- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Many disease-causing proteins have multiple pathogenic mechanisms, and conventional inhibitors struggle to reliably disrupt more than one. Targeted protein degradation (TPD) can
eliminate the protein, and thus all its functions, by directing a cell’s protein turnover machinery towards it. Two established strategies either engage catalytic E3 ligases or drive uptake
towards the endolysosomal pathway. Here we describe CYpHER (CatalYtic PH-dependent Endolysosomal delivery with Recycling) technology with potency and durability from a catalytic mechanism
that shares the specificity and straightforward modular design of endolysosomal uptake. By bestowing pH-dependent release on the target engager and using the rapid-cycling transferrin
receptor as the uptake receptor, CYpHER induces endolysosomal delivery of surface and extracellular targets while re-using drug, potentially yielding increased potency and reduced off-target
tissue exposure risks. The TfR-based approach allows targeting to tumors that overexpress this receptor and offers the potential for transport to the CNS. CYpHER function was demonstrated
in vitro with EGFR and PD-L1, and in vivo with EGFR in a model of EGFR-driven non-small cell lung cancer. SIMILAR CONTENT BEING VIEWED BY OTHERS EXTRACELLULAR TARGETED PROTEIN DEGRADATION:
AN EMERGING MODALITY FOR DRUG DISCOVERY Article 07 December 2023 LIPID-MEDIATED INTRACELLULAR DELIVERY OF RECOMBINANT BIOPROTACS FOR THE RAPID DEGRADATION OF UNDRUGGABLE PROTEINS Article
Open access 10 July 2024 LYSOSOME-TARGETING CHIMAERAS FOR DEGRADATION OF EXTRACELLULAR PROTEINS Article 29 July 2020 INTRODUCTION Contemporary targeted therapeutics aim to modulate the
activity of a particular target, usually a protein, that has a defined role in disease pathology. This modulation is often the disruption of protein function, most commonly seen by enzyme
inhibition (e.g., kinase inhibitors) or steric blocking (e.g., antibodies). These conventional inhibitors and blockers can disrupt a defined function, and often add beneficial pleiotropic
effects, such as protein homeostasis disruption1 or altered target trafficking2,3. Nevertheless, these drugs are often insufficient to meaningfully and durably alter disease pathology. For
one, many targets exhibit multiple functions, and inhibiting one function can leave the others available for potentiating pathologic signaling. Second, the nature of many of these inhibitors
leaves them particularly vulnerable to cellular adaptation and mutational resistance that diminishes drug durability. An exemplary group of protein targets is receptor tyrosine kinases
(RTKs) in cancer, as they can potentiate growth, differentiation, and survival signaling4. Many involve a mechanism for activation that involves multimerization and cross-phosphorylation at
the cell surface. As such, they not only function as kinases, but as kinase substrates mediating signal transduction. Tyrosine kinase inhibitors (TKIs) and antibodies are typical
RTK-targeted therapeutics. TKIs can disrupt kinase activity of RTKs5, but they do not block the RTKs from being substrates for other kinases. As many RTKs function through both homo- and
heterodimerization with other RTKs, this leads to resistance via upregulation of other partners6. As an additional liability, point mutations can often directly or indirectly disrupt TKI
binding7. Conversely, antibodies can alter the multimerization tendencies of RTKs, but with kinase function retained, target or heterodimer partner upregulation (or gain-of-function
mutation) becomes a common resistance mechanism8, with increased total membrane kinase activity compensating for disrupted multimerization. Altogether, these inhibitors can be effective for
a period of time, but the myriad mechanisms for functional bypass typically render their efficacy transient. In order to simultaneously disrupt all of a target’s disease-associated
functions, targeted protein degradation (TPD) can be used. Expertly summarized in a recent review9, TPD leverages cells’ mechanisms for target turnover, altering the kinetics of this process
for specific targets. This is mainly through formation of ternary complexes between target protein and degradation effector. For intracellular (e.g., molecular glue and PROTAC)10 and some
extracellular (e.g., AbTAC and REULR)11,12 molecules, this is through E3 ligase recruitment, inducing ubiquitin-mediated target degradation by the proteasome or lysosome. This benefits from
a catalytic mechanism of action (the drug is not expended in the process) but can be complicated by finding an E3 ligase expressed in the target tissue that can be induced to interact in an
effective orientation with the target for functional ubiquitination. Meanwhile, small molecules (e.g., molecular glue and PROTAC) also have many of the same point mutational resistance
liabilities of TKIs. Other approaches used for surface and extracellular soluble targets (e.g., LYTAC, ATAC, KineTAC)13,14,15 engage surface protein trafficking systems. Often targeting
membrane sugar receptors (cation-independent mannose-6-phosphate receptor [CI-M6PR] or asialoglycoprotein receptor [ASGPR]) but also cytokine receptors (e.g., CXCR7), these are designed to
“hitch a ride” with the uptake receptor into the cell before target release in the endolysosomal system. These tend to be biologics, benefiting from modular design and greater target
specificity, but the drug follows the target through its trafficking, limited to stoichiometric (as opposed to catalytic) activity. Transferrin receptor (TfR) is another such uptake receptor
with potential in extracellular TPD (eTPD) approaches. Its normal role is facilitating uptake of iron-loaded transferrin (holoTF)16. Upon uptake and endosomal maturation, which involves
acidification to roughly pH 5.5, transferrin releases its iron but remains bound to TfR, returning to the surface with its receptor. The process takes ~10–20 min16, repeating dozens to
hundreds of times over the protein’s lifetime17. Our work concerns the development of eTPD molecules that mimic the behavior of transferrin, as they are engineered for reduced binding
affinity at endosomal pH to permit target release and subsequent trafficking through the endolysosomal system. They also contain a TfR-binding end with no such pH sensitivity, which permits
the molecules to return to the surface to take in additional targets. This catalytic activity increases potency (multiple target molecules eliminated by a single drug), permits retained
activity after drug is cleared from extracellular space, and should reduce the disruptive effects of shed, soluble variants of a membrane target (that can otherwise act as a decoy for
conventional antibodies)18, since the soluble form would simply represent one round of uptake and endosomal release. Using TfR as the uptake receptor presents numerous advantages,
particularly for CNS disease and oncology. TfR is commonly upregulated on a wide variety of solid tumors19,20, presumably to accommodate the increased iron demands in rapidly-dividing cells,
and its overexpression often correlates with disease severity20,21. This overexpression compared to healthy tissue should concentrate the drug in the tumor tissue, improving the therapeutic
window. Also, cancer cells are highly dependent on TfR for growth22, so a potential resistance mechanism of receptor downregulation is avoided by using TfR. In addition, TfR is well known
as a mediator of blood-brain barrier transcytosis23, having been used to deliver biologic molecules to the brain parenchyma24,25. A drug whose mechanism of action includes TfR engagement has
the potential to enable specific depletion of targets in the CNS, an area of high unmet medical need. We note that, during the preparation of this work, another group also highlighted the
advantages of TfR-based TPD for oncology (TransTACs)26, but designed the molecules to be degraded in the endosome to avoid (rather than harness) TfR-based recycling for catalytic activity.
Here, we present CYpHER (CatalYtic PH-dependent Endolysosomal delivery with Recycling), an eTPD technology that combines the specificity and modularity of an endolysosomal trafficking
approach with a catalytic mechanism. We demonstrate the development of CYpHER molecules, catalytic target uptake and elimination, suppressing signaling and growth of cancer cells, and in
vivo pharmacokinetics and activity. We discuss the steps for engineering molecules with these characteristics, assays for demonstrating surface target elimination and uptake of multiple
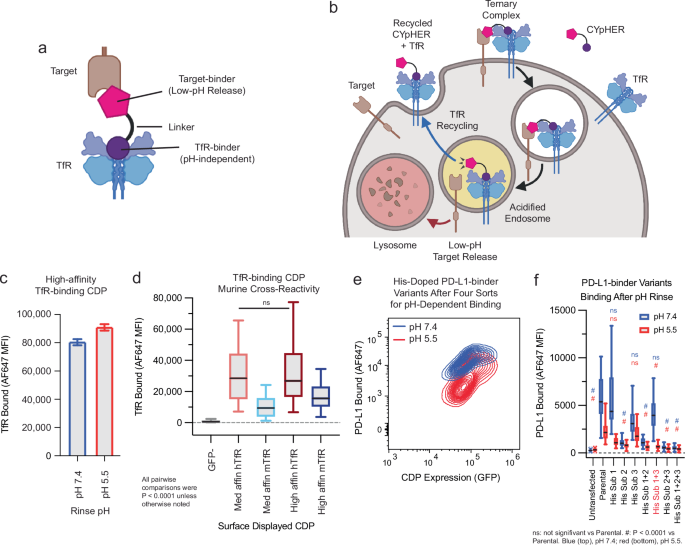
targets per drug molecule, and then discuss potential advantages and utilities of this approach. RESULTS THE CYPHER CONCEPT The core CYpHER concept is illustrated in Fig. 1a, b. A simplistic
diagram of a CYpHER molecule (Fig. 1a) includes a pH-dependent target-binder linked to a recycling uptake receptor-binder (here, a TfR binder). After CYpHER has initiated ternary complex
formation on the cell surface (Fig. 1b), the target and CYpHER molecule are brought into the cell in TfR-containing clathrin-coated vesicles. Following the trafficking of TfR, the vesicle
then joins endosomes that begin acidifying16. The target-binders are engineered to release the target at low pH, permitting the TfR:CYpHER complex to be trafficked back to the surface where
the target-binding end of CYpHER is free to bind and traffic additional target molecules. Meanwhile, the released target undergoes intracellular trafficking which includes lysosomal delivery
and subsequent degradation. To construct CYpHER molecules, we began with two cystine-dense peptide (CDP) miniproteins that we’ve recently characterized separately for other in vivo
functionalities25,27. CDPs are of interest for incorporating into multifunctional biologics due to their small size, potential for high affinity, and high inherent protease-resistance28. We
made use of a TfR-binding CDP allelic series25 that maintains binding at reduced pH (Fig. 1c), has murine cross-reactivity (Fig. 1d), and has demonstrated access to the CNS25. Then we
further engineered a PD-L1-binding CDP27 for enhanced pH-dependent release by generating a library of variants that each contained up to two histidine substitutions, and surface-display
screened to enrich for variants that conferred strong PD-L1 binding at pH 7.4 but reduced PD-L1 binding after a low-pH rinse (Fig. 1e). Three singleton histidine substitutions were
significantly enriched, and when testing them as singletons and combinations, one combination (His Sub 1 + 3) was found to confer PD-L1-binding almost as well as the parental variant at pH
7.4 but conferred substantially less PD-L1 binding after a low-pH rinse (Fig. 1f). A pilot molecule comprised of the pH-dependent PD-L1-binding CDP fused via flexible Gly-Ser linker to a
high affinity TfR-binding CDP (Supplementary Fig. 1A) was used to stain 293T cells transfected with PD-L1-GFP and TfR-RFP (or with TfR-RFP alone). Subsequent cell staining for a 6xHis tag on
the pilot molecule and gating for double negative, GFP+/RFP−, GFP−/RFP+, or GFP+/RFP+ cells indicated increased cell staining when one or the other surface binding partner is overexpressed
with a massive increase in cell staining when both are overexpressed (24–42× vs one or the other) (Supplementary Fig. 1B), confirming cooperative targeting of TfR and target. ENGINEERING
CYPHER CANDIDATES Additional PD-L1-targeting CYpHER candidates were generated (Fig. 2a). Both candidates contained an Fc domain with a high-affinity TfR binder fused to its C-terminus by a
Gly-Ser linker. One candidate (CT-4212-1) also contained an N-terminal fusion (via Gly-Ser linker) with the pH-dependent PD-L1-binding CDP, while the other candidate (CT-4212-3) used a rigid
linker from human IgA to fuse the PD-L1-binding CDP to the TfR binder’s C-terminus. The two designs were to test whether a given binder could function at the N- or C-terminus of a CYpHER.
The rigid linker in CT-4212-3 is to prevent formation of aberrant cystines between the two CDPs. Both molecules were tested on polyclonal 293T, H1650, and MDA-MB-231 cell populations
transduced (via lentivirus) to express PD-L1-GFP. In all three populations, PD-L1 was trafficked from the surface by CYpHER (Fig. 2b), as seen in both microscopy and flow cytometry assays
(using a non-competitive PD-L1 antibody [clone 22C3], Fig. 2c–e)29. Flow cytometric quantitation of per-cell GFP levels demonstrated substantial and consistent surface PD-L1 removal (Fig.
2f, h, j), as well as overall flow-assessed PD-L1-GFP reduction in all cases (Fig. 2g, i, k). The degree of total PD-L1-GFP reduction varied by cell line and CYpHER. The highest-expressing
population, 293T-PDL1-GFP, experienced the largest degree of GFP reduction. This could be caused by better cooperative CYpHER accumulation when more target is present, by saturation of the
recycling pathway, or both. The CYpHER platform is amenable to any target for which surface or soluble elimination would benefit patients beyond simply binding or blocking one site. One such
target, with dual roles as both kinase and kinase substrate (as well as other cell-surface-driven functions)30, is EGFR. It is implicated as a driver of several common and deadly cancers,
including glioblastoma, lung cancer, head and neck cancer, and colon cancer6,31,32,33. It is primarily targeted by TKIs or monoclonal antibodies (mAbs). Patients can benefit for a time, but
resistance to these treatments inevitably emerges and remains an area of urgent need for new therapies34. These resistance mechanisms are often either a point mutation to reduce TKI activity
or an upregulation of another EGFR heterodimerization partner like HER2, HER3, or c-Met. Elimination of EGFR from the surface would drastically reduce EGFR-associated signaling, including
kinase-independent signaling35,36, as it primarily occurs at the plasma membrane. An EGFR-binding VHH nanobody37 was engineered into a CYpHER component through similar means to the
PD-L1-binding CDP. In one method, a pool of variants with His substitutions was screened in mammalian surface display28 through four rounds of enrichment; two rounds enriched for high
binding at neutral pH (pH 7.4), while the other two enriched for low binding at early endosomal pH (pH 5.5). In the final round of sorting, populations with low (Supplementary Fig. 2A) or
high binding at pH 5.5 (Supplementary Fig. 2B) were collected. The primary variant in the pool enriched for low binding in low pH loses roughly half its bound EGFR in tetravalent
(streptavidin) stain conditions upon low-pH rinse (Supplementary Fig. 2C), while the dominant variant from the population with high binding at low pH does not have this property
(Supplementary Fig. 2D). Fc fusions to each of these molecules, when used as ligands in surface plasmon resonance experiments, verified these properties (Supplementary Fig. 2, E, F); the
variant engineered for pH-dependent EGFR release demonstrated higher affinity at neutral pH (_K_D = 16.2 ± 0.1 nM at pH 7.4) compared to low pH (_K_D = 61.2 ± 0.3 nM at pH 5.8) and the other
variant demonstrating slightly lower affinity at neutral pH (_K_D = 10.5 ± 0.1 nM at pH 7.4) compared to low pH (_K_D = 7.8 ± 0.07 nM at pH 5.8). Both of these represent >10-fold higher
affinity vs the reported affinity of the parental nanobody38. The pH-dependent release nanobody was named EGFR Nanobody v1. As an additional allele identification strategy, singleton His
scanning (i.e. single His substitutions tested one at a time) was performed on the nanobody in CDRs 1 and 3, as these CDRs are primarily implicated in EGFR-binding38. One variant (His Sub
10) was identified in mammalian surface display that lost roughly half its bound EGFR in tetravalent stain conditions upon low-pH rinse (Supplementary Fig. 2G); this was named EGFR Nanobody
v2. ADAPTING A NATIVE LIGAND FOR CYPHER In building the platform, it was apparent that the target-binding domain needn’t be an exogenous molecule. As a great many disease-associated target
proteins do so by signal transduction mediated by ligand-binding, the ligand presents itself as a natural target-binder that can be adapted, through engineering and affinity/pH maturation,
for CYpHER incorporation. In the case of EGFR, EGF itself (a naturally pH-dependent binder which is also a CDP)39 can be used after engineering it to disable signal transduction
capabilities. Rosetta protein modeling software40,41 was used to design EGF variants that bind to EGFR Domain III in the absence of Domain I, after which studying the EGF:EGFR co-crystal
structure (PDB: 1IVO)42 was used to identify mutations predicted to disrupt Domain I interaction. The end result was a dominant-negative EGF variant a) that binds to Domain III in the
absence of Domain I, b) whose Domain I interaction is disrupted to the degree that Domain I impairs binding, and c) that retains the pH-dependent binding of the parent molecule
(Supplementary Fig. 3). The details for this design effort are found in the Supplemental Methods. The final variant from this process, EGFd1.5.36, was then used in CYpHER molecules. EGFR
CYPHER INDUCES EGFR SURFACE CLEARANCE AND ELIMINATION A candidate EGFR CYpHER, CT-1212-1, was produced using a high-affinity TfR-binding CDP and EGFR Nanobody v1 (Fig. 3a–c). The molecule
demonstrated good expression and assembly with negligible aggregate after capture from supernatant and buffer exchange. 293T cells expressing a variable level of EGFR-GFP, expected to
undergo target internalization upon CYpHER treatment similar to PD-L1-GFP (Fig. 3d), were dosed and analyzed by microscopy and flow cytometry (staining with non-competitive anti-EGFR clone
199.12)43 for total EGFR-GFP and surface EGFR (Fig. 3e). As a population, cells demonstrated >80% reduction of surface EGFR and ~50% reduction of total EGFR-GFP signal at 24 h, which was
validated by Western blot of lysate from flow-sorted, viable cells (Fig. 3f). Viable cells were studied because dead cells and debris are not metabolically active and cannot drive
endolysosomal trafficking of targets. The flow profile was not “shifted” en-masse by CYpHER treatment; instead, much of the reduction occurred in those cells with the highest initial EGFR
levels. Partitioning flow profiles by surface EGFR supported this observation (Fig. 3g), where the cells with the most surface EGFR experienced the greatest proportional loss. Meanwhile,
time-course experiments demonstrated that surface EGFR clearance is rapid (near maximal effect after 1 h), consistent with rapid TfR-mediated uptake, while EGFR-GFP signal loss takes more
time, likely due to the slower kinetics of lysosomal degradation vs surface internalization (Fig. 3h, i). This was corroborated by labeling 293T-EGFR-GFP cells with LAMP1-RFP via baculovirus
(BacMam 2.0, Invitrogen) for 24 h and then treating cells with DyLight 755-conjugated CT-1212-1 for 0, 1, or 4 h (Fig. 3j). Lysosomal RFP signal overlapped with EGFR-GFP foci more often and
more intensely at 4 h than at 1 h. CYpHER signal was also more intense overall at 4 h than at 1 h. The CYpHER signal at 4 h was within several intracellular compartments, including the
lysosome, indicating some of the CYpHER being trafficked alongside EGFR-GFP to the lysosome and/or a portion of TfR undergoing normal lysosomal turnover bringing CYpHER with it. We focus
much of our protein trafficking data on surface clearance instead of total protein elimination. First, removal of EGFR from the plasma membrane separates it from access to both ligand and
downstream signaling modulators like KRas. Second, after endosomal release, actual elimination vs recycling of protein is highly context-dependent, involving cell-specific recycling kinetics
and saturable trafficking modulators44. Third, proteins in the process of synthesis can be detected as total protein but can neither signal effectively nor be accessed by CYpHER. We
explored surface EGFR clearance rates on a panel of non-small cell lung cancer (NSCLC) cell lines which include various drug-resistance mechanisms that can occur in patients: H1650 (EGFR
with exon 19 deletion and PTEN knockout), H1975 (EGFR with L858R and T790M mutations [the latter rendering it resistant to 1st generation EGFR TKIs] along with activating G118D mutation in
PIK3CA), A549 (wild type EGFR with activated KRas G12S), and H358 (wild type EGFR with activated KRas G12C). They represent a range of total surface levels and ratios of EGFR and TfR (the
latter measured with non-competitive clone OKT9)45 (Fig. 4a). All four lines responded to CYpHER for surface EGFR clearance (Fig. 4b), with rapid kinetics (1 h) and to a much greater degree
than seen with cetuximab, a molecule known to induce surface clearance and overall protein reduction via induction of ubiquitin-mediated uptake2,3. We also confirmed that CYpHER remains
detectible on the cell surface 24 h after media exchange, where cetuximab does not (Fig. 4c). CYPHERS WITH ANY OF THE THREE ENGINEERED EGFR BINDERS CLEAR SURFACE EGFR The other two
engineered EGFR binders were also incorporated into CYpHER molecules (Fig. 4d). The same Fc with a high-affinity TfR-binding CDP (separated by Gly-Ser linker) was the starting point. The
nanobodies were fused via a Gly-Ser linker to the N-terminus of the Fc domain as was done in CT-4212-1; the nanobody’s N-terminus is at the EGFR Domain III interface38, so this format is
optimal for EGFR binding. As with EGFR Nanobody v1 and CT-1212-1, EGFR Nanobody v2 was incorporated into CT-6212-1. Conversely, the C-terminus of EGF is adjacent to the EGFR Domain III
interface42, so fusion via its N-terminus is optimal; it was incorporated in a similar format to that of CT-4212-3, producing CT-5212-3. Surface EGFR levels in A549 cells were reduced after
24 h CYpHER treatment with all 3 designs, including after media exchange and growth without drug for another 24 h (“Withdrawal”) (Fig. 4e); as EGFR turnover in the absence of ligand is
fairly rapid (~6–10 h on most cell lines)46, this suggested retention of activity via catalytic mechanism. All three CYpHER molecules were still present on the surface after a 24 h drug
withdrawal (Fig. 4f). CT-1212-1 was tested for up to 3 days in the four NSCLC cell lines (A549, H1975, H358, and H1650). All four cancer lines had rapid (within 1 h) reduction of surface
EGFR, which was reduced by 55–81% of untreated after 24 h (Fig. 4g and Supplementary Fig. 4); this reduction was maintained for at least 3 days. Additionally, 24 h without drug after 24 h of
treatment (“withdrawal”) still yielded surface EGFR levels markedly lower than untreated cells; in A549 and H1975 cells, this effect is durable out to 3 days after drug withdrawal, at which
point CYpHER is still observed on the cell surface (Supplementary Fig. 5). Different cell lines are expected to have different uptake and rebound kinetics, so this phenomenon is not assumed
to be universal. We also evaluated surface TfR levels (Supplementary Fig. 6). In A549 and H1975 cells, mild fluctuations that may represent drug-dependent effects on TfR expression and/or
trafficking, but are not persistent with continuous drug exposure, are apparent. In the lines with the highest TfR levels (H358 and H1650), a more substantial and sustained reduction of
surface TfR occurred that took 24 h to reach these low levels and largely returned to normal after 24 h of withdrawal. Their higher levels of surface TfR may facilitate CYpHER-induced
multimerization, which is known to alter TfR trafficking47. We also tested whether we could observe a hook effect. This phenomenon has been documented for some bispecific TPD molecules,
wherein target depletion is blunted if the drug concentration is so high that separate drug molecules occupy each respective partner (preventing ternary complex formation)48. At all doses of
CT-1212-1, CT-5212-3, and CT-6212-1 evaluated (2–200 nM), surface EGFR levels on A549 cells were reduced compared to untreated cells after 24 h of treatment (Fig. 4h). The degree of EGFR
reduction by CT-1212-1 was modestly blunted at 200 nM compared to lower doses, whereas CT-5212-3 and CT-6212-1 show minimal variation over this concentration range. This suggests that the
effect, if any, is mild and the nature of the molecule (binder and/or modular organization) may have some impact. Effects may also differ by cell line, target, and metabolic state.
CYPHER-DRIVEN EGFR INTRACELLULAR SEQUESTRATION We evaluated EGFR trafficking in a knock-in A549-EGFR-GFP cell line using CT-1212-1 and variants thereof (Fig. 5a) that used heterodimeric Fc
domains to alter EGFR- and TfR-binding valence. All four molecules that contained at least one EGFR-binder and at least one TfR-binder drove EGFR-GFP from the membrane into intracellular
compartments (Fig. 5b), with flow assays demonstrating this effect to be dose-dependent in 3 of 4 drugs and mostly retained by all after 24 h drug withdrawal (Fig. 5c). Control molecules
containing a non-EGFR-binding nanobody or a non-TfR-binding CDP did not induce trafficking (Fig. 5d); this trafficking is not associated with a reduction in total EGFR-GFP in this line
(Supplementary Fig. 7). We confirmed the rapid activity of the mechanism, showing this internalization phenotype after only 20 min of treatment (Fig. 5e). We also tested holoTF competition,
as our TfR-binding CDP binds to the same site on TfR as transferrin25. Both EGFR-GFP internalization (Fig. 5f) and surface EGFR clearance (Fig. 5g) were retained in the presence of human
holoTF, with only mild suppression of activity at 1000x molar levels of holoTF versus CYpHER. CYPHER CATALYTIC TARGET UPTAKE Our experiments with drug withdrawal strongly suggest a catalytic
mechanism of action where one drug molecule can induce uptake of multiple target molecules, as we see both retention of activity and retention of CYpHER molecules on the surface of cells
after 24 h drug withdrawal. We developed an assay to further evaluate catalytic uptake of soluble cargo (Fig. 6a and Supplementary Fig. 8). We first demonstrated that cells specifically take
up soluble target:CYpHER complexes. Cells were exposed to CYpHER saturated with fluorescently labeled target for 2 h, permitting time for target uptake and for CYpHER (via TfR) to cycle in
and out of the cell several times. This yielded specific target uptake (Supplementary Fig. 8, bars 1 vs 2 [2 h] and bars 3 vs 5 [24 h]). Next, the assay was modified to look for uptake of
newly introduced target after the initial 2 h uptake followed by removal of all soluble CYpHER from the system. In step 1 of this assay, cells are exposed to CYpHER saturated with unlabeled
target for 2 hrs. Then the cells are thoroughly rinsed to remove all soluble molecules and then exposed to fluorescent soluble target alone for 24 h. Any fluorescent target uptake during
these 24 h, in excess of that seen by cells that were not pre-exposed to CYpHER, is due to the catalytic activity of CYpHER molecules that have already cycled and released their
non-fluorescent cargo (Supplementary Fig. 8, bars 3 vs 4). With this assay, using various CYpHER designs (Fig. 6b), we saw catalytic CYpHER-driven uptake of cargo with all CYpHER molecules
and cell lines tested. Fluorescent soluble EGFRvIII uptake via CT-1212-1 in A549, H1650, H1975, and H358 cell lines (Fig. 6c), was seen, with catalytic uptake (normalized to passive uptake
without CYpHER, which is likely via pinocytosis) at levels that vary by cell line. The degree of catalytic uptake also changed dependent on the nature of the target-binder (Fig. 6d).
Additionally, as demonstrated using PD-L1 CYpHERs and soluble PD-L1, the format of the CYpHER (Fig. 6e) had an impact. In all cases, uptake of soluble target in excess of passive uptake was
seen, demonstrating a catalytic mechanism of action for CYpHER. PHARMACODYNAMIC EFFECTS OF CYPHER IN VITRO Having established target depletion, we tested how CYpHER alters EGFR-mediated
signaling and cell growth (Fig. 7). Using various CYpHER and control molecules (Fig. 7a), we investigated ligand-induced signaling, where both surface clearance and competitive binding to
EGFR could have impacts. Stimulating CYpHER-treated cells with EGF produced no increased EGFR phosphorylation in CT-1212-1-treated cells (Fig. 7b), suggesting any remaining EGFR on the
surface is blocked by the drug or is otherwise signaling-incapable. The valence-altering variants (CT-1211-1, CT-1112-1, CT-1111-1) had the same effect, but control molecules CT-1232-1
(non-TfR-binding CDP) and CT-3212-1 (non-EGFR-binding nanobody) did not, demonstrating that both TfR- and EGFR-binding functions are necessary together to block ligand-induced activation of
EGFR. CYpHER containing the EGF variant was also tested (Fig. 7c); in contrast to the nanobody-containing CYpHER series, CT-5212-3-treated cells showed some residual EGFR phosphorylation in
response to EGF, suggesting the EGF variant on the CYpHER provides less complete blockade of the EGF-binding site of the residual surface-resident EGFR. Further affinity maturation of the
variant may improve its ability to competitively inhibit EGF stimulation under these conditions. Meanwhile, the EGF variant itself (in the form of Fc fusion CT-5200-6) did not induce EGFR
phosphorylation, confirming its capacity as a dominant-negative EGF. As head and neck cancer (e.g., HNSCC) and lung cancer (e.g., NSCLC) are commonly treated with EGFR-targeting agents6,33,
CT-1212-1 was tested for growth suppression in several relevant cell lines: head and neck cancer line A431 (with a massive duplication of the EGFR gene), and the four NSCLC lines H1650,
H1975, A549, and H358. All of these lines were tested alongside clinical EGFR-targeted drugs cetuximab (EGFR mAb), gefitinib (1st generation EGFR TKI), and osimertinib (3rd generation EGFR
TKI) (Fig. 7d–i). CT-1212-1 suppressed growth across all cancer types and mutational profiles tested. Moreover, It had higher potency than the three clinical comparators in all five lines,
with the exception of comparable potency with osimertinib in H1975 cells containing the T790M mutation against which osimertinib was originally developed49. The potency of CYpHER in KRas
mutant lines A549 (G12S) and H358 (G12C) led us to ask to what degree EGFR signaling and/or iron uptake disruption is contributing to the cell growth inhibition (Supplementary Fig. 9).
Reducing TfR-binding valence or affinity modestly reduced potency. Using ferric ammonium citrate (FAC), a cell-penetrant chelated iron supplement, FAC prevented the CT-1212-1 growth
disruption on A549 cells. Despite expressing mutant KRas G12S, A549 cells still respond to 1 ng/mL EGF (an amount consistent with tumor parenchyma levels50) in the media by demonstrating a
migratory phenotype alongside increased growth, both of which were completely or partially (respectively) suppressed by CT-1212-1 but not CT-3212-1 (control that binds TfR but not EGFR) when
FAC is present. Additionally, we tested a colon cancer cell line with wild type KRas and high, but not constitutively-active, EGFR expression51,52, since such colon cancers are commonly
treated with EGFR-targeting biologics53. It demonstrates growth disruption in response to CT-1212-1 but not cetuximab, similar to the NSCLC lines, but its growth is still partially disrupted
when FAC is present. These experiments provide additional context to CYpHER growth disruption, in that cells in vitro can be highly sensitive to iron uptake disruption, but CYpHER can still
disrupt EGFR-based growth and migration. As many EGFR-targeted therapeutics (mAbs and TKIs) demonstrate skin toxicities due to the sensitivity of keratinocytes to EGFR suppression7,54,
CT-1212-1 was also tested in primary human dermal keratinocytes (Supplementary Fig. 10). Examining TfR and EGFR on these cells, TfR levels were much lower on these normal cells than on the
cancer lines we tested, while EGFR levels are high (Supplementary Fig. 10A). Surface EGFR clearance was observed with CT-1212-1, but with much slower kinetics than seen in the cancer lines
(likely due to much lower surface TfR), and no reduction in surface TfR was seen (Supplementary Fig. 10B). In primary keratinocyte growth inhibition assays (Supplementary Fig. 10C),
CT-1212-1 demonstrated similar properties to cetuximab, while the TKIs had similar profiles as their activity on the cancer cell lines. Comparative potencies on cancer and keratinocytes
suggest CYpHER could have advantages in sparing keratinocytes from growth disruption while still impairing cancer cell growth (Supplementary Fig. 10D). In light of CYpHER’s interactions with
TfR and because some TfR-targeting biologics have impacted reticulocyte levels55,56, we chronically dosed female athymic nude mice with CT-1222-1 for 4 weeks, twice weekly, at doses from 15
μg to 1000 μg. Looking at red blood cells, reticulocytes, total white blood cells, neutrophils, and lymphocytes 48 h after the final dose, blood cell counts were not significantly altered
(Supplementary Fig. 11). The mice also did not demonstrate any body weight loss, gross physical responses, or drug-related behavioral changes. Thus, there were no signs that chronic CYpHER
exposure in vivo yielded gross toxicity or hematopoietic side effects, in spite of any impact by CYpHER on TfR levels or iron homeostasis in cancer lines in vitro. IN VIVO CYPHER
PHARMACOKINETICS AND PHARMACODYNAMICS To investigate the in vivo pharmacokinetic (PK) properties of the CYpHER candidates, we dosed NCr nu/nu mice with 1.5 mg/kg CT-1212-1, CT-1222-1,
CT-1211-1, and CT-1232-1 (Fig. 8a). The nanobody does not cross-react with murine EGFR, so only the murine cross-reactive TfR-binding CDP and the Fc domain would be expected to influence PK.
CT-1212-1 has two high-affinity TfR-binding CDPs, CT-1222-1 has two medium-affinity TfR-binding CDPs, CT-1211-1 has one high-affinity TfR-binding CDP, and CT-1232-1 has no TfR-binding
capability (Fig. 8a). As measured by ELISA (Fig. 8b), serum levels of the CYpHER molecules demonstrated serum half-lives between 41 and 88 hrs at this dose, with the longest belonging to the
non-TfR-binding molecule CT-1232-1. It is likely that TfR binding increases clearance, a phenomenon seen in other studies55,57, even with the Fc otherwise extending serum residence.
Considering that CYpHERs exhibited potency on cancer cells at concentrations as low as 0.2 nM and EGFR surface clearance at concentrations as high as 200 nM, the PK data indicates that serum
levels in a therapeutic range may be readily attained with infrequent dosing. The biodistribution to the target tissue (e.g., tumor), as well as the durability of activity given catalytic
target clearance in cells even when CYpHER is removed from extracellular fluid, are still to be investigated. We next tested mice implanted with flank H1975 xenografts and treated for 8 days
with CYpHER for any observed pharmacodynamic (PD) effects (Fig. 8c). Female athymic nude mice were implanted with 5×106 H1975 cells. After 21 days of growth, mice received three doses of
CYpHER on days 0 (enrollment day), 3, and 7. CYpHERs and doses administered were: CT-1212-1 450 μg/dose; CT-1212-1 150 μg/dose; CT-1212-1 30 μg/dose; CT-1222-1 150 μg/dose; and CT-5212-3 150
μg/dose. On day 8, tumors were harvested for Western blotting and histology. Western blotting (Fig. 8d, e) demonstrated a reduction (_P_ = 0.04, as normalized to actin) in total EGFR by
CT-1222-1 150 μg/dose, and a trend towards EGFR reduction (_P_ = 0.11) by CT-1212-1 450 μg/dose; pY1068 blotting was not significantly different between the groups (Supplementary Fig. 12).
Histology for EGFR and Ki67 (Fig. 8f, g) demonstrated two phenomena. First, most fields from the CT-1212-1 450 μg/dose and CT-1222-1 150 μg/dose tumors demonstrated altered localization of
EGFR (Fig. 8f), visible as marked reduction of membrane EGFR (DAB stain) relative to intracellular levels. Second, automated Ki67 quantitation (Fig. 8g) demonstrated that 8 days of treatment
caused a reduction in the proliferation marker Ki67 (percent Ki67 positivity) in the CT-1212-1 450 μg/dose (_P_ = 0.003), CT-1212-1 150 μg/dose (_P_ = 0.0004), and CT-1222-1 150 μg/dose
(_P_ = 0.0002) groups vs vehicle. We will note that the ineffectiveness of CT-5212-3 at reducing proliferation suggests that bivalent high-affinity TfR-binding is not sufficient for
disrupting H1975 flank tumor growth. As the nanobody-based CYpHERs that reduced proliferation also were more effective at reducing EGF-mediated EGFR phosphorylation than CT-5212-3 (Fig. 7b,
c), the EGFR-inhibition capabilities of CYpHER may be driving the effect in vivo. However, CT-5212-3, which has a murine cross-reactive EGFR binder, may also have altered biodistribution
dynamics, which could impact tumor accumulation. These, and other variables, are of interest for future studies. DISCUSSION Catalytic TPD via CYpHER is a promising approach to durably
depleting disease-driving proteins, capable of altering target trafficking and cell behavior both in vitro and in vivo. CYpHER leverages TfR, a protein whose rapid recycling kinetics provide
activity across many tumor and cell types. Furthermore, TfR is overexpressed and required for cancer cell growth, increasing potential tumor accumulation and reducing the risk of acquired
drug resistance. TfR also delivers cargo across the blood-brain barrier, adding CNS proteins as potential targets. Through engineering the target-binding end for pH-dependent release, CYpHER
is not reliant on other enzymes for function, and its catalytic activity permits one drug molecule to clear multiple target molecules with more durable function. Lastly, the drug molecules
are proteins produced by standard recombinant expression, requiring no chemical modifications and using binder modalities found in other clinically-approved molecules. CYpHER is a promising
approach for cancers driven by RTKs, exemplified by the inexorable challenge of EGFR-driven cancer. Our pilot studies demonstrate a pharmacodynamic effect in tumors, but more studies are
necessary to establish potential efficacy in tumor growth inhibition. Meanwhile, the CYpHER platform can approach numerous difficult targets in oncology and CNS disease. The ErbB family of
RTKs (e.g., EGFR, HER2, HER3) are all associated with driving cancer and/or inflammation in various tissues and settings, alongside other growth factor and cytokine receptors (e.g., c-Met,
FGF receptors, IGF-1 receptors, interleukin receptors). Similar to EGFR, all feature multiple functions like homodimerization, heteroassociation, kinase function, and/or scaffolding for
signal transduction. Protein elimination is the only way to completely neutralize all possible disease-causing functions. For CNS conditions, access to the CNS via TfR-mediated transcytosis
offers exciting possibilities such as clearing neurodegeneration-associated misfolded proteins (e.g., amyloid, tau, huntingtin) or their inflammatory mediators. CNS metastasis is also a
common cause for cancer progression, and TfR-mediated CNS access may prevent this mechanism of recurrence. In conclusion, CYpHER adds a powerful entry to the TPD field. With catalytic
functionality, broad target applicability, good assembly and production, potent and durable alteration of target trafficking, and demonstrable in vivo activity, proteins for which
traditional targeted therapeutics have struggled may be approachable, with promising implications to our most insidious and intransigent diseases. METHODS STUDY DESIGN This work describes
the conception, engineering, and applications of CYpHER, a catalytic targeted protein degradation technology using a recycling receptor (here, TfR), evaluating target trafficking and
elimination in tissue culture and in murine tumors. The work complies with all relevant ethical regulations. Cyclera Therapeutics oversaw compliance with biosafety regulations for in vitro
work. Animal work compliance described in the pharmacokinetics and tumor implantation sections below. RECOMBINANT PROTEINS, ANTIBODIES, AND CO-STAINS/SECONDARY ANTIBODIES Identities of
catalog item recombinant proteins and antibodies can be found in the Supplementary Information. EGF VARIANT DESIGN USING ROSETTA A previously-published co-crystal structure containing EGF
and EGFR (PDB 1IVO) was processed to separate EGF (chain C) and EGFR domain III (chain A, residues 311-510). They were combined into a single PDB, which was used as the input for
Rosettascripts40 using proprietary XML scripts optimized for CDP redesign. 1000 unique variants were designed and scored using an interface analysis script, with 488 that had favorable
scoring parameters incorporated into a mammalian surface display screening library. The steps for screening the binders and selectively mutagenizing the variants to identify a
dominant-negative variant for CYpHER incorporation can be found in the Supplementary Information. MAMMALIAN SURFACE DISPLAY Surface display flow cytometry was based on previously described
protocols (62), with details found in Supplementary Information. The library screens for identifying and maturing EGFR-binding nanobody and EGF variants were each conducted once. RECOMBINANT
PROTEIN PRODUCTION AND ANALYSIS Pilot CYpHER (Supplementary Fig. 1) molecule was produced as previously described27; in short, 293 FreeStyle cells (Thermo Fisher R79007) were transduced
with constructs driving expression of the protein of interest at a multiplicity of infection ≈10 and grown until terminal volume (~30 mL) in FreeStyle 293 Expression Medium (Invitrogen
12338018) until harvest, 0.22-μm sterile filtration, and IMAC Ni-affinity chromatography (Cytiva, 17525501) purification. All other CYpHER molecules were produced by transient expression in
suspension HEK293 cells (ThermoFisher GeneArt) and purified either via Ni-NTA pull-down as previously described27 or by Protein A columns (Cytiva 28985254 [pre-packed Protein A columns] and
28903059 [buffer kit]) as per manufacturer’s protocol. Proteins were buffer exchanged (Sephadex G25 desalting columns, Cytiva 17085101) into PBS and aliquoted for storage at −80 °C. SDS-PAGE
(4–12% Bis-Tris 1 mm thickness, ThermoFisher NP0321BOX or NP0323BOX) was run with MES buffer (ThermoFisher NP0002) at 180 V for 50 min prior to Coomassie stain. SE-HPLC was performed on
Agilent instrumentation using a TSKgel G3000SWXL column (Tosoh 08541). Mobile phase was 50 mM acetate pH 5.0, 100 mM NaCl, 100 mM arginine, 5% EtOH. Flow rate for the run was 0.5 mL/min. 100
μg protein was loaded. SURFACE PLASMON RESONANCE (SPR) INTERACTION ANALYSES, MICROSCOPY, WESTERN BLOTTING, CELL VIABILITY DOSE RESPONSE TESTING, IMMUNOHISTOCHEMISTRY, AND ELISA The details
for the SPR, microscopy, Western blotting, cell viability dose response tests, immunohistochemistry, and ELISA can be found in the Supplementary Information. CANCER CELL LINE AND PRIMARY
KERATINOCYTE SURFACE PROTEIN FLOW CYTOMETRY Detailed protocols can be found in the Supplementary Information. In brief, cells for surface protein analysis were lifted, pelleted, and stained
for 30 min on ice with 10 nM of the appropriate antibody (anti-PD-L1, anti-EGFR, anti-TfR, or anti-human Fc), all of which were chosen to bind a site non-competitive with the CYpHER binding
site. Fluorescent co-stains were included, permitting flow cytometry quantitation of surface protein after cells were rinsed. Note: where Y axes in figures are denoted with “vs PBS”, “vs
Untreated”, or “vs No CYpHER”, it means values were normalized to the mean of that sample (PBS, Untreated, or No CYpHER) prior to plotting. CATALYTIC SOLUBLE PROTEIN UPTAKE The detailed
protocol is available in the Supplementary Information. In brief, cells in 24 well plates were treated for 2 h with 20 nM biotinylated EGFRvIII with or without 5 nM CYpHER. After 2 h, cells
were rinsed twice with PBS and then exposed to 10 nM biotinylated EGFRvIII and 10 nM iFluor 647-conjugated monovalent streptavidin. After 24 h, cells were lifted and analyzed by flow
cytometry for fluorescence in the 647 channel, normalizing averages of cells treated with both EGFRvIII and CYpHER in Step 1 to cells only treated with EGFRvIII in Step 1. IN VIVO
PHARMACOKINETIC ANALYSIS PK work was performed at Charles River Laboratories with approval from their internal review board. For each test article, 12 female NCr nu/nu mice (8–10 weeks of
age) received single IV doses of 1.5 mg/kg test article in PBS (or only PBS vehicle). Females were used for ease in re-housing and to reduce waste in the breeding scheme (males used for
strain maintenance). Further development experiments will use both male and female mice. In groups of 3, mice were bled at 10 min, 30 min, 1 h, 2 h, 4 h, 8 h, 24 h, 48 h, 96 h, and 168 h. 12
mice produced these samples: one trio of mice was bled at 10 min, 4 h, and 96 h; one trio was bled at 30 min, 8 h, and 168 h; one trio was bled at 1 h and 24 h; and one trio was bled at 2 h
and 48 h. Serum samples were snap-frozen and stored at −80 °C until analysis using an in-house ELISA method. IN VIVO TUMOR IMPLANTATION AND DOSAGE Tumor implantation and dosage was
performed at Seattle Children’s Research Institute. All mice were maintained in accordance with the National Institutes of Health Guide for the Care of Laboratory Animals with approval from
the Seattle Children’s Research Institute, Institutional Animal Care and Use Committee (protocol ACUC00682). 7-week-old female Athymic Nude mice (Foxn1nu) were purchased from Inotiv
Laboratories (#069) and housed under specific pathogen free conditions. Females were used for ease in re-housing and to reduce waste in the breeding scheme (males used for strain
maintenance). Further development experiments will use both male and female mice. NCI-H1975 lung tumor cells were purchased from ATCC (CRL-5908) and verified human pathogen and mycoplasma
free. CYpHER proteins CT-1212-1, CT-1222-1, and CT-5212-3 were formulated in phosphate-sucrose buffer and confirmed to meet endotoxin specifications. 5 ×106 tumor cells in PBS were
inoculated in the subcutaneous space on the right flank of seven weeks old mice. Study enrollment was done en mass on day 0, 21 days after tumor implantation when the average tumor volume
per group was 275 mm3. Six mice were randomly assigned to each treatment group, normalizing for equal starting tumor volume. Vehicle or therapeutic were administered as a 200 μL bolus via
tail vein injection on days 0, 3, and 7. Tumor volume and body weight were recorded on days 0, 2, 4, and 7. The study ended on day 8. Mice were removed from the study early if ulcerations
developed on the tumor surface. Tumors did not reach IACUC-determined maximal size (1500 mm3). For blood counts, mice bearing H1975 xenografts were treated twice per week IV with CT-1212-1
or vehicle for four weeks. 48 h after the final dose, mice were euthanized and blood collected. Blood was kept on ice and shipped overnight to IDEXX BioAnalytics (West Sacramento, CA) for
complete blood count (CBC) analysis. All mice were group-housed with unrestricted mobility and free access to food and water for the duration of study. The housing room is maintained at
68–79 °F with 30–70% relative humidity on a 12-h light dark cycle. STATISTICS AND REPRODUCIBILITY All comparisons within a given plot or panel were performed simultaneously. Measurements
involved distinct samples; the same sample was not measured repeatedly. No data were excluded from analyses. Flow cytometry and microscopy experiments were performed once or twice as
described in figure legends; when once, the experiments were compared to similar, but not identical, experiments (e.g., same cell line and treatment but different timepoints) for
reproducibility assessment before inclusion in the manuscript. In vitro growth suppression experiments were performed once (A431, H358, SW48) or twice (A549, H1975, H1650, primary
keratinocytes); when twice, both datasets produced similar data so one was used in analysis. Experiments consisted of 8-9 concentrations dosed in triplicate to arrive at reported potencies,
calculated using Graphpad Prism v10, details for which can be found in the Supplementary Information. All animals in both the PK and tumor xenograft studies were female. No statistical
method was used to predetermine sample size. Investigators were blinded to test article identity for the PK study, while the tumor xenograft study was unblinded. For the tumor xenografts,
enrollment was done when the average tumor volume per group was 275 mm3, randomly assigning mice to each treatment group, normalizing for equal starting tumor volume. For the ELISA, all
murine serum samples were measured at two dilutions (1:100 and 1:1000) in technical triplicate, using whichever dilution produced interpolated values closest to the middle of the linear
portion of the ten-point standard curve (technical triplicate at all concentrations) as the actual values. Full information on statistical tests used, significance values, and N per sample,
are found in the Supplementary Data. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY
All data associated with this study are present in the paper, Supplementary Information and Supplementary Data. Additional information and/or materials related to this study, including
recombinant proteins and datasets, will be made available through a material transfer agreement upon request to the corresponding author. Source data are provided with this paper. REFERENCES
* Chang, M. T. et al. Identifying transcriptional programs underlying cancer drug response with TraCe-seq. _Nat. Biotechnol._ 40, 86–93 (2022). Article PubMed CAS Google Scholar * Dai,
W. et al. Cetuximab inhibits oral squamous cell carcinoma invasion and metastasis via degradation of epidermal growth factor receptor. _J. Oral. Pathol. Med._ 43, 250–257 (2014). Article
PubMed CAS Google Scholar * Lu, Y. et al. Epidermal Growth Factor Receptor (EGFR) Ubiquitination as a Mechanism of Acquired Resistance Escaping Treatment by the Anti-EGFR Monoclonal
Antibody Cetuximab. _Cancer Res._ 67, 8240–8247 (2007). Article PubMed CAS Google Scholar * Du, Z. & Lovly, C. M. Mechanisms of receptor tyrosine kinase activation in cancer. _Mol.
Cancer_ 17, 58 (2018). Article PubMed PubMed Central Google Scholar * Abourehab, M. A. S., Alqahtani, A. M., Youssif, B. G. M. & Gouda, A. M. Globally Approved EGFR Inhibitors:
Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. _Molecules_ 26, 6677 (2021). Article PubMed PubMed Central CAS Google Scholar
* Tomasello, C. et al. Resistance to EGFR inhibitors in non-small cell lung cancer: Clinical management and future perspectives. _Crit. Rev. Oncol. Hematol._ 123, 149–161 (2018). Article
PubMed Google Scholar * Fu, K., Xie, F., Wang, F. & Fu, L. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance. _J. Hematol. Oncol._
15, 173 (2022). Article PubMed PubMed Central Google Scholar * Reslan, L., Dalle, S. & Dumontet, C. Understanding and circumventing resistance to anticancer monoclonal antibodies.
_mAbs_ 1, 222–229 (2009). Article PubMed PubMed Central Google Scholar * Wells, J. A. & Kumru, K. Extracellular targeted protein degradation: an emerging modality for drug discovery.
_Nat. Rev. Drug Discov._ https://doi.org/10.1038/s41573-023-00833-z (2023). * Schapira, M., Calabrese, M. F., Bullock, A. N. & Crews, C. M. Targeted protein degradation: expanding the
toolbox. _Nat. Rev. Drug Discov._ 18, 949–963 (2019). Article PubMed CAS Google Scholar * Cotton, A. D., Nguyen, D. P., Gramespacher, J. A., Seiple, I. B. & Wells, J. A. Development
of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. _J. Am. Chem. Soc._ 143, 593–598 (2021). Article PubMed PubMed Central CAS Google
Scholar * Siepe, D. H., Picton, L. K. & Garcia, K. C. Receptor Elimination by E3 Ubiquitin Ligase Recruitment (REULR): A Targeted Protein Degradation Toolbox. _ACS Synth. Biol._ 12,
1081–1093 (2023). Article PubMed PubMed Central CAS Google Scholar * Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. _Nature_ 584, 291–297
(2020). Article ADS PubMed PubMed Central CAS Google Scholar * Leusmann, S., Ménová, P., Shanin, E., Titz, A. & Rademacher, C. Glycomimetics for the inhibition and modulation of
lectins. _Chem. Soc. Rev._ 52, 3663–3740 (2023). Article PubMed PubMed Central CAS Google Scholar * Pance, K. et al. Modular cytokine receptor-targeting chimeras for targeted
degradation of cell surface and extracellular proteins. _Nat. Biotechnol._ 41, 273–281 (2023). Article PubMed CAS Google Scholar * Mayle, K. M., Le, A. M. & Kamei, D. T. The
intracellular trafficking pathway of transferrin. _Biochim. Biophys. Acta Gen. Subj._ 1820, 264–281 (2012). Article CAS Google Scholar * Jonker, C. T. H. et al. Accurate measurement of
fast endocytic recycling kinetics in real time. _J. Cell Sci._ jcs.231225, https://doi.org/10.1242/jcs.231225 (2019). * Awuah, P., Bera, T. K., Folivi, M., Chertov, O. & Pastan, I.
Reduced Shedding of Surface Mesothelin Improves Efficacy of Mesothelin-Targeting Recombinant Immunotoxins. _Mol. Cancer Therapeutics_ 15, 1648–1655 (2016). Article CAS Google Scholar *
Essaghir, A. & Demoulin, J.-B. A Minimal Connected Network of Transcription Factors Regulated in Human Tumors and Its Application to the Quest for Universal Cancer Biomarkers. _PLoS ONE_
7, e39666 (2012). Article ADS PubMed PubMed Central CAS Google Scholar * Shen, Y. et al. Transferrin receptor 1 in cancer: a new sight for cancer therapy. _Am. J. Cancer Res_ 8,
916–931 (2018). PubMed PubMed Central CAS Google Scholar * Rahman, S. A., Yokoyama, M., Nishio, S. & Takeuchi, M. Flow cytometric evaluation of transferrin receptor in transitional
cell carcinoma. _Urol. Res._ 25, 325–329 (1997). Article PubMed CAS Google Scholar * Meyers, R. M. et al. Computational correction of copy number effect improves specificity of
CRISPR–Cas9 essentiality screens in cancer cells. _Nat. Genet_ 49, 1779–1784 (2017). Article PubMed PubMed Central CAS Google Scholar * Johnsen, K. B., Burkhart, A., Thomsen, L. B.,
Andresen, T. L. & Moos, T. Targeting the transferrin receptor for brain drug delivery. _Prog. Neurobiol._ 181, 101665 (2019). Article PubMed CAS Google Scholar * Kariolis, M. S. et
al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. _Sci. Transl. Med._ 12, eaay1359 (2020). Article PubMed CAS
Google Scholar * Crook, Z. R. et al. A TfR-Binding Cystine-Dense Peptide Promotes Blood–Brain Barrier Penetration of Bioactive Molecules. _J. Mol. Biol._ 432, 3989–4009 (2020). Article
PubMed PubMed Central CAS Google Scholar * Zhang, D. et al. _Transferrin Receptor Targeting Chimeras (TransTACs) for Membrane Protein Degradation_.
http://biorxiv.org/lookup/doi/10.1101/2023.08.10.552782, https://doi.org/10.1101/2023.08.10.552782 (2023). * Crook, Z. R. et al. Ex silico engineering of cystine-dense peptides yielding a
potent bispecific T cell engager. _Sci. Transl. Med._ 14, eabn0402 (2022). Article PubMed PubMed Central CAS Google Scholar * Crook, Z. R., Sevilla, G. P., Mhyre, A. J. & Olson, J.
M. Mammalian Surface Display Screening of Diverse Cystine-Dense Peptide Libraries for Difficult-to-Drug Targets. In _Genotype Phenotype Coupling_ (eds. Zielonka, S. & Krah, S.) vol. 2070
363–396 (Springer US, 2020). * Lawson, N. L. et al. Mapping the binding sites of antibodies utilized in programmed cell death ligand-1 predictive immunohistochemical assays for use with
immuno-oncology therapies. _Mod. Pathol._ 33, 518–530 (2020). Article PubMed CAS Google Scholar * Thomas, R. & Weihua, Z. Rethink of EGFR in Cancer With Its Kinase Independent
Function on Board. _Front. Oncol._ 9, 800 (2019). Article PubMed PubMed Central Google Scholar * Troiani, T. et al. Therapeutic value of EGFR inhibition in CRC and NSCLC: 15 years of
clinical evidence. _ESMO Open_ 1, e000088 (2016). Article PubMed PubMed Central Google Scholar * Eskilsson, E. et al. EGFR heterogeneity and implications for therapeutic intervention in
glioblastoma. _Neuro Oncol._ 20, 743–752 (2018). Article PubMed CAS Google Scholar * Boeckx, C. et al. Anti-Epidermal Growth Factor Receptor Therapy in Head and Neck Squamous Cell
Carcinoma: Focus on Potential Molecular Mechanisms of Drug Resistance. _Oncologist_ 18, 850–864 (2013). Article PubMed PubMed Central CAS Google Scholar * Passaro, A., Jänne, P. A.,
Mok, T. & Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. _Nat. Cancer_ 2, 377–391 (2021). Article PubMed CAS Google Scholar * Weihua, Z. et al. Survival of
Cancer Cells Is Maintained by EGFR Independent of Its Kinase Activity. _Cancer Cell_ 13, 385–393 (2008). Article PubMed PubMed Central Google Scholar * Katreddy, R. R. et al. Targeted
reduction of the EGFR protein, but not inhibition of its kinase activity, induces mitophagy and death of cancer cells through activation of mTORC2 and Akt. _Oncogenesis_ 7, 5 (2018). Article
PubMed PubMed Central Google Scholar * Roovers, R. C. et al. Efficient inhibition of EGFR signalling and of tumour growth by antagonistic anti-EGFR Nanobodies. _Cancer Immunol.
Immunother._ 56, 303–317 (2007). Article PubMed CAS Google Scholar * Schmitz, K. R., Bagchi, A., Roovers, R. C., van Bergen en Henegouwen, P. M. P. & Ferguson, K. M. Structural
Evaluation of EGFR Inhibition Mechanisms for Nanobodies/VHH Domains. _Structure_ 21, 1214–1224 (2013). Article PubMed PubMed Central CAS Google Scholar * French, A. R., Tadaki, D. K.,
Niyogi, S. K. & Lauffenburger, D. A. Intracellular Trafficking of Epidermal Growth Factor Family Ligands Is Directly Influenced by the pH Sensitivity of the Receptor/Ligand Interaction.
_J. Biol. Chem._ 270, 4334–4340 (1995). Article PubMed CAS Google Scholar * Fleishman, S. J. et al. RosettaScripts: A Scripting Language Interface to the Rosetta Macromolecular Modeling
Suite. _PLoS One_ 6, e20161 (2011). Article ADS PubMed PubMed Central CAS Google Scholar * Leaver-Fay, A. et al. Rosetta3. in _Methods in Enzymology_ vol. 487, 545–574 (Elsevier,
2011). * Ogiso, H. et al. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. _Cell_ 110, 775–787 (2002). Article PubMed CAS Google
Scholar * Cochran, J. R., Kim, Y.-S., Olsen, M. J., Bhandari, R. & Wittrup, K. D. Domain-level antibody epitope mapping through yeast surface display of epidermal growth factor receptor
fragments. _J. Immunological Methods_ 287, 147–158 (2004). Article PubMed CAS Google Scholar * Ahn, G. et al. Elucidating the cellular determinants of targeted membrane protein
degradation by lysosome-targeting chimeras. _Science_ 382, eadf6249 (2023). Article PubMed PubMed Central CAS Google Scholar * Panaccio, M. et al. Heterogeneity of the human transferrin
receptor and use of anti‐transferrin receptor antibodies to detect tumours in vivo. _Immunol. Cell Biol._ 65, 461–472 (1987). Article PubMed Google Scholar * Greig, M. J. et al. Effects
of Activating Mutations on EGFR Cellular Protein Turnover and Amino Acid Recycling Determined Using SILAC Mass Spectrometry. _Int. J. Cell Biol._ 2015, 1–8 (2015). Article ADS Google
Scholar * Niewoehner, J. et al. Increased Brain Penetration and Potency of a Therapeutic Antibody Using a Monovalent Molecular Shuttle. _Neuron_ 81, 49–60 (2014). Article PubMed CAS
Google Scholar * Chamberlain, P. P. & Hamann, L. G. Development of targeted protein degradation therapeutics. _Nat. Chem. Biol._ 15, 937–944 (2019). Article PubMed CAS Google Scholar
* Cross, D. A. E. et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. _Cancer Discov._ 4, 1046–1061 (2014). Article PubMed
PubMed Central CAS Google Scholar * Pinilla-Macua, I., Grassart, A., Duvvuri, U., Watkins, S. C. & Sorkin, A. EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis
in tumors in vivo. _eLife_ 6, e31993 (2017). Article PubMed PubMed Central Google Scholar * Yuan, Z. et al. An A13 Repeat within the 3′-Untranslated Region of Epidermal Growth Factor
Receptor (EGFR) Is Frequently Mutated in Microsatellite Instability Colon Cancers and Is Associated with Increased EGFR Expression. _Cancer Res._ 69, 7811–7818 (2009). Article PubMed
PubMed Central CAS Google Scholar * Ashraf, S. Q. et al. Direct and immune mediated antibody targeting of _ERBB_ receptors in a colorectal cancer cell-line panel. _Proc. Natl Acad. Sci.
USA_ 109, 21046–21051 (2012). Article ADS PubMed PubMed Central CAS Google Scholar * Morris, V. K. et al. Treatment of Metastatic Colorectal Cancer: ASCO Guideline. _JCO_ 41, 678–700
(2023). Article Google Scholar * Pastore, S., Lulli, D. & Girolomoni, G. Epidermal growth factor receptor signalling in keratinocyte biology: implications for skin toxicity of tyrosine
kinase inhibitors. _Arch. Toxicol._ 88, 1189–1203 (2014). Article PubMed CAS Google Scholar * Couch, J. A. et al. Addressing Safety Liabilities of TfR Bispecific Antibodies That Cross
the Blood-Brain Barrier. _Sci. Transl. Med_. 5, 183ra57 (2013). * Ogama, Y. et al. Phase 1 Clinical Trial of PPMX‐T003, a Novel Human Monoclonal Antibody Specific for Transferrin Receptor 1,
to Evaluate Its Safety, Pharmacokinetics, and Pharmacodynamics. _Clin. Pharm. Drug Dev._ 12, 579–587 (2023). Article CAS Google Scholar * Arguello, A. et al. Molecular architecture
determines brain delivery of a transferrin receptor–targeted lysosomal enzyme. _J. Exp. Med._ 219, e20211057 (2022). Article PubMed PubMed Central CAS Google Scholar Download references
ACKNOWLEDGEMENTS The authors wish to thank Pauline Bariola and Jacob Felcyn for development of the SE-HPLC methodology, Jeannette Bannink for help with ELISA development and PK data
interpretation and analysis, Akinsola Oyelakin for help with tissue processing, Andrew J. Mhyre for in vivo study discussions, and Connor Burns for slide scanning. Figures 1A, B, 2B, 3D,
6A, 8C, and Supplementary Fig. 1A were created with Biorender. The project was supported by NIH grant R01 CA223674 (to J.M.O.); Run of Hope; Project Violet; Seattle Children’s Research
Institute startup funds; Blaze Bioscience, Inc; and Cyclera Therapeutics, Inc. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Cyclera Therapeutics Inc, Seattle, WA, 98115, USA Zachary R.
Crook, Gregory P. Sevilla & Natalie W. Nairn * Blaze Bioscience Inc, Seattle, WA, 98109, USA Zachary R. Crook, Gregory P. Sevilla, Pamela Young, Tinh-Doan Phi & Natalie W. Nairn *
Clinical Research Division, Fred Hutchinson Research Center, Seattle, WA, 98109, USA Zachary R. Crook, Gregory P. Sevilla, Jason Price & James M. Olson * Ben Towne Center for Childhood
Cancer Research, Seattle Children’s Research Institute, Seattle, WA, 98105, USA Emily J. Girard & James M. Olson * NW Biosensor, Seattle, WA, 98103, USA Monique L. Howard Authors *
Zachary R. Crook View author publications You can also search for this author inPubMed Google Scholar * Gregory P. Sevilla View author publications You can also search for this author
inPubMed Google Scholar * Pamela Young View author publications You can also search for this author inPubMed Google Scholar * Emily J. Girard View author publications You can also search for
this author inPubMed Google Scholar * Tinh-Doan Phi View author publications You can also search for this author inPubMed Google Scholar * Monique L. Howard View author publications You can
also search for this author inPubMed Google Scholar * Jason Price View author publications You can also search for this author inPubMed Google Scholar * James M. Olson View author
publications You can also search for this author inPubMed Google Scholar * Natalie W. Nairn View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
Z.R.C., J.P., J.M.O, and N.W.N. conceptualized the research. Z.R.C., G.P.S., P.Y., E.J.G., T-D.P., and M.H. designed the experiments. Z.R.C., G.P.S., P.Y., E.J.G., T-D.P., and M.L.H.
performed the experiments and/or contributed to data analysis. Z.R.C. and M.L.H. produced figures. J.M.O. and N.W.N. contributed to funding acquisition. Z.R.C., G.P.S., E.J.G., J.M.O., and
N.W.N. supervised researchers. Z.R.C., E.J.G., M.L.H., J.M.O., and N.W.N. wrote the manuscript. All authors contributed to review of the manuscript. CORRESPONDING AUTHOR Correspondence to
Natalie W. Nairn. ETHICS DECLARATIONS COMPETING INTERESTS Cyclera Therapeutics Inc. retains intellectual property rights to the technology described in this manuscript. Z.R.C., G.P.S., and
N.W.N. own stock in and are or were employees of Cyclera. J.M.O. owns stock in and is an advisor of Cyclera. Z.R.C., J.M.O., and N.W.N. are inventors on patent applications for this
technology. Z.R.C., G.P.S., P.Y., T-D.P., and N.W.N were previously employees of Blaze and may own stock in Blaze. E.J.G., M.L.H., and J.P. have no competing interests. PEER REVIEW PEER
REVIEW INFORMATION _Nature Communications_ thanks Sonia Levi, who co-reviewed with Andrea Moro; Qiwei Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of
this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 PEER REVIEW FILE SOURCE DATA SOURCE
DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial
use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or
other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Crook, Z.R., Sevilla,
G.P., Young, P. _et al._ CYpHER: catalytic extracellular targeted protein degradation with high potency and durable effect. _Nat Commun_ 15, 8731 (2024).
https://doi.org/10.1038/s41467-024-52975-2 Download citation * Received: 25 March 2024 * Accepted: 27 September 2024 * Published: 09 October 2024 * DOI:
https://doi.org/10.1038/s41467-024-52975-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative