- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Biocatalysis is an attractive approach for the synthesis of chiral pharmaceuticals and fine chemicals, but assessing and/or improving the enantioselectivity of biocatalyst towards
target substrates is often time and resource intensive. Although machine learning has been used to reveal the underlying relationship between protein sequences and biocatalytic
enantioselectivity, the establishment of substrate fitness space is usually disregarded by chemists and is still a challenge. Using 240 datasets collected in our previous works, we adopt
chemistry and geometry descriptors and build random forest classification models for predicting the enantioselectivity of amidase towards new substrates. We further propose a heuristic
strategy based on these models, by which the rational protein engineering can be efficiently performed to synthesize chiral compounds with higher ee values, and the optimized variant results
in a 53-fold higher _E_-value comparing to the wild-type amidase. This data-driven methodology is expected to broaden the application of machine learning in biocatalysis research. SIMILAR
CONTENT BEING VIEWED BY OTHERS A META-LEARNING APPROACH FOR SELECTIVITY PREDICTION IN ASYMMETRIC CATALYSIS Article Open access 16 April 2025 COMBINING CHEMISTRY AND PROTEIN ENGINEERING FOR
NEW-TO-NATURE BIOCATALYSIS Article 12 January 2022 ROBUST ENZYME DISCOVERY AND ENGINEERING WITH DEEP LEARNING USING CATAPRO Article Open access 20 March 2025 INTRODUCTION Owing to the high
efficiency, excellent selectivity and environmentally benign reaction conditions, biocatalysis and biotransformation have become an important and powerful strategy in asymmetric
synthesis1,2,3,4,5,6. Along with the increasing discovery of new enzymes and the development of protein engineering strategies, substrate scope and catalytic performance for biocatalysis are
improving. However, the conventional “trial-and-error” protocol of biocatalysis research is very laborious and requires extensive experience of researchers7,8,9. It usually spends several
months or even years on the discovery and engineering of a satisfactory biocatalyst. Among the various reaction functions of biocatalysis, enantioselectivity has received almost the most
attention10,11. Prediction on the enantioselectivity of a protein toward a target substrate will be able to greatly accelerate the establishment of a biocatalytic reaction system. Although a
number of computational methods7,8,9,10,11,12,13,14 have been developed to simulate a biocatalytic reaction, efforts to predict the enantioselectivity of biocatalysis usually fail because a
small free energy difference out of the valid accuracy range of widely-used computational method can lead to a large change in enantiomeric excess values1. Further improvement in the
accuracy of free energy calculation requires unaffordable computational expense. In recent years, machine learning (ML) has emerged as a powerful and effective tool for biocatalytic property
prediction and protein engineering15,16,17,18,19,20,21,22,23,24,25,26,27,28. The success of a ML predictor depends critically on data acquisition and feature extraction. A large amount of
protein sequence/structure information and biocatalysis-related reaction kinetic parameters can be obtained from open-source databases (e.g., PDB29, UniProt30 and BRENDA31). However, the
lack of information on biocatalytic enantioselectivity as well as the difficulty of enantioselectivity data measurement has seriously impeded the ML study of enzyme enantioselectivity. To
our best knowledge, only a few ML predictors have been reported to establish the relationship between reaction enantioselectivity and enzyme sequence/structure, including an epoxide
hydrolase32, a nitric oxide dioxygenase33, an imine reductase34, an amine transaminase35 and an ene-reductase36. Although these predictors enable the construction of protein fitness
landscapes, the important role of substrates is usually ignored16. It remains a challenge to (1) collect a sufficient amount of reliable data, (2) build predictors that fully describe the
relationship between substrates structure and biocatalytic enantioselectivity, and (3) effectively design enzyme variants with higher enantioselectivity assisted by ML predictors. Amidases
(EC 3.5.1.X) are a class of cofactorless enzymes capable of hydrolyzing amide groups to produce acid products. Amidase-containing microbial whole cells or isolated amidases have been widely
used and have successfully hydrolyzed a large number of amide substrates, making them one of the most versatile enzymes for the potential production of pharmaceuticals and commodity
chemicals, such as clausena alkaloids, aza-nucleoside analogs and chiral non-natural amino acids37. Since the late 1990s, using nitrile hydratase/amidase-containing _Rhodococcus
erythropolis_ AJ270 whole cells as a catalyst, our group have systematically investigated and reported the kinetic resolution or desymmetrization of a variety of racemic or prochiral
substrates to yield a series of chiral carboxamides and carboxylic acids38,39,40,41,42,43. In particular, when a nitrile substrate is catalyzed in tandem by nitrile hydratase and amidase
from whole cells, the nitrile hydratase typically exhibits rather low enantioselectivity, while the amidase shows dominant enantioselectivity. The ee values of the products of these
biotransformations therefore faithfully reflect the enantioselectivity of the amidase. Such continuous explorations also provide hundreds of reliable and comparable data for the building of
corresponding machine learning model. Herein we report ML classification models based on our collected data as well as “chemistry” and “geometry” descriptors to establish the underlying
relationship between substrate structure and reaction enantioselectivity. This model is capable of predicting amidase-catalytic enantioselectivity towards new substrates and thus can be used
for rapid feasibility assessment of reaction route in a heuristic way. With the help of ML, we also characterized the substrate structure and catalytic property relationship and
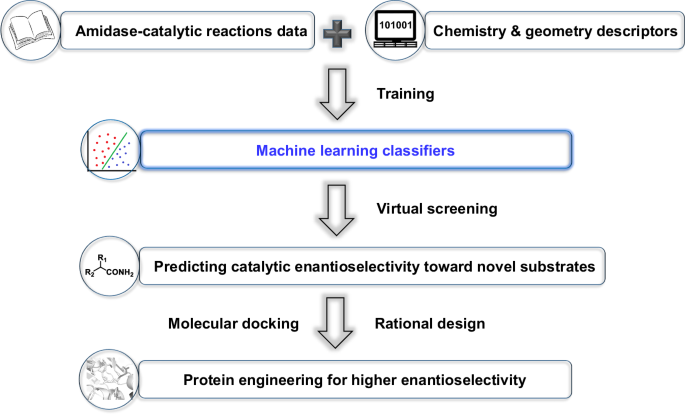
successfully applied it to the rational design of variants for better catalytic enantioselectivity (Fig. 1). RESULTS AND DISCUSSION DATA COLLECTION Firstly, we summarized and collected the
reactions of 240 substrates catalyzed by _Rhodococcus erythropolis_ AJ270 in our previous research, including 160 kinetic resolution reactions and 80 desymmetrization reactions. Most of the
reactions have been reported in journals38,39,40,41,42,43, while a small number of reactions with negative results have been published in PhD theses (See Supplementary Source Data). In order
to standardize the enantioselectivity characterization of kinetic resolution reactions and desymmetrization reactions, the ee values of products and/or the recovered substrates were
transformed to _E_ (Enantiomeric ratio) values44 and then represented by \(\Delta \Delta {G}^{\ne }\) according to \(\Delta \Delta {G}^{\ne }=-{RT}{\mathrm{ln}}E\). All attempts to construct
a regression model failed, giving a poor R2 value as 0.354 on the test set (see Figure S2, SI). It is not surprising on account of the relatively small size of the present dataset, which is
prone to the overfitting problem. The classification model was therefore considered for further research. All reactions in the dataset were classified into “positive” and “negative” based
on whether the values of \(-\Delta \Delta {G}^{\ne }\) were larger than (or equal to) 1.86, 2.40 or 3.00 kcal/mol (corresponding to ee values of products equal to 80%, 90% or 95% at 303 K,
respectively). For example, under the criterion of 2.40 kcal/mol, 143 samples with \(-\Delta \Delta {G}^{\ne }\) ≥ 2.40 kcal/mol were defined as positive, and the remaining 97 samples were
negative. MODEL TRAINING Two types of descriptors developed by Barnard et al.45 were adopted in this work. The first type can be obtained according to a vocabulary of molecular “cliques”
that were derived from the molecular structure of substrate46. The second type can be calculated as the histograms of weighted atomic-centered symmetry functions47,48. The former is more
relevant to the chemistry information about functional groups of substrates, and the latter focuses on the three-dimensional geometry of substrates. A feature selection process was
implemented in prior to training. Four classification models, that is, random forest (RF), support vector machine (SVM), logistics regression (LR), and gradient boosted decision tree (GBDT),
were built on the basis of five-fold cross-validation. Their performance was evaluated based on the accuracy, precision, recall, _F_-score and the area under receiver operating
characteristic curve (AUC). All ML algorithms were performed with the Scikit-learn library49. The geometry optimizations on substrates were implemented with Gaussian 09 software package50.
The performance of different ML classifiers were listed in Table S3. RF, LR and GBDT are able to achieve _F_-scores above 0.8 on the test set under the classification criterion of 2.40. On
account of the highest _F_-score and the smallest number of selected descriptors, the RF classifier was employed hereafter and rebuilt under other criteria. In order to check the robustness
of ML predictions, 30 RF classifiers with different random seeds were rebuilt under each criterion. The results were collected in Table 1. It can be seen that the performance was good under
the criteria of 1.86 and 2.40, but the _F_-score decreased below 0.8 under the criterion of 3.00. It is far from perfect but still acceptable in this work, since the ML classifier acts as a
heuristic tool in prior to experiments. Two rigorous data splitting strategies were further applied by leaving all molecules involving bromine (denoted as “strategy 1”) or a five-membered
ring (denoted as “strategy 2”) out of the training set. The ML classifier under either of these two splitting schemes can achieve an acceptable level of accuracy (Table S4). FEATURE
IMPORTANCE ANALYSIS The feature importance can be analyzed based on the mean decrease in impurity (MDI)51 of RF classifiers as well as the SISSO feature compositions52 that distinguish
positive reactions (higher enantioselectivity) from negative ones (lower enantioselectivity). The raw data of MDI and SISSO were shown in Figs. S3 and S4, respectively. For example, it can
be seen in Figure S4(b) that a substrate with large values of three specific descriptors (denoted as SFR54, SFR69 and SFR94) has a higher tendency to be “positive”, that is, \(-\Delta \Delta
{G}^{\ne }\) ≥ 2.40 kcal/mol and ee ≥ 90%. Some descriptors extracted by SISSO such as SFR55, SFR94 and SFR54 also appear in Figure S3, which agrees well with the feature importance
analyzes based on MDI. Most of important features belong to the atomic-centered symmetry functions (ACSFs). Based on the raw data, we further explored chemical information, that is, which
functional groups or fragments are more relevant to the enantioselectivity of reactions, by mapping the extracted ACSFs to the pre-defined type of atoms53,54 at the center. More
computational details can be seen in Algorithm S1 and Table S5, SI. As shown in the importance scores in Figure S5, some specific atom types, such as the H bonded to aliphatic C with 2
electron-withdraw groups, the aliphatic sp2 N with two connected atoms, and the sp3 C in square systems, may have more significant impact on the biocatalytic enantioselectivity of
substrates. PREDICTION AND TESTING TOWARD NEW SUBSTRATES To demonstrate the ability of machine learning models to assist in the construction of enantioselective amidase catalytic system for
the preparation of important chiral pharmaceuticals, we chose racemic 2-hydroxy-2-phenylacetamide 1A and meso cyclopentane-1,2-dicarboxamide 3 as model substrates, which can be biocatalyzed
to chiral mandelic acids55 and disubstituted cyclopentane56, respectively. Three classifiers under different classification criteria were used together to predict the range of \(-\Delta
\Delta {G}^{\ne }\) for a given substrate. Substrate 3 (Fig. 2B) was predicted to have a probably high enantioselectivity of this reaction, that is, \(-\Delta \Delta {G}^{\ne }\) ≥ 3.00
kcal/mol. On the contrary, the prediction result of substrate 1A (Fig. 2A) was \(-\Delta \Delta {G}^{\ne }\) < 1.86 kcal/mol, implying the potentially low ee values of its product. To
validate the accuracy of these predictions, we synthesized and experimentally measured the ee values of their reaction products. Substrates 1A and 3 were readily prepared from simple
compounds according to the literature method (see SI). Wild-type amidase-containing _Escherichia coli_ whole cells were able to efficiently catalyze the kinetic resolution of the substrate
1A within 5 min under very mild conditions (neutral phosphate buffer, 30 °C). To facilitate the isolation and detection of the product, the carboxylic acid was alkylated with benzyl bromide
under base conditions and finally the recovered amide 1A and benzyl ester 2A were obtained with ee values of 61% and 52%, respectively, resulting in \(-\Delta \Delta {G}^{\ne }\) of only
1.05 kcal/mol (Fig. 2A). Following a similar approach, the desymmetrization of substrate 3 gave benzyl ester 4 with 97% ee value, indicating that the \(-\Delta \Delta {G}^{\ne }\) value of
this reaction is up to 3.39 kcal/mol (Fig. 2B). Both of the above experimental results were in agreement with ML predictions, which demonstrates the reliability of our constructed predictor.
It is able to significantly reduce the time for substrate synthesis and biotransformation experiments compared to the conventional research strategy. VIRTUAL SCREENING The core of
conventional protein engineering approaches to enhance the poor enantioselectivity biocatalysis toward 1A is protein engineering based on directed evolution and high-throughput screening.
Instead, our strategy in the present work focuses on the substrates at the beginning and consists of two steps. First, the ML predictor is used to predict the enantioselectivity toward a
series of substrates with a similar structure to 1A. The ensemble of 30 individual RF classifiers, each of which was rebuilt with a different random seed, was applied under each
classification criterion. The result was labeled as positive when more than half of the predictions were positive, otherwise it was labeled as negative. Based on the diverse results obtained
using ML, we would carefully examine the substituent effect on the substrates, expecting to reveal the key factors that influence the enantioselectivity. Second, the rules revealed for the
effect of substituents on enantioselectivity can be applied to assist in rational design of protein variants, which in traditional asymmetric synthesis methodologies usually require
extensive wet laboratory experimental studies to reveal38,40, thus reducing the need for mutation and screening efforts. Specifically, the aromatic group of substrate 1A is the key site for
chiral recognition with amidase. In order to comprehensively investigate the effect of the aromatic group on the catalytic performance of biocatalysis, we fine-tuned the structure of the
aromatic group on 1A to design its chemical analogs 1B-L (Fig. 3). The results in the first step are summaried in Fig. 3. When the phenyl group of 1A was replaced by benzyl (1B) or
phenylethyl group (1C), the ML-predicted values of enantioselectivity of both remain low (\(-\Delta \Delta {G}^{\ne }\) < 1.86 kcal/mol). To investigate the effect of substituents
attached on the phenyl ring, a series of substrates 1D-I containing an electron-donating methyl group or an electron-withdrawing bromine group in the _ortho_-, _meta_- or _para_-position
were virtually designed. According to ML predictions, the substrate 1 H with a methyl substituent in the _para_-position of the phenyl group exhibits a higher enantioselectivity (\(-\Delta
\Delta {G}^{\ne }\) > 1.86 kcal/mol) in comparison with the _ortho_- and _meta_-substituted analogs (1E and 1 F). Furthermore, the substrate 1I with a _para_-bromo substituent achieves
the highest predicted \(-\Delta \Delta {G}^{\ne }\) value, which is larger than 2.40 kcal/mol. The tendency suggests that substituents in the _para_-position may be relevant to high
enantioselectivity. Three additional substrates 1J-L with a _para_-substituent were further examined. The ML-predicted values of \(-\Delta \Delta {G}^{\ne }\) were both larger than 1.86
kcal/mol, leading to better enantioselectivity again when the _para_-position of the phenyl group of substrate 1 contains a substituent. MOLECULAR DOCKING AND RATIONAL DESIGN To bridge the
impact of _para_-substituents of substrates on stereoselectivity and the design of enzyme variants with higher enantioselectivity, we performed molecular docking of (_R_)- and
(_S_)-enantiomers of substrate 1A or 1I into the catalytic cavity of amidase. The computational details of molecular docking can be seen in Section 4, SI. As shown in Fig. 4A, B, substrates
(_R_)−1A and (_S_)−1A exhibits different binding modes, in which the _para_-position of (_R_)−1A and (_S_)−1A are close to residue I198 and W328 with a distance of 2.7 and 3.3 Å,
respectively. This steric blocking was unfavorable for substrate-enzyme recognition. Similar to the binding mode of (_S_)−1A, the steric phenyl group of (_S_)−1I also extends into the
substrate tunnel with a distance to W328 as 3.3 Å (see Fig. 4C). However, substrate (_R_)−1I failed to dock into the catalytic cavity, suggesting that its steric para-substituted phenyl
group may be too close to I198 to form the similar binding mode of (_R_)−1A. Molecular docking of the (_R_)- and (_S_)-enantiomers of other substrates 1B-L into the catalytic cavity of
amidase also demonstrated similar binding rule (Figure S8). It inspires us to mutate residue I198 and/or W328 of amidase to enhance its enantioselectivity toward substrate 1A, which is the
key point to the second step of our rational-design strategy. PROTEIN ENGINEERING Following the above biocatalysis rules, we implemented protein mutation to shrink its binding cavity of
(_R_)−1A (residue I198) or to broaden its binding cavity of (_S_)-1A (W328). Three variants encoding for the substitutions I198H, I198F, and W328F of wild-type amidase were first created.
The hydrolase of racemic 1A was then measured and shown in Fig. 5. All variants displayed higher enantioselectivity with increasing _E_-values (5.7 for wild-type, 12.8 for I198H, 16.6 for
I198F, 13.5 for W328F). Moreover, the double variant I198F/W328F performed the best enantioselectivity with a 53-fold higher _E_-value (i.e., 303.5) in comparison with the wild-type amidase.
With the help of ML prediction of the enantioselectivity of substrates and the deep analysis based on molecular docking, we finally designed new variants and effectively achieved a dramatic
increase in the enantioselectivity of amidase-catalysis. WET EXPERIMENTAL VALIDATION TOWARD NEW SUBSTRATES In the end, we experimentally measured the hydrolytic enantioselecivity toward
substrates 1B-K to confirm the accuracy of ML. As listed in Table 2, most of experimental results (9 out of 12) are consistent with ML predictions shown in Fig. 3. The enantioselectivity of
substrates 1E and 1 L was overestimated, whereas the enantioselectivity of 1 H was underestimated. The incorrect prediction regarding 1 H and 1 L may be related to the proximity of their
measured \(-\Delta \Delta {G}^{\ne }\) values (2.02 kcal/mol for 1 H and 1.83 kcal/mol for 1 L) to the classification threshold (1.86 kcal/mol) of ML predictor. The disagreement has no
influence on the structure-property relationship of substrates observed by ML. Regardless, the present ML model is able to capture biocatalysis rules such as the beneficial effect of the
_para_-substituents on enantioselectivity, which further assists us in rational protein engineering for highly enantioselective biocatalytic synthesis of chiral compounds. There is still
much room to improve this research in our future work. One is how to enhance and exploit the interpretability of features used in machine learning. In protein engineering and enzyme design,
the chemical composition and stereostructure of substrates typically have a critical impact on the reaction, making interpretable features essential26,57. Some other descriptors, which have
been encoded using deep neural networks58,59 or designed for organic catalytic reactions’ enantioselectivity60, can be employed as better candidates in our future works. In the present
research, however, more attention is paid to improve the traditional variant design strategy based on wet experiments and substrate engineering38,40, and rationally designing enzyme variants
through ML-assisted virtual screening of substrates, which requires a set of features with good and balanced performance. Therefore, we applied a specific combination of chemical
descriptors and 3D geometry descriptors. Another is how to collect and integrate data and features involving amidase variants into existing ML models, so as to build/upgrade them to ML
models describing the correlation between substrates structure, variants structure and catalytic stereoselectivity, and to explore their application in accelerating protein engineering
studies. In conclusion, based on the collection of experimental biocatalytic data and the well-adopted descriptors of substrates, we have developed machine-learning classification models to
predict the amidase-catalytic enantioselectivity toward new substrates. We further applied it to investigate the key structural factors of enantioselectivity and demonstrated the observed
structure-property rule in the guiding of reaction route design and protein variants design. We believe that this study will shed light on the ML-assisted substrate design and protein
engineering in biocatalysis. METHODS MATERIALS All the restriction enzymes were purchased from Thermo Fisher Scientific. High fidelity PCR DNA-polymerase, and dNTPs were purchased from
Vazyme Biotech Co., Ltd. PCR primers were synthesized and DNA sequencing was conducted by TsingKe Biotech Co., Ltd. Other common biochemical and media components were obtained from standard
commercial sources and used directly. The plasmid pET22b for amidase expression is gifted from Yapeng Chao and Shijun Qian from Institute of Microbiology, Chinese Academy of Sciences. All
the biochemical and commercial chemicals were used without further purification. The protocol of the synthesis of substrates and characterization data of compounds are given in SI. DATASET
CONSTRUCTION The whole dataset was classified into “positive” (higher enantioselectivity) and “negative” (lower enantioselectivity) according to \(\Delta \Delta {G}^{\ne }\), which is the
difference of activation Gibbs free energies between two processes for the generation of _R_- and _S_-products. Three classification criteria, \(-\Delta \Delta {G}^{\ne }\) = 1.86, 2.40 or
3.00 kcal/mol (corresponding to ee = 80%, 90% or 95% at 303 K), were used in this work. The numbers of positive and negative samples were listed in Table S1. The dataset under each criterion
was respectively divided into training (80%) and test sets (20%) with stratified random sampling. In order to address the class imbalance problem, we performed a random oversampling method
to randomly duplicate samples in the minority class before ML training. DESCRIPTORS One type of descriptors was derived from the molecular structure of substrates. First, the whole structure
was represented by the SMILES string and decomposed into fragments, which was also called as “cliques”. Second, a vocabulary of molecular cliques can be created. As shown in Figure S1,
there are 32 cliques extracted from this dataset and indexed as the _i_-th clique (_i_ = 1, 2, …, 32). Finally, a 32-dimensional one-hot vector was defined and converted into 32 descriptors.
For a given compound, the _i_-th component of the vector represents the number of the _i_-th clique that appears in this molecule. Another type of descriptors was obtained based on the
weighted atomic-centered symmetry functions (wACSFs). The radial and angular wACSFs centered at atom _i_ are defined as $${W}_{i}^{{rad}}={\sum}_{j\ne i}{Z}_{i}{e}^{-\eta
{\left({r}_{{ij}}-\mu \right)}^{2}}{f}_{{ij}}$$ (1) $${W}_{i}^{{ang}}={\sum}_{k\ne i,j}{\sum}_{j\ne i}{Z}_{j}{Z}_{k}(1+\lambda \cos {\theta }_{{jik}}){e}^{-\eta {\left({r}_{{ij}}-\mu
\right)}^{2}}{e}^{-\eta {\left({r}_{{ik}}-\mu \right)}^{2}}{e}^{-\eta {\left({r}_{{jk}}-\mu \right)}^{2}}{f}_{{ij}}\, {f}_{{ik}}\, {f}_{{jk}}$$ (2) where \({r}_{{ij}}\) is the distance
between atom _i_ and _j_, \({\theta }_{{jik}}\) is the angle that consists of atom _i_, _j_ and _k_, \({Z}_{i}\) denotes the atomic number of atom _i_, _η_, _μ_ and _λ_ are hyperparameters
of symmetry functions, and \({f}_{{ij}}\) is the cutoff function expressed as $${f}_{{ij}}=\left\{\begin{array}{c}\dfrac{1}{2}\left[\cos \left(\frac{\pi
{r}_{{ij}}}{{R}_{c}}\right)+1\right]{{\rm{if}}}\, {r}_{{ij}}\le {R}_{c}\\ 0,\hfill{{\rm{otherwise}}}\end{array}\right.$$ (3) Here _R__c_ is the pre-defined cutoff radius, which was 6.0 Å in
this work; the value of _λ_ was set as 1 or −1 for different angular symmetry functions; _η_ and _μ_ were determined as $$\eta=\frac{1}{2{\left(\Delta r\right)}^{2}}$$ (4) and
$$\mu=0.5\;{{{\text{\AA}} }}+n\Delta r,\, n=0,1,2\ldots,N-1$$ (5) where $$\Delta r=\frac{{R}_{c}-1.5\;{{{\text{\AA}} }}}{N-1}$$ (6) and _N_ is the number of wACSFs centered at atom _i_. Note
that different values of _N_ can be applied to radial and angular symmetry functions, denoted as \({N}_{{rad}}\) and \({N}_{{ang}}\), respectively. Since different substrates in the dataset
usually have different numbers of atoms or elements, a histogram scheme is used to regularize symmetry functions, leading to the histogram-wACSFs as “geometry” descriptors. The number of
bins to build the histogram is another hyperparameter (denoted as \({N}_{{bin}}\)). In brief, the molecular clique descriptors reflect the “chemistry” of substrates, while the
histogram-wACSF descriptors capture the three-dimensional information about substrates. Three hyperparameters in histogram-wACSFs, that is, \({N}_{{rad}}\), \({N}_{{ang}}\) and
\({N}_{{bin}}\), should be tuned. The geometry of substrate was optimized in vacuum using the B3LYP density functional61,62,63 and 6-31 + + G(d,p) basis set. Note that “descriptor” was also
called as “feature” in this paper. FEATURE SELECTION Feature selection in prior to ML training was designed as follows. First, the features with a variance lower than a given threshold
(e.g., 0.025) after normalization were removed. Second, the Pearson correlation map between the remaining features was calculated. If the coefficient of a feature pair is larger than a given
threshold (e.g., 0.98), one of the features is removed. Third, recursive feature elimination64 (RFE) was performed to filter the remaining features. After several attempts, we employed
support vector machine as the estimator of RFE according to the final performance of ML classification model with the selected features. This procedure was implemented under the
classification criteria of 1.86 and 2.40. However, under the criterion of 3.00, the third step was omitted. Instead, after the second step, the correlation coefficients between the remaining
features and the training labels were examined, removing the features with a coefficient lower than a given threshold (e.g., 0.15). HYPERPARAMETERS The dataset under the classification
criterion of 2.40 was used to search the best hyperparameters (Table S2). First, the hyperparameters of four classifiers, that is, random forest (RF), support vector machine (SVM), logistics
regression (LR), and gradient boosted decision tree (GBDT), were tuned with a five-fold cross-validated grid-search on the training set. Second, these classifiers were retrained on the
training set with the above optimized hyperparameters and evaluated on the test set. The RF model was selected as the best classifier. Finally, the RF model was rebuilt under two other
classification criteria (1.86 and 3.00) with the same procedure, except for the fixed hyperparameters in histogram-wACSFs (\({N}_{{rad}}\), \({N}_{{ang}}\) and \({N}_{{bin}}\)). EVALUATION
ON PERFORMANCE The quality of ML classifiers is always evaluated using the accuracy, precision, recall, _F_-score and the area under receiver operating characteristic curve (AUC) (Table S3).
They are defined as $${{\rm{accuracy}}}=\frac{{{\rm{TP}}}+{{\rm{TN}}}}{{{\rm{TP}}}+{{\rm{FP}}}+{{\rm{TN}}}+{{\rm{FN}}}}$$ (7)
$${{\rm{precision}}}=\frac{{{\rm{TP}}}}{{{\rm{TP}}}+{{\rm{FP}}}}$$ (8) $${{\rm{recall}}}=\frac{{{\rm{TP}}}}{{{\rm{TP}}}+{{\rm{FN}}}}$$ (9) and $${F}_{\beta }=\frac{(1+{\beta }^{2})\times
{{\rm{precision}}}\times {{\rm{recall}}}}{{\beta }^{2}\times {{\rm{precision}}}+{{\rm{recall}}}}$$ (9) Here TP, FP, FN, and TN represent the number of positive samples correctly classified,
the number of negative samples that are misclassified as positive, the number of positive samples that are misclassified as negative, and the number of negative samples correctly classified,
respectively. The value of _β_ in _F_-score determines the relative importance of precision and recall on the evaluation. In this work, it was set to be 1 as usual. A receiver operating
characteristic curve is a plot of \(\frac{{{\rm{TP}}}}{{{\rm{TP}}}+{{\rm{FN}}}}\) in function of \(\frac{{{\rm{FP}}}}{{{\rm{TN}}}+{{\rm{FP}}}}\). A larger area under this curve (AUC)
indicates better classification performance. PROTEIN ENGINEERING AND EXPRESSION The PCR mixture (50 μL) contained 25 μL 2 × Phanta Max Master Mix, 17 μL H2O, 2 μL DMSO, 2 μL (about 50 ng)
template DNA and 2 μL (about 10 μM) each primer mix. The PCR was performed as follows: (i) 98 °C, 30 s; (ii) 30 cycles: 98 °C, 10 s; 50-72 °C, 30 s; 72 °C, 0.5 min/kbp; (iii) 72 °C, 2 min.
The resulting PCR product was directly treated with the kinase, ligase & _Dpn_I (KLD enzyme mix) (100 μL mL−1; NEB) at room temperature for 30 minutes and then used for the
transformation of chemically competent _E. coli_ TOP10 cells. After confirming the introduced mutation(s) by single colonies sequence detection, the plasmids were used for the transformation
into chemical competent _E. coli_ BL21(DE3) cells by the heat shock method. Primers used in this work include I198H Fw (AAGGCGGATCGATCCGGCACCCGGCGGCAT), I198H Rv
(CCGCAGAATGCCGCCGGGTGCCGGATCGAT), I198F Fw (AAGGCGGATCGATCCGGTTCCCGGCGGCAT), I198F Rv (CCGCAGAATGCCGCCGGGAACCGGATCGAT), W328F Fw (ATCTGCATGCTTTCCACATCTTTAACGTGATCGCC) and W328 Rv
(CCGTCCGTGGCGATCACGTTAAAGATGTGGAAAG). They are also listed in Table S6. The pre-cultures were prepared by inoculating 5 mL of Luria-Bertani (LB) broth (composed of 1% Tryptone, 1% NaCl, and
0.5% yeast extract) containing 100 μg/mL ampicillin with a single colony of _E. coli_ BL21 (DE3)65. Following overnight incubation at 37 °C with shaking at 220 rpm, the pre-cultures were
diluted 1:100 into 300 mL of LB medium supplemented with ampicillin and cultured until the optical density at 600 nm reached approximately 0.6–0.8. After cooling at 4 °C for 30 minutes,
protein expression was induced by the addition of 300 μM isopropyl-β-D-thiogalactopyranoside (IPTG), followed by further incubation for 6 hours at 25 °C. The cells were collected by
centrifugation at 7100 g for 5 minutes at 4 °C, and the supernatant was discarded. The cell pellets were re-suspended in phosphate buffer (0.1 M, pH 7.0) and stored at −20 °C. All resulting
variant sequences were verified through DNA sequencing. GENERAL PROCEDURE FOR THE BIOTRANSFORMATIONS OF SUBSTRATES 1 AND 3 CATALYZED BY AMIDASE-CONTAINING OR VARIANT-CONTAINING _E. COLI_ In
an Erlenmeyer flask (150 mL) with a screw cap a suspension of _E. coli_ cells (0.05−0.5 g wet weight) in aqueous phosphate buffer (pH 7.0, 0.1 M, 25 mL) was activated at 37 °C for 0.5 h.
Substrates 1A-L or 3 (0.5 mmol) was dissolved in aqueous phosphate buffer (pH 7.0, 0.1 M, 25 mL) and added in one portion, and the resulting mixture was incubated at 37 °C with orbital
shaking (220 rpm). The reaction process was monitored using TLC method. After a period of time, the reaction was quenched by removing microbial cells through a celite pad filtration. The
filtration cake was washed consecutively with water (3 × 15 mL) and ethyl acetate (3 × 30 mL). The organic phase of filtrate was separated and dried with anhydrous Na2SO4, and then was
removed under vacuum. The residue was chromatographed on a silica gel column with ethyl acetate as the mobile phase to give amide (_R_)−1A-L or 3. The aqueous phase was evaporated under
vacuum, giving a waxy solid which is a mixture of acid product and salt. The residue was dissolved in DMF (5 mL) followed by the addition of K2CO3 (0.25 mmol, 1 equiv.) and benzyl bromide
(0.5 mmol, 2 equiv.). The mixture was stirred at room temperature overnight, and the reaction was then quenched by adding water (20 mL). Extraction with ethyl acetate (3 × 15 mL) and dried
over anhydrous NaSO4. After removing the solvent under vacuum, the crude mixture was purified by flash column chromatography using a mixture of petroleum ether and ethyl acetate (10:1 v/v)
as the mobile phase to give benzyl esters (_S_)−2A-L or 4. Enantiomeric excess values were obtained from HPLC analysis using columns coated with chiral stationary phases. REPORTING SUMMARY
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY All the source data generated in this study have been
deposited in Supplementary Information files. The X-ray crystallographic coordinate for structure of (_R_)-1H reported in this study has been deposited at the Cambridge Crystallographic Data
Center (CCDC), under deposition number 2224210. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. The
supplementary methods for synthesis and characterization, crystallography, NMR, HPLC studies and additional data supporting the findings of this study are available in Supplementary
Information files. The training data used in this study are provided in the Source Data File. All data are available from the corresponding author upon request. Source data are provided with
this paper. CODE AVAILABILITY The source code employed for generating descriptors and training ML models in this research are available at
https://github.com/ZYChen33/ML-assisted-amidase-catalytic-enantioselectivity-prediction-and-rational-design and https://doi.org/10.5281/zenodo.1375970066. REFERENCES * Faber, K. et al.
_Biotransformations in Organic Chemistry: A Textbook, 7th,_ pp 442 (Springer, Berlin, 2018). * Hanefeld, U., Hollmann, F. & Paul, C. E. Biocatalysis making waves in organic chemistry.
_Chem. Soc. Rev._ 51, 594–627 (2022). Article CAS PubMed Google Scholar * Wu, S. et al. Biocatalysis: enzymatic synthesis for industrial applications. _Angew. Chem. Int. Ed._ 60, 88–119
(2021). Article ADS CAS Google Scholar * Yi, D. et al. Recent trends in biocatalysis. _Chem. Soc. Rev._ 50, 8003–8049 (2021). Article CAS PubMed PubMed Central Google Scholar *
Winkler, C. K., Schrittwieser, J. H. & Kroutil, W. Power of biocatalysis for organic synthesis. _ACS Cent. Sci._ 7, 55–71 (2021). Article CAS PubMed PubMed Central Google Scholar *
Devine, P. N. et al. Extending the application of biocatalysis to meet the challenges of drug development. _Nat. Rev. Chem._ 2, 409–421 (2018). Article Google Scholar * Buller, R. et al.
From nature to industry: harnessing enzymes for biocatalysis. _Science_ 382, eadh8615 (2023). Article CAS PubMed Google Scholar * Hossack, E. J., Hardy, F. J. & Green, A. P. Building
enzymes through design and evolution. _ACS Catal._ 13, 12436–12444 (2023). Article CAS Google Scholar * Miller, D. C., Athavale, S. V. & Arnold, F. H. Combining chemistry and protein
engineering for new-to-nature biocatalysis. _Nat. Synth._ 1, 18–23 (2022). Article ADS PubMed PubMed Central Google Scholar * Qu, G. et al. The crucial role of methodology development
in directed evolution of selective enzymes. _Angew. Chem. Int. Ed._ 59, 13204–13231 (2020). Article CAS Google Scholar * Adams, J. P. et al. Biocatalysis: a pharma perspective. _Adv.
Synth. Catal._ 361, 2421–2432 (2019). Article CAS Google Scholar * Quesne, M. G. et al. Advances in sustainable catalysis: a computational perspective. _Front. Chem._ 7, 182 (2019).
Article ADS CAS PubMed PubMed Central Google Scholar * Klinman, J. P., Offenbacher, A. R. & Hu, S. Origins of enzyme catalysis: experimental findings for C-H activation, new
models, and their relevance to prevailing theoretical constructs. _J. Am. Chem. Soc._ 139, 18409–18427 (2017). Article CAS PubMed PubMed Central Google Scholar * Lonsdale, R., Harvey,
J. N. & Mulholland, A. J. A practical guide to modelling enzyme-catalysed reactions. _Chem. Soc. Rev._ 41, 3025–3038 (2012). Article CAS PubMed PubMed Central Google Scholar * Yang,
J., Li, F.-Z. & Arnold, F. H. Opportunities and challenges for machine learning-assisted enzyme engineering. _ACS Cent. Sci._ 10, 226–241 (2024). Article CAS PubMed PubMed Central
Google Scholar * Ao, Y.-F. et al. Data-driven protein engineering for improving catalytic activity and selectivity. _ChemBioChem_ 25, e202300754 (2024). Article CAS PubMed Google Scholar
* Markus, B. et al. Accelerating biocatalysis discovery with machine learning: a paradigm shift in enzyme engineering, discovery, and design. _ACS Catal._ 13, 14454–14469 (2023). Article
CAS PubMed PubMed Central Google Scholar * Kouba, P. et al. Machine learning-guided protein engineering. _ACS Catal._ 13, 13863–13895 (2023). Article CAS PubMed PubMed Central Google
Scholar * Dou, B. et al. Machine learning methods for small data challenges in molecular science. _Chem. Rev._ 123, 8736–8780 (2023). Article CAS PubMed PubMed Central Google Scholar
* Wittmund, M., Cadet, F. & Davari, M. D. Learning epistasis and residue coevolution patterns: current trends and future perspectives for advancing enzyme engineering. _ACS Catal._ 12,
14243–14263 (2022). Article CAS Google Scholar * Jiang, Y., Ran, X. & Yang, Z. J. Data-driven enzyme engineering to identify function-enhancing enzymes. _Protein Eng. Des. Sel._ 36,
gzac009 (2023). Article PubMed Google Scholar * Sapoval, N. et al. Current progress and open challenges for applying deep learning across the biosciences. _Nat. Commun._ 13, 1728 (2022).
Article ADS CAS PubMed PubMed Central Google Scholar * Hie, B. L. & Yang, K. K. Adaptive machine learning for protein engineering. _Curr. Opin. Struct. Biol._ 72, 145–152 (2022).
Article CAS PubMed Google Scholar * Lovelock, S. L. et al. The road to fully programmable protein catalysis. _Nature_ 606, 49–58 (2022). Article ADS CAS PubMed Google Scholar * Cui,
Y., Sun, J. & Wu, B. Computational enzyme redesign: large jumps in function. _Trends Chem._ 4, 409–419 (2022). Article CAS Google Scholar * Xu, Y. et al. Deep dive into machine
learning models for protein engineering. _J. Chem. Inf. Model._ 60, 2773–2790 (2020). Article CAS PubMed Google Scholar * Volk, M. J. et al. Biosystems design by machine learning. _ACS
Synth. Biol._ 9, 1514–1533 (2020). Article CAS PubMed Google Scholar * Mazurenko, S., Prokop, Z. & Damborsky, J. Machine learning in enzyme engineering. _ACS Catal._ 10, 1210–1223
(2020). Article CAS Google Scholar * Berman, H. M. et al. The protein data bank. _Nucleic Acids Res._ 28, 235–242 (2000). Article ADS CAS PubMed PubMed Central Google Scholar *
UniProt Consortium, The UniProt: the universal protein knowledgebase in 2023. _Nucleic Acids Res._ 51, D523–D531 (2023). Article Google Scholar * Chang, A. et al. BRENDA, the ELIXIR core
data resource in 2021: new developments and updates. _Nucleic Acids Res._ 49, D498–D508 (2021). Article CAS PubMed Google Scholar * Cadet, F. et al. A machine learning approach for
reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes. _Sci. Rep._ 8, 16757 (2018). Article ADS PubMed PubMed Central
Google Scholar * Wu, Z. et al. Machine learning-assisted directed protein evolution with combinatorial libraries. _Proc. Natl Acad. Sci. USA._ 116, 8852–8858 (2019). Article ADS CAS
PubMed PubMed Central Google Scholar * Ma, E. J. et al. Machine-directed evolution of an imine reductase for activity and stereoselectivity. _ACS Catal._ 11, 12433–12445 (2021). Article
CAS Google Scholar * Ao, Y.-F. et al. Structure- and data-driven protein engineering of transaminases for improving activity and stereoselectivity. _Angew. Chem. Int. Ed._ 62, e202301660
(2023). Article CAS Google Scholar * Clements, H. D. et al. Using data science for mechanistic insights and selectivity predictions in a non-natural biocatalytic reaction. _J. Am. Chem.
Soc._ 145, 17656–17664 (2023). Article CAS PubMed PubMed Central Google Scholar * Wu, Z. et al. Amidase as a versatile tool in amide-bond cleavage: from molecular features to
biotechnological applications. _Biotechnol. Adv._ 43, 107574 (2020). Article CAS PubMed Google Scholar * Ao, Y.-F. et al. Reversal and amplification of the enantioselectivity of
biocatalytic desymmetrization toward meso heterocyclic dicarboxamides enabled by rational engineering of amidase. _ACS Catal._ 11, 6900–6907 (2021). Article CAS Google Scholar * Hu, H.-J.
et al. Modification of the enantioselectivity of biocatalytic _meso_-desymmetrization for synthesis of both enantiomers of _cis_−1,2-disubstituted cyclohexane by amidase engineering. _Adv.
Synth. Catal._ 363, 4538–4543 (2021). Article CAS Google Scholar * Hu, H.-J. et al. Enantioselective biocatalytic desymmetrization for synthesis of enantiopure _cis_−3,4-disubstituted
pyrrolidines. _Green. Synth. Catal._ 2, 324–327 (2021). Article Google Scholar * Hu, H.-J. et al. Highly efficient biocatalytic desymmetrization of _meso_ carbocyclic 1,3-dicarboxamides: a
versatile route for enantiopure 1,3-disubstituted cyclohexanes and cyclopentanes. _Org. Chem. Front._ 6, 808–812 (2019). Article CAS Google Scholar * Ao, Y.-F. et al. Biocatalytic
desymmetrization of prochiral 3-aryl and 3-arylmethyl glutaramides: different remote substituent effect on catalytic efficiency and enantioselectivity. _Adv. Synth. Catal._ 360, 4594–4603
(2018). Article CAS Google Scholar * Wang, M.-X. Enantioselective biotransformations of nitriles in organic synthesis. _Acc. Chem. Res._ 48, 602–611 (2015). Article CAS PubMed Google
Scholar * Janes, L. E., Kazlauskas, R. J. & Quick, E. a fast spectrophotometric method to measure the enantioselectivity of hydrolases. _J. Org. Chem._ 62, 4560–4561 (1997). Article
CAS Google Scholar * Barnard, T. et al. Less may be more: an informed reflection on molecular descriptors for drug design and discovery. _Mol. Syst. Des. Eng._ 5, 317–329 (2020). Article
CAS Google Scholar * Jin, W., Barzilay, R. & Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. _Proceedings of the 35th International Conference on
Machine Learning,_ PMLR 80, 2323–2332 (2018). * Gastegger, M. et al. wACSF—Weighted atom-centered symmetry functions as descriptors in machine learning potentials. _J. Chem. Phys._ 148,
241709 (2018). Article ADS CAS PubMed Google Scholar * Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. _Phys.
Rev. Lett._ 98, 146401 (2007). Article ADS PubMed Google Scholar * Pedregosa, F. et al. Scikit-learn: machine learning in python. _J. Mach. Learn. Res._ 12, 2825–2830 (2011). MathSciNet
Google Scholar * Gaussian 09, Revision D.01, Frisch, M. J. et al. Gaussian, Inc., Wallingford CT, (2013). * Breiman, L. Random forests. _Mach. Learn._ 45, 5–32 (2001). Article Google
Scholar * Ouyang, R. et al. SISSO: a compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates. _Phys. Rev. Mater._ 2, 083802
(2018). Article CAS Google Scholar * Case, D. A. et al. AMBER18, University of California, San Francisco, (2018). * Wang, J. et al. Development and testing of a general amber force field.
_J. Comput. Chem._ 25, 1157–1174 (2004). Article CAS PubMed Google Scholar * Singh, R. V. & Sambyal, K. Green synthesis aspects of (_R_)-(-)-mandelic acid; a potent pharmaceutically
active agent and its future prospects. _Crit. Rev. Biotechnol._ 43, 1226–1235 (2023). Article CAS PubMed Google Scholar * Borzilleri, R. M., Weinreb, S. M. & Parvez, M. Total
synthesis of the unusual marine alkaloid (-)-Papuamine utilizing a novel imino ene reaction. _J. Am. Chem. Soc._ 117, 10905–10913 (1995). Article CAS Google Scholar * Tahil, G. et al.
Stereoisomers are not machine learning’s best friends. _J. Chem. Inf. Model._ 64, 5451–5469 (2024). Article PubMed Google Scholar * Walters, W. P. & Barzilay, R. Applications of deep
learning in molecule generation and molecular property prediction. _Acc. Chem. Res._ 54, 263–270 (2021). Article CAS PubMed Google Scholar * Schütt, K. T. et al. SchNetPack 2.0: a neural
network toolbox for atomistic machine learning. _J. Chem. Phys._ 158, 144801 (2023). Article ADS PubMed Google Scholar * Reid, J. P. & Sigman, M. S. Holistic prediction of
enantioselectivity in asymmetric catalysis. _Nature_ 571, 343–348 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Becke, A. D. Density-functional exchange-energy
approximation with correct asymptotic behavior. _Phys. Rev. A: ., Mol., Opt. Phys._ 38, 3098–3100 (1988). Article ADS CAS Google Scholar * Lee, C., Yang, W. & Parr, R. G. Development
of the Colle-Salvetti correlation-energy formula into a functional of the electron density. _Phys. Rev. B: Condens. Matter Mater. Phys._ 37, 785–789 (1988). Article ADS CAS Google
Scholar * Becke, A. D. Density-functional thermochemistry. III. the role of exact exchange. _J. Chem. Phys._ 98, 5648–5652 (1993). Article ADS CAS Google Scholar * Guyon, I. et al. Gene
selection for cancer classification using support vector machines. _Mach. Learn._ 46, 389–422 (2002). Article Google Scholar * Xue, Z. et al. Overexpression of a recombinant amidase in a
complex auto-inducing culture: purification, biochemical characterization, and regio- and stereoselectivity. _J. Ind. Microbiol. Biotechnol._ 38, 1931–1938 (2011). Article CAS PubMed
Google Scholar * Li, Z.-L. et al. ML-assisted-amidase-catalytic-enantioselectivity-prediction-and-rational-design. https://doi.org/10.5281/zenodo.13759700 (2024). * The PyMOL molecular
graphics system, version 2.3.0. Schrödinger, LLC. New York, (2019). Download references ACKNOWLEDGEMENTS Financial supports from the National Key Research and Development Program of China
(2019YFA0709400 to LS), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0960302 to YFA), the National Natural Science Foundation of China (22193041 to LS,
21977098 to YFA, 22120102005 to XC) and the Fundamental Research Funds for the Central Universities to LS are gratefully acknowledged. We are grateful to Prof. Mei-Xiang Wang for providing
the training data. AUTHOR INFORMATION Author notes * These authors contributed equally: Zi-Lin Li, Shuxin Pei. AUTHORS AND AFFILIATIONS * Beijing National Laboratory for Molecular Sciences,
CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China Zi-Lin Li, Teng-Yu Huang, Xu-Dong Wang, Qi-Qiang Wang, De-Xian
Wang & Yu-Fei Ao * University of Chinese Academy of Sciences, Beijing, China Zi-Lin Li, Teng-Yu Huang, Qi-Qiang Wang, De-Xian Wang & Yu-Fei Ao * Key Laboratory of Theoretical and
Computational Photochemistry of Ministry of Education, College of Chemistry, Beijing Normal University, Beijing, China Shuxin Pei, Ziying Chen, Lin Shen & Xuebo Chen * Yantai-Jingshi
Institute of Material Genome Engineering, Yantai, China Lin Shen & Xuebo Chen * Shandong Laboratory of Yantai Advanced Materials and Green Manufacturing, Yantai, China Xuebo Chen Authors
* Zi-Lin Li View author publications You can also search for this author inPubMed Google Scholar * Shuxin Pei View author publications You can also search for this author inPubMed Google
Scholar * Ziying Chen View author publications You can also search for this author inPubMed Google Scholar * Teng-Yu Huang View author publications You can also search for this author
inPubMed Google Scholar * Xu-Dong Wang View author publications You can also search for this author inPubMed Google Scholar * Lin Shen View author publications You can also search for this
author inPubMed Google Scholar * Xuebo Chen View author publications You can also search for this author inPubMed Google Scholar * Qi-Qiang Wang View author publications You can also search
for this author inPubMed Google Scholar * De-Xian Wang View author publications You can also search for this author inPubMed Google Scholar * Yu-Fei Ao View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS Y.F.A. and L.S. conceived the project and supervised the work with Q.Q.W., D.X.W., and X.C. Data collection and dataset building
was performed by Y.F.A., S.P., T.Y.H., and X.D.W. The ML model was designed and built by S.P., Z.C., L.S., and Y.F.A. Biocatalytic experiments were performed by Z.L.L. Protein engineering
was designed and performed by Y.F.A. and Z.L.L., Y.F.A., L.S., and S.P. wrote the manuscript, which was edited and approved by all authors. CORRESPONDING AUTHORS Correspondence to Lin Shen,
Xuebo Chen or Yu-Fei Ao. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Arkadij
Kummer, Eric Ma and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE
REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International
License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived
from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line
to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS
ARTICLE CITE THIS ARTICLE Li, ZL., Pei, S., Chen, Z. _et al._ Machine learning-assisted amidase-catalytic enantioselectivity prediction and rational design of variants for improving
enantioselectivity. _Nat Commun_ 15, 8778 (2024). https://doi.org/10.1038/s41467-024-53048-0 Download citation * Received: 26 February 2024 * Accepted: 30 September 2024 * Published: 10
October 2024 * DOI: https://doi.org/10.1038/s41467-024-53048-0 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
:max_bytes(150000):strip_icc():focal(734x329:736x331)/jack-dennis-quaid-984c538a1a4044299f63ead008f18283.jpg)







