._ Transgenerational transmission of post-zygotic mutations suggests symmetric contribution of first two blastomeres to human
germline. _Nat Commun_ 15, 9117 (2024). https://doi.org/10.1038/s41467-024-53485-x Download citation * Received: 05 May 2023 * Accepted: 10 October 2024 * Published: 23 October 2024 * DOI:
https://doi.org/10.1038/s41467-024-53485-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
:max_bytes(150000):strip_icc():focal(373x649:375x651)/pepper-teigen-1-346cf8c6df824f91adc320d9c09a9c6b.jpg)




