- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Microglia play important roles in brain development and homeostasis by removing dying neurons through efferocytosis. Morphological changes in microglia are hallmarks of many
neurodegenerative conditions, such as Niemann-Pick disease type C. Here, NPC1 loss causes microglia to shift from a branched to an ameboid form, though the cellular basis and functional
impact of this change remain unclear. Using zebrafish, we show that NPC1 deficiency causes an efferocytosis-dependent expansion of the microglial gastrosome, a collection point for engulfed
material. In vivo and in vitro experiments on microglia and mammalian macrophages demonstrate that NPC1 localizes to the gastrosome, and its absence leads to cholesterol accumulation in this
compartment. NPC1 loss and neuronal cell death synergistically affect gastrosome size and cell shape, increasing the sensitivity of NPC1-deficient cells to neuronal cell death. Finally, we
demonstrate conservation of cholesterol accumulation and gastrosome expansion in NPC patient-derived fibroblasts, offering an interesting target for further disease investigation. SIMILAR
CONTENT BEING VIEWED BY OTHERS LOSS OF NPC1 ENHANCES PHAGOCYTIC UPTAKE AND IMPAIRS LIPID TRAFFICKING IN MICROGLIA Article Open access 24 February 2021 TREX1 IS REQUIRED FOR MICROGLIAL
CHOLESTEROL HOMEOSTASIS AND OLIGODENDROCYTE TERMINAL DIFFERENTIATION IN HUMAN NEURAL ASSEMBLOIDS Article Open access 21 December 2023 LOSS OF CLN3 IN MICROGLIA LEADS TO IMPAIRED LIPID
METABOLISM AND MYELIN TURNOVER Article Open access 22 October 2024 INTRODUCTION Efferocytosis, the phagocytosis of dying cells, plays a crucial role in various biological processes,
including development, tissue homeostasis and numerous diseases. Microglia, the major efferocytes of the brain, must efficiently digest engulfed neurons, and process the resulting breakdown
products, through mechanisms that are poorly understood. Recent data indicate that such internal processes become limiting in contexts where microglia engulf large numbers of dead neurons,
such as during brain development and neurodegenerative disease1,2. In these contexts, microglia also adopt a similar gene expression signature that is characterized by the upregulation of
genes involved in phagocytosis and lipid metabolism and transport3,4 reviewed in ref. 5). Indeed, the processing of lipids originating from neuronal degradation appears to be particularly
challenging for microglia as in many neurodegenerative conditions these cells often contain abnormal lipid inclusions6,7,8,9. One such condition is Niemann-Pick disease type C (NPC), caused
by mutations in the NPC intracellular cholesterol transporter 110, resulting in the accumulation in endo-lysosomal compartments of cholesterol, its derivatives and glycolipids11,12,13,14. In
NPC, these endo-lysosomal defects have been linked with the loss of Purkinje cells15,16,17 via mechanisms that are compatible with neuronal apoptosis18,19,20 and necroptosis21,22. Microglia
have been shown to also express _NPC1_ and _NPC2_23,24,25 and to display impaired lipid processing in NPC25,26,27. These cells show the acquisition of a DAM-like signature25 and a
disease-associated ameboid morphology characterized by swelling and the loss of long extensions, a phenotype that gets stronger as the disease progresses27,28,29,30. The cellular mechanisms
directing this branched-to-ameboid transition remain elusive. It is unclear whether this transformation is solely prompted by lysosomal defects or if the efferocytic function of microglia
also plays a role. Indeed, there is a significant knowledge gap regarding how microglia process lipids, particularly when these cells are under high phagocytic stress. An intriguing player
in efferocytosis in microglia and macrophages is the gastrosome, a single compartment that serves as a point of convergence for all phagosomes containing engulfed apoptotic material2. This
compartment is characterized by a set of unique features such as a distinct molecular signature, being LAMP1 positive and Cathepsin D negative, and an electron-lucent lumen containing
membrane fragments2. Furthermore, it has been observed to expand in response to increased cell death and because of mutations in SLC37A2, a putative Glucose-6-phosphate transporter
responsible for phagosomal shrinkage2. In this study, in vivo imaging and perturbation in the zebrafish identify the gastrosome as an important link between neuronal cell death, cholesterol
transport defects in NPC and microglial morphological changes. We show that NPC1 localizes to the gastrosome, which becomes the major site of cholesterol accumulation in the absence of this
transporter. The dynamic expansion of the gastrosome leads to NPC-deficient microglia adopting an ameboid phenotype, which can be further enhanced by increasing neuronal apoptosis, providing
a mechanistic explanation for this known hallmark of NPC disease progression. We show that NPC1 loss renders microglia more sensitive to increased neuronal apoptosis, leading to the cell
death of microglia themselves, a finding that has important implications for our understanding of NPC disease. Finally, we demonstrate cholesterol accumulation in the gastrosome of
fibroblasts derived from NPC patients, which could have significant implications for understanding the cellular mechanisms underlying this and other neurodegenerative diseases. RESULTS LOSS
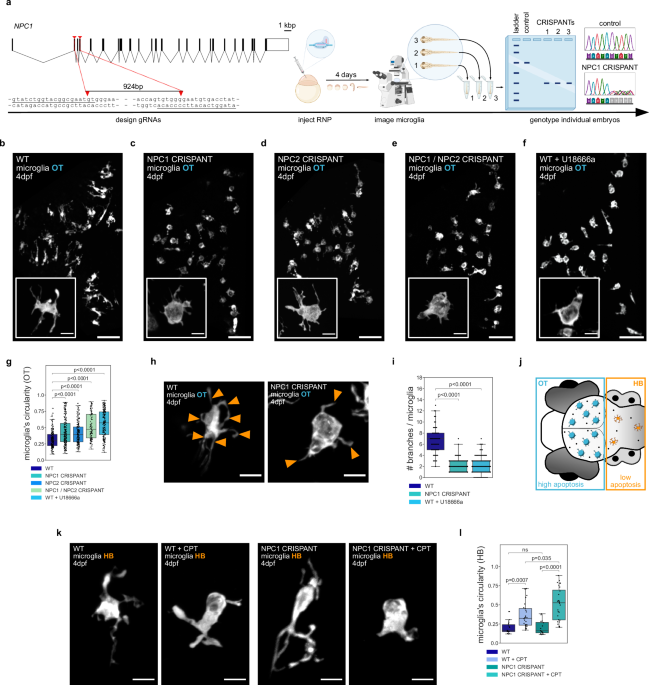
OF NPC1 RESULTS IN PHAGOCYTIC MICROGLIA ADOPTING AN AMEBOID MORPHOLOGY We investigated the role of NPC1 in microglia using a CRISPR/Cas9 strategy31 see schematics in Fig. 1a) to generate F0
somatic _NPC1_ mutants (referred to as CRISPANTs and NPC1 defective) directly in the background of fluorescently labeled microglia (_Tg(mpeg1.1:EGFP-CAAX)_)2 or (_TgBAC(csf1ra:GAL4-VP16)_,
_Tg(UAS-E1B:NTR-mCherry)_)32. Guide RNAs (gRNAs) were designed to create a 924bp-long deletion in the region spanning from exon 2 to exon 6 in _NPC1_ locus and were injected together with
Cas9 into one-cell stage zebrafish eggs. Embryos were examined four days after injection to investigate the role of NPC1 in microglia that populate the optic tectum (OT), as these cells are
phagocytically active due to neuronal developmental apoptosis33. Microglia in these larvae showed an ameboid morphology, being more circular with fewer cellular extensions (Fig. 1b, c; Fig.
1g; Fig. 1h, i; Supplementary Movie 1), a phenotype that is characteristic of NPC27,28,29,30. Genomic (Supplementary Fig. 1a) and Western Blot analysis (Supplementary Fig. 1b, c; see
Material and Methods) confirmed editing of the _NPC1_ locus and a significant protein loss in NPC1 deficient embryos. We further investigated the role of NPC1 in microglia by using the NPC1
inhibitor U18666a34 and by targeting NPC2, known to work together with NPC1 in cholesterol transport35,36. Deletion in _NPC2_ locus resulted in ameboid microglia (Fig. 1d, g), and embryos
with double NPC1/NPC2 deficiency exhibited a phenotype similar to that of single NPC1 or NPC2 deficient embryo (Fig. 1e, g). In addition, wild-type embryos displayed ameboid microglia 4–6 h
after incubation with the NPC1 inhibitor U18666a (Fig. 1f, g, i). We also examined a published _NPC1_ allele (_npc1__hg37_ 37) and found that these embryos were also characterized by the
presence of ameboid microglia (Supplementary Fig. 1d). As shown in mice26, fish embryos deficient in NPC1 also exhibited upregulation of pro-inflammatory markers such as Interleukin-1
(_IL1_) and Nuclear factor NF-kappa-B subunit 2 (_NFKB2_)38, and to a lesser extent Interleukin-6 (_IL6_)39,40 (Supplementary Fig. 1g). Interestingly, in the absence of microglia, as
observed in _irf8__(−/−)_ mutants41 that are also NPC1 deficient, the expression of these markers increased further, indicating a potential anti-inflammatory role for microglia in NPC1
deficient embryos (Supplementary Fig. 1g). We next took advantage of brain-wide imaging in the zebrafish model to investigate potential regional differences in the microglial phenotype. This
revealed that compared to ameboid microglia found in the optic tectum (OT), microglia in the adjacent hindbrain (HB) maintained their typical branched appearance (see schematics in Fig. 1j;
Supplementary Fig. 1e, f). First, loss of NPC1 has no impact on the overall rate of neuronal apoptosis and neuronal removal at this stage, as shown by quantifying neuronal cell death in
NPC1 deficient brains that also lack microglia (Supplementary Fig. 1h, see Material and Methods41) and the rate of phagosome formation in NPC1 deficient microglia (Supplementary Fig. 1i; see
Material and Methods42). However, compared to the HB, the OT is known to display higher levels of developmental neuronal apoptosis33,43, suggesting that NPC1 might exert its influence
specifically on phagocytically active microglia. We tested this hypothesis by taking three different approaches. First, we increased apoptosis in the HB using Camptothecin (CPT), an
established compound that kills proliferating neurons2,44. Adding CPT significantly increased the rate of neuronal cell death, affecting both wild-type and NPC1 deficient microglia (Fig. 1k,
l). However, the NPC1 deficient microglia underwent a more pronounced change (Fig. 1k, l). Second, we used a genetic approach based on nitroreductase (NTR) expression in HB neurons
(_Tg(NBT:DLexPR::NTR-mCherry)_, referred to as HB-NTR-mCherry; see schematic in Supplementary Fig. 1j)33,45,46. While NTR-based induction of apoptosis did not have a detectable effect on the
morphology of HB microglia in wild-type controls, in the absence of NPC1, HB microglia became more ameboid (Supplementary Fig. 1k and l). Finally, we did the opposite experiment and reduced
neuronal efferocytosis by knocking down BAI1, a phagocytic receptor active in microglia46. In NPC1-deficient embryos, this led to a shift in the microglia morphology from ameboid to more
ramified (Supplementary Fig. 1m, n). Thus, our data reveal that NPC1 loss impacts microglia morphology, with these cells adopting an ameboid phenotype. Cells exposed to high neuronal
apoptosis show this phenotype, while others adopt it when neuronal apoptosis is increased locally, indicating that the effect is more prevalent in phagocytically active microglia. NPC1
DEFICIENCY LEADS TO THE EXPANSION OF THE GASTROSOME IN PHAGOCYTIC MACROPHAGES AND MICROGLIA Imaging of NPC1 and NPC2 deficient microglia revealed the presence of a large vesicle that
occupied most of their soma (Fig. 2a, b, Supplementary Movie 2), a feature absent in control cells (Fig. 2a, b; Supplementary Movie 2). This vesicle exhibited hallmarks of the gastrosome,
as, for example, its size fluctuated over time in response to fusions with all incoming phagosomes (Fig. 2c, d, Supplementary Movie 32). Moreover, the inhibition of efferocytosis through
BAI1 knock-down reduced the diameter of the enlarged vesicle observed in NPC1-deficient cells (Supplementary Fig. 2a), linking the size of this vesicle to the phagocytic activity of
microglia. In RAW 264.7 mammalian macrophages (referred to as RAW macrophages) the gastrosome can be visualized by feeding these cells with fluorescently tagged apoptotic HeLa cells
(referred to as HeLa-fed RAW macrophages)2. Indeed, following feeding, the gastrosome becomes both fluorescent and light-scattering, a characteristic that allows us to differentiate it from
other HeLa cell inclusions (Fig. 2e, orange arrowhead2). When we treated HeLa-fed RAW macrophages with U18666a to inhibit NPC1, the labelled compartment expanded significantly (Fig. 2e, f;
orange arrowhead), supporting the hypothesis that the large vesicle found in the absence of NPC1 is indeed the gastrosome. Moreover, this compartment displayed the typical molecular and
ultrastructural features of the gastrosome. It was LAMP1 positive (Fig. 2e) and Cathepsin D negative (Supplementary Fig. 2b, c) and had an electron-lucent lumen that contained membrane
fragments (Fig. 2g, h2). In comparison, lysosomes in U18666a treated HeLa-fed RAW macrophages are smaller and electron-dense (Fig. 2g; the gastrosome and lysosomes are marked by black and
colored boxes, respectively). Moreover, an impact of NPC1 knockout on autophagy appears unlikely, as the gastrosome is negative for LC3B, a canonical autophagy marker (Supplementary Fig. 2d,
e). Taken together these data indicate that lack of the NPC1 cholesterol transporter in phagocytic macrophages and microglia leads to the expansion of the gastrosome, suggesting a possible
role for this compartment in NPC1-mediated cholesterol trafficking. NPC1 LOSS LEADS TO CHOLESTEROL AND GM1 GANGLIOSIDE ACCUMULATION IN THE GASTROSOME The absence of NPC1 is known to result
in the accumulation of unesterified cholesterol and gangliosides within endo-lysosomal compartments14. Embryos lacking NPC1 exhibited cholesterol accumulation (Supplementary Fig. 3a) as
shown by using TopFluor cholesterol and large needle-shaped crystals, a recognized sign of cholesterol overload47, colocalizing with the microglia (Supplementary Fig. 3b). At higher
magnification, these crystals were found to be inside the gastrosome (green arrowhead in Fig. 3c) that also contained high TopFluor cholesterol levels (orange arrowhead in Fig. 3b) and was
surrounded by TopFluor cholesterol-rich small vesicles (yellow arrows in Fig. 3b), likely to be endo-lysosomal compartments11,12,13,14. The accumulation of cholesterol in the gastrosome is
linked to efferocytosis. Indeed, knocking down BAI1, which reduces efferocytosis, resulted in a reduction in both the size of the gastrosome (Supplementary Fig. 2a) and its TopFluor
cholesterol content (Supplementary Fig. 3c)46. Next, we turned to RAW macrophages and treated these cells with U18666a to inhibit NPC1. As expected, 24 hours after treatment, filipin
staining48 showed unesterified cholesterol accumulation in a puncta-like pattern, consistent with previous research (Fig. 3n, p, blue arrow)11,12,13,14. Interestingly, feeding
U18666a-treated RAW macrophages with apoptotic HeLa cells led to a dramatic change in unesterified cholesterol distribution characterized by an additional buildup in the expanded gastrosome
(Fig. 3p, indicated by an orange arrowhead). Lipidomic analysis further supported these data, showing that free (or unesterified) cholesterol and esterified cholesterol accumulated upon NPC1
inhibition in both RAW macrophages and HeLa-fed RAW macrophages (Supplementary Fig. 3d, Supplementary Table 1 and 2). To test the involvement of the gastrosome in lipid handling we
exploited the previous finding that the gastrosome expands when the Glucose-6-phosphate transporter SLC37A2 is absent2. In SLC37A2 mutants, the expanded gastrosome was characterized by a
distinct bubble-like morphology (Fig. 3d, g2) and contained low GM1 ganglioside levels (Fig. 3j, l). In contrast, it appeared looser (Fig. 3e, g) with elevated GM1 ganglioside levels in the
absence of NPC1 (Fig. 3i, l). Interestingly, in double NPC1 and SLC37A2 embryos, microglia were characterized by one expanded gastrosome with features of both single knockouts, namely a
bubble-like shape (Fig. 3f, g) and accumulation of GM1 gangliosides (Fig. 3k, l), indicating that both SLC37A2 and NPC1 play distinct yet complementary roles in regulating gastrosome
morphology and handling of sugars and lipids. Similarly, HeLa-fed RAW macrophages mutant for SLC37A2 showed increased cholesterol levels in the gastrosome after inhibition of NPC1 (Fig. 3q,
r, s) highlighting a potential role for this compartment in NPC1-mediated cholesterol handling. Interestingly, real-time imaging revealed normal phagosomal behavior and shrinkage in NPC1
deficient microglia (Supplementary Fig. 4a, b, c, Supplementary Movie 4), supporting the hypothesis of NPC1 might exert its function downstream of phagosomal shrinkage at the level of the
gastrosome. Next, we utilized a CRISPR-mediated approach to tag the endogenous NPC1 protein in both zebrafish and RAW macrophages to investigate its expression and localization on the
gastrosome. As expected NPC1 is expressed in RAW macrophages and localized on small vesicles that are also LAMP1 positive (Fig. 4a49). Feeding these RAW macrophages with apoptotic HeLa cells
revealed localization of NPC1 also on the gastrosome (Fig. 4b, orange arrowhead) which expanded upon NPC1 inhibition using U18666a (Fig. 4b, orange arrowhead). In line with this, the
endogenous tagging of NPC1 in zebrafish revealed expression in microglia and localization of this transporter on the gastrosome, which expanded when embryos were treated with U18666a (Fig.
4c). Additionally, NPC1 was observed on smaller vesicles likely of endo-lysosomal nature (Fig. 4c and Supplementary Fig. 4d). NPC1 IMPACT ON THE GASTROSOME IS CONSERVED IN NIEMANN-PICK TYPE
C HUMAN PATIENT-DERIVED CELLS We next asked if the mechanism linking NPC1 to changes in gastrosomal morphology was conserved in human Niemann-Pick type C syndrome. We began by first
establishing the impact of NPC1 inhibition on the gastrosome in human microglia, using the cell line HMC350. Feeding these cells fluorescent apoptotic HeLa cells resulted in the accumulation
of the labeled material in a compartment that had defining hallmarks of the gastrosome (Supplementary Fig. 5a, c). In addition, this compartment contained high cholesterol levels
(Supplementary Fig. 5d) and expanded upon U18666a treatment (Supplementary Fig. 5b), pointing to the high conservation of cholesterol sorting mechanisms in human microglia. Next, we obtained
two NPC human patient-derived fibroblast lines with different mutations in _NPC1_ (GM18393, referred to as NPC-P1 and GM17919, referred to as NPC-P2) and one healthy individual control
(GM03652, referred to as HI). As the size of the gastrosome is coupled to efferocytosis, we first determined the ability of fibroblasts to engulf apoptotic cells. Twenty-four hours after
exposure to fluorescently labeled apoptotic HeLa cells, we observed the accumulation of fluorescence in the gastrosome, as defined using standard criteria (Fig. 5a–d). A conspicuous
ultrastructural difference between the gastrosome in immune cells and fibroblasts was the elevated content within the lumen of the fibroblast gastrosome, suggesting that fibroblasts exhibit
a lower efficiency in digesting apoptotic material compared to professional phagocytes such as microglia and macrophages (Fig. 5d compare with Fig. 2g, h). Nevertheless, the size of the
gastrosome was increased in HeLa-fed NPC-P1 and NPC-P2 cells when compared to HI control cells (Fig. 5a, c, dark blue violin plot), as was the accumulation of cholesterol in the gastrosome
(Fig. 5e, indicated by an orange arrowhead and Fig. 5g, dark blue box plot) and in the endo-lysosomal pool (Fig. 5e, marked by blue box and Fig. 5f, dark blue box plot). Both features of NPC
patient derived cells could be recapitulated in HI fibroblasts treated with U18666a, supporting the conclusion that these gastrosomal changes in patient-derived cells are due to reduced
NPC1 activity (Fig. 5a, c, e, f, g, dark blue plots). To determine if the gastrosomal expansion observed in HeLa-fed patient fibroblasts is due to cholesterol trafficking defects, we
conducted experiments using 2-hydroxypropyl-γ-cyclodextrin (HPγCD)51,52. This drug has shown efficacy in reducing cholesterol accumulation in fibroblasts from NPC patients by activating the
transcription factor EB (TFEB), boosting cholesterol transport to the endoplasmic reticulum and autophagy53. Incubation of HeLa-fed HI fibroblasts with HPγCD did not affect gastrosomal size
(Fig. 5c, light blue violin plot) nor cholesterol levels (Fig. 5e, f, g, light blue box plots). In contrast, NPC1 deficient fibroblast lines exhibited a gastrosomal size reduction (Fig. 5c,
light blue violin plot), decreased cholesterol levels in the endo-lysosomal pool (Fig. 5e, marked by blue box and Fig. 5f, light blue box plot), and a noticeable downward trend of
cholesterol levels in the gastrosome (Fig. 5e, indicated by an orange arrowhead and Fig. 5g, light blue box plot). While we cannot rule out the possibility that the impact of HPγCD on the
gastrosome is indirect and potentially mediated by its influence on upstream components of the phagocytic pathway, such as lysosomes, the modifications observed in gastrosome size and
content resulting from HPγCD strongly suggest a substantial involvement of this vesicle in lipid metabolism. These observations could offer mechanistic explanations for the poorly understood
late-onset phenotypes observed in NPC and other lysosomal storage disorders, where cells have been shown to expand and form vacuoles (for review see ref. 54). CHANGES IN THE GASTROSOME AND
INCREASED CHOLESTEROL ACCUMULATION UNDERLIE NPC1’S IMPACT ON MICROGLIAL MORPHOLOGY Next, we investigated the impact of NPC1 activity on the gastrosome and its influence on microglial
morphology and function. The transition from branched to ameboid morphology in microglia caused by NPC1 inhibition via U18666a (Fig. 1f, g, i) could be reversed by changing the medium and
rinsing off the NPC1 inhibitor. This led to deflation of the gastrosome and microglia to regain their branched morphology within a few hours (Supplementary Fig. 6a, b–d and Supplementary
Movie 5 and 6). This suggests a direct link between NPC1 activity and the physical characteristics of the gastrosome as well as microglial shape. Quantifying gastrosome size, cholesterol
content, and microglial morphology before (T0) and after (T1) washing off the drug further validated this relationship (Fig. 6a–e). Resumed NPC1 activity led to reduced cholesterol levels in
the gastrosome, decreased vesicle size, and the recovery of the microglial morphology, indicating a causal connection between these features. As upon NPC1 inhibition, the gastrosome becomes
a significant site of cholesterol accumulation with a dramatic effect on microglial morphology, we wondered if boosting cholesterol trafficking in NPC1-deficient microglia using HPγCD would
lead to improved microglial phenotypes. First, treating wild-type embryos with HPγCD had no effect on cell morphology (Fig. 6f, h), however, HPγCD treatment of NPC1 deficient embryos
reduced the size of the gastrosome (Fig. 6g, i) and the severity of the ameboid microglia phenotype (Fig. 6g, h), supporting our hypothesis that the ameboid microglial phenotype observed in
phagocytic NPC deficient cells is linked to cholesterol buildup and expansion of the gastrosome. Next, we tested how NPC1 deficient microglia respond to challenges, particularly heightened
neuronal death, a characteristic of later stages of NPC disease27,28,29,30. To increase neuronal cell death, we used Camptothecin (CPT), an established inducer of neuronal apoptosis44,2.
Wild-type microglia responded by adopting a more ameboid morphology (Fig. 6j, k and Supplementary Fig. 6e). Notably, CPT administration to NPC1-deficient microglia intensified the ameboid
morphology (Fig. 6j, k and Supplementary Fig. 6e), further strengthening the link between NPC1 loss and the rate of neuronal apoptosis on microglial morphology. The experiments also revealed
a significant increase in the size of the gastrosome (Fig. 6l), especially notable in NPC1-deficient microglia, indicating a synergistic effect between NPC1 loss and increased phagocytosis
on gastrosomal expansion. We next leveraged our ability to monitor microglial dynamics in intact living zebrafish brains to investigate the subsequent impact of high neuronal cell death and
NPC1 deficiency on microglia. While CPT treatment alone does not affect microglial viability in wild-type, in NPC1 deficient embryos, CPT-induced neuronal apoptosis led to a further
reduction in the already low number of microglia compared to wild-type (Fig. 6m). Live imaging of CPT-treated NPC1 deficient embryos revealed that microglial reductions can be explained by
the death of these cells, which are subsequently cannibalized via efferocytosis by surrounding microglia (Fig. 6n, Supplementary Fig. 6h, Supplementary Movie 7). We tested whether the death
of NPC1-deficient microglia depends on efferocytosis by reducing neuronal uptake via BAI1 knockdown, which led to increased microglial survival (Supplementary Fig. 6g). Similarly, improving
cholesterol trafficking using HPγCD showed a trend toward fewer microglia dying (Supplementary Fig. 6f). Thus, the loss of NPC1 renders microglia highly sensitive to neuronal cell death. In
response to high neuronal apoptotic levels, phagocytic microglia die and are eliminated by other microglia. This finding can have implications for understanding microglial responses in NPC
and other neurodegenerative diseases. DISCUSSION In the absence of the cholesterol transporter NPC1, microglia are known to undergo a branched-to-ameboid transition. This investigation
unveils the role of the gastrosome in linking neuronal cell death and cholesterol transport defects with morphological changes in microglia in NPC disease. While not excluding the potential
roles of NPC1 in other endo-lysosomal system compartments, this work uncovers a function for NPC1 in regulating cholesterol trafficking at the level of the gastrosome during efferocytosis.
The transmembrane transporter NPC1 localizes to the gastrosome, acting as a player in preventing cholesterol accumulation within this compartment downstream of neuronal engulfment.
Furthermore, it shows the exquisite sensitivity of the gastrosomal shape and size to alterations in lipid transport. The expansion of the gastrosome in the absence of NPC1’s activity
coincides with microglia becoming ameboid, but when these cells regain NPC1 function in U18666a wash-out experiments, the gastrosome undergoes a reduction in size and the microglia
re-establish their complex, branched morphology. This suggests the existence of a link between the gastrosome and microglial morphology. As cholesterol serves as a fundamental component of
the plasma membrane, crucial for maintaining its structural integrity and fluidity (for review see ref. 55), NPC1-mediated cholesterol transport at the level of the gastrosome might enable
microglia to form branches and sustain a high rate of neuronal engulfment. Therefore, understanding the signaling pathways and molecular interactions downstream of NPC1 activity becomes very
important. Further research into these pathways could provide valuable insights into how cholesterol affects microglia and their functions, elucidating the broader implications of
cholesterol dysregulation in neurodegenerative diseases and other conditions associated with defective neuronal clearance. Interestingly, we show that in response to elevated neuronal
apoptosis, phagocytic NPC1 deficient microglia undergo cell death and are subsequently cleared by other microglia. This finding has implications for understanding microglial responses in NPC
as the death of these cells could effectively reduce the brain’s ability to clear dead neurons, leading to the accumulation of neuronal corpses and increased inflammation. Several
mechanisms could be responsible for the death of microglia in this context. One is the presence of large cholesterol crystals in these cells (Fig. 3c and Supplementary Fig. 3b), which have
been shown to damage cells by physically disrupting their integrity56,57. Alternatively, microglia could undergo pyroptotic death, as suggested by high IL1 expression observed in NPC1
deficient embryos58. The discovery of the gastrosome and its central role in lipid handling post-engulfment brings to light a critical aspect of efferocytosis, namely the collection of
apoptotic material into a single compartment. This suggests a coordinated strategy in dealing with engulfed cellular material, potentially specialized for certain molecules like lipids. On
the other hand, this compartment might also serve a more general function handling a wider range of different products resulting from the breakdown of apoptotic material. Indeed, while we
have demonstrated that the gastrosome contains lipids whose transport depends on NPC1, this compartment may also contain additional important molecules. In this context, it is crucial to
understand the role of the gastrosome in relation to other key components of the phagocytic pathway, such as, lysosomes and LAMP1-labeled non-degradative compartments that lack lysosomal
hydrolases and have been identified in neurons59. It is well-established that the vesicular components of the phagocytic pathway undergo constant fusion-fission events and share common
molecular markers and structural features. However, the gastrosome stands out because it is a single compartment characterized by electron-lucent properties while lysosomes, telolysosomes,
and other non-degradative vesicles are typically numerous, small, and electron-dense60. This distinction adds depth to our understanding of intracellular processing and lysosomal storage
disorders. The lack of Cathepsin D observed in the gastrosome strongly supports a scenario in which this compartment is non degradative fulfilling a storage function, which will be further
explored. Efferocytosis-dependent collection of cholesterol into one compartment is also found in human non-myeloid cells, such as fibroblasts, demonstrating the remarkable conservation of
this compartment and its function across different cell types in vertebrates. This collecting function of the gastrosome is also reminiscent of similar processes occurring in single-celled
organisms. For example, in malaria plasmodium, where small hemoglobin digestive compartments fuse, creating a single food vacuole with known storage and transport functions (for review see
ref. 61). Hence, the gastrosome might be compared to a food vacuole seen in single-celled organisms, emerging as an optimal hub for lipid sensing and trafficking within phagocytic cells.
Finally, this study highlights the importance of NPC1 in phagocytic microglia, as interfering with its function under phagocytic stress expands the gastrosome affecting microglial morphology
and viability. In NPC disease, microglia are exposed to increasingly high levels of neuronal cell death, suggesting that NPC1’s localization on the gastrosome could be relevant for
understanding this disease and developing treatments. METHODS ANIMAL HANDLING Zebrafish (_Danio rerio_) were raised, maintained, and bred according to standard procedures as described in
“Zebrafish – A practical approach” (Nüsslein-Volhard, 2012). All experiments were performed on embryos younger than 5dpf, in accordance with the European Union Directive 2010/62/EU and local
authorities (Kantonales Veterinäramt; Fishroom licence TVHa Nr. 178). Live embryos were kept in E3 buffer at 28–30 °C until the desired developmental stage. Pigmentation was prevented
during experiments by treating the embryos with 0.003% N-phenylthiourea (PTU) (Sigma-Aldrich, St. Louis, US-MO, #P7629) from 1dpf onwards. Embryos were anaesthetized during mounting
procedures and experiments using 0.01% tricaine (Sigma-Aldrich, #A5040). FISH LINES The following transgenic animals were used in this study: (_Tg(mpeg1.1:EGFP-CAAX)_)2,
(_TgBAC(csf1ra:GAL4-VP16)_, _Tg(UAS-E1B:NTR-mCherry)_)32, (_Tg(NBT:DLexPR::NTR-mCherry)_46. The following mutant lines were used in this study: _slc37a2__NY007_ 2, _irf8__st95_ 41,
_npc1__hg37_ 37. CELL LINES RAW 264.7 mouse macrophages (ATCC) and HeLa Kyoto H2B-mCherry cells (Mitocheck consortium, EMBL) were cultured as previously described2, in DMEM (Gibco) plus 10%
FBS (Gibco) and in DMEM plus 1% Penicillin/Streptomycin (Gibco) and 1% GlutMAX (Gibco) respectively. Human microglia cell line 3 (HMC3) was obtained from ATCC and cultured in Minimum
Essential Medium (MEM) (Gibco) supplemented with 1% sodium pyruvate (Gibco) and 10% FBS (Gibco). Human patients’ fibroblasts (GM03652, GM18393, GM17919) were obtained from the NHGRI Sample
Repository for Human Genetic Research at the Coriell Institute for Medical Research (informed consent obtained by the provider) and cultured in Minimum Essential Medium (MEM) with NEAA
(Gibco) supplemented with 1% GlutMAX (Gibco) and 15% FBS (Gibco). CRISPR KNOCK-OUT IN ZEBRAFISH gRNAs (_NPC1_: 5’-GTATCTGGTACGGCGAATGT-3’, 5’-ATAGGTCACATTCCCCACAC-3’; _NPC2_:
5’-GGACTACAGAGTGCTCGGCG-3’, 5’-GGTAGACGGAAAAGTAGTTC-3’) were designed using the CRISPR/Cas9 target online predictor CHOPCHOP and ordered from IDT as crRNA. crRNA was mixed in a 1:1 ratio
with tracrRNA (IDT) and incubated at 95 °C for 5 min to obtain a 10 µM gRNA solution. gRNAs for either _NPC1_ or _NPC2_ were mixed in a 1:1 ratio and incubated at 37 °C for 20 min to form a
RNP complex with in-house Cas9 protein (Protein Expression and Purification Facility, EMBL, Heidelberg, Germany) and injected into one cell stage zebrafish eggs. Embryos were raised to 4
days post fertilization and experiments were performed. Afterwards, genomic DNA was extracted with QuickExtract™ DNA Extraction Solution (Lucigen) to confirm genomic deletions by PCR
(primers _NPC1_: 5’-TGGCTGCCAATGGTTACAGG-3’, 5’-ACCATCCAAACTAAGCGTATAGCA-3’; primers _NPC2_: 5’-AGTGTACGCATATTCGCTGT-3’, 5’-TAAAGCAACTCACCACTGCT-3’). CRISPR KNOCK-OUT IN RAW 264.7 MOUSE
MACROPHAGES gRNAs targeting the fifth and seventh exon of _SLC37A2_ were designed using the CRISPR/Cas9 target online predictor CHOPCHOP. The template for the sgRNAs
(5’-TTACTACCTCTCGGCTGGAA-3’, 5’-TTCAGGCGGGGATTCATCAT-3’) were produced by designing primers containing the T7 promotor. Amplicons were transcribed using the MEGAshortscript T7 transcription
kit (Ambion). RAW 264.7 mouse macrophages were transfected using Viromer CRISPR kit (Lipocalyx) with gRNAs and in-house Cas9 protein (Protein Expression and Purification Facility, EMBL,
Heidelberg, Germany) in a 1:1 ratio. Cells were diluted and seeded as single cells to obtain monoclonal lines. Successful _SLC37A2_ targeting was confirmed by PCR and scoring for the
expected phenotype by imaging cells fed with apoptotic material. CRISPR/Cas9-based editing generated a trans-heterozygous line characterized by several deletions in exons 5 (5 bp) and 7 (3
and 117 bp). CRISPR KNOCK-IN IN ZEBRAFISH A sgRNA (5’-ACGCTAAAATTGCTGCATCT-3’) was designed using the CRISPR/Cas9 target online predictor tool CRISPR scan (https://www.crisprscan.org) and
ordered from IDT as crRNA. crRNA was mixed in a 1:1 ratio with tracrRNA (IDT) and incubated at 95 °C for 5 min to obtain 10 μM gRNA solution. NPC1-mNG PCR fragment was used as the homology
recombination template. gRNA was mixed with PCR fragment and in-house Cas9 protein (Protein Expression and Purification Facility, EMBL, Heidelberg, Germany). The mix was incubated at 37 °C
for 20 min to form a RNP complex that was injected into zebrafish eggs at the one cell stage. Embryos were raised to 3dpf and screened on a fluorescence microscope. Experiments were
performed at 4dpf. CRISPR KNOCK-IN IN RAW 264.7 MOUSE MACROPHAGES sgRNAs (_LAMP1_: 5’-CATGTTGATCCCCATTGCTG-3’; _NPC1_: 5’-CCACTTACGAGCGCTACAGA-3’) were designed using the CRISPR/Cas9 target
online predictor CHOPCHOP and ordered from IDT as crRNA. crRNA was mixed in a 1:1 ratio with tracrRNA (IDT) and incubated at 95 °C for 5 min to obtain a 100 μM gRNA solution. LAMP1-EGFP or
NPC1-EGFP PCR fragments were used as homology recombination templates. RAW 264.7 mouse macrophages were transfected using Cell Line Nucleofector™ Kit V (Lonza) with gRNA, PCR fragment and
HiFi Cas9 protein (IDT). Cells were sorted as single cells to obtain EGFP positive monoclonal lines. Successful endogenous tagging was confirmed by flow cytometry, light microscopy, PCRs and
sequencing. MORPHOLINO KNOCKDOWN To reduce phagocytosis of the optic tectum microglia BAI1 morpholino (CTAGAACTCTAACACACTTACTCAT) was injected into the yolk of zebrafish eggs at the one
cell stage as previously described46. Embryos were raised to 4dpf, mounted and imaged. IN VITRO PHAGOCYTOSIS ASSAY HeLa Kyoto H2B-mCherry were seeded on a 60 mm dish and left until adhered.
Cells were exposed to UV light (150mJ/cm2) using Stratalinker 1800 and left for at least 8 h in the incubator (5% CO2, 37 °C). Apoptotic cells were collected, centrifuged, 4.2 ml of the
supernatant was removed and cells were resuspended in the remaining 800 μl of the supernatant. Concentrated apoptotic cells were added on separately seeded phagocytic cells (either RAW 264.7
mouse macrophages, human microglia HMC3 or human patients’ fibroblasts) and phagocytosis was allowed for the next 24 h before fixation. Such samples are referred to as HeLa-fed. Consistent
with previous work on the gastrosome2, HeLa cells were used because they stably express fluorescent proteins that allow for tracking apoptotic cargo when these cells undergo apoptosis and
are engulfed by phagocytes. CHEMICAL PERTURBATIONS U18666A 4dpf zebrafish embryos were incubated in 200 μM U18666a (Sigma-Aldrich, #U3633) solution for 6 h, or 3dpf zebrafish embryos were
incubated in 100 μM U18666a solution overnight at RT. Embryos were mounted and imaged in the drug. For wash out experiments, the drug was removed from the medium before imaging. RAW 264.7
mouse macrophages and human microglia HMC3 or human patients’ fibroblasts were incubated with 7 μM and 50 μM U18666a, respectively, in the growth medium for 24 h before fixation.
CAMPTOTHECIN To induce higher levels of apoptosis in zebrafish embryos, Camptothecin (CPT) (Sigma-Aldrich, #C9911) was used. Embryos were placed in 1 μM CPT solution in E3 medium with 1%
(v/v) DMSO for 7-8 h before mounting and imaging. METRONIDAZOLE To induce higher levels of apoptosis specifically in the hindbrain, _Tg(NBT:DLexPR::NTR-mCherry)_ zebrafish embryos were
incubated with 10 mM MTZ (Sigma, #M3761) solution in E3 medium with 0.2% (v/v) DMSO for 24 h. Embryos were washed extensively with E3 before mounting and imaging.
2-HYDROXYPROPYL-Γ-CYCLODEXTRIN (HPΓCD) To prevent the NPC1 deficient microglia phenotype, 2-hydroxypropyl-γ-cyclodextrin (HPγCD) (Sigma-Aldrich, #H125) was used. 2.5dpf zebrafish embryos
were placed in 1 M HPγCD in E3 medium for 24 h before mounting and imaging. Human patients’ fibroblasts were incubated with 100 mM HPγCD in the growth medium for 24 h before fixation.
TOPFLUOR CHOLESTEROL INJECTIONS IN ZEBRAFISH To label cholesterol, TopFluor cholesterol (Avanti Polar Lipids, #810255 P) was injected into the yolk of zebrafish eggs at the one cell stage as
previously described in ref. 37. Embryos were raised to 4dpf, mounted and imaged. ACRIDINE ORANGE STAINING IN ZEBRAFISH To visualize apoptotic cells in vivo, embryos were stained with
acridine orange (Sigma-Aldrich, #A6014). The larvae were incubated in the dark for 1 h at 28 °C in 10 μg/ml acridine orange solution in E3 and washed extensively before imaging. GM1
GANGLIOSIDE STAINING IN ZEBRAFISH To visualize GM1 gangliosides, a group of glycophospholipids, Cholera Toxin subunit B (CtxB recombinant) Alexa Fluor 488 conjugate (Invitrogen, #C34775)
staining was performed. 4dpf zebrafish embryos were fixed in 4% PFA for 1 h at RT, permeabilized in acetone for 20 min at −20 °C, washed in 0.1% Triton-PBS and incubated in blocking buffer
(1% BSA / 5% normal goat serum / 1% Triton-X-100 / PBS) for 1 hour at RT. Afterwards, larvae were placed in 10 μg/ml CtxB solution in a blocking buffer overnight at 4 °C. Embryos were washed
extensively in 0.1% Triton-PBS before mounting and imaging. FILIPIN STAINING Cells were fixed for 10 min with 4% PFA, washed three times with 1xPBS and stained with filipin (Sigma-Aldrich,
#SAE0088) for 2 h at 0.05 mg/ml final concentration in the dark. Afterwards cells were washed three times with PBS and imaged directly. IMMUNOHISTOCHEMISTRY IN CELL CULTURE Cells were fixed
for 10 min in 4% PFA solution, washed with PBS and permeabilized for 7 min with 0.2% Triton-X-100/PBS solution. After 1 h of blocking solution (3% BSA / 0.2% Triton-X-100 / PBS), cells were
incubated with primary antibody in blocking solution overnight at 4 °C: LAMP1 (Abcam, 1:100, ab25245, anti-mouse), LAMP1 (Abcam, 1:1000, ab24170, anti-human), Cathepsin D (Abcam, 1:100,
ab75852, anti-mouse), Cathepsin D (Abcam, 1:200, ab72915, anti-human), LC3B (Novus Biologicals, 1:200, nb100-2220, anti-mouse). Cells were washed three times with 1xPBS and incubated for 1 h
with fluorescently labeled secondary antibody in blocking solution at RT (1:500 or 1:1000, Thermo Fisher Scientific, Abcam). Cells were mounted using a Vectashield Kit (Vector Laboratories,
Inc., Burlingame, CA) with DAPI or stained with 300 nM DAPI for 5 min and directly imaged in PBS. IMMUNOHISTOCHEMISTRY IN ZEBRAFISH Embryos at 4dpf were fixed in 4% PFA and processed for
whole-mount antibody staining as previously described in ref. 62. The primary mouse 4C4 monoclonal antibody (gift from Valérie Wittamer laboratory, 1:500) and secondary Alexa Fluor
647-conjugated anti-mouse (Abcam, 1:500, ab150107) antibodies were used. WESTERN BLOT AND PROTEIN EXTRACTION IN ZEBRAFISH 50 4dpf wild-type and 50 4dpf NPC1 CRISPANT zebrafish embryos were
anesthetized in tricaine and placed in 170 μl of RIPA buffer supplemented with protease inhibitor cocktail cOmplete EDTA-free tablets (Roche, #11836170001). After rotating for 30 min at 4
°C, samples were sonicated (3 x 5s) with Bandelin Electronic Sonoplus HD 2070 Homogenisator (Bandelin) and lysates were cleared by centrifugation. Samples were mixed with 4x NuPAGE Lithium
Dodecyl Sulphate (LDS) buffer and electrophoresed on a 4–12% Bis-Tris NuPAGE gel (Invitrogen). Proteins were transferred to nitrocellulose membrane (Invitrogen) using XCell II Blot module
(Invitrogen). After blocking with 5% milk saturation buffer, blots were incubated overnight at 4 °C with anti-NPC1 primary antibody (Abcam, 1:1000, ab134113) and for 1 h at RT with
anti-rabbit secondary antibody HRP-conjugated (Promega, 1:2500, W4018). Proteins were revealed using Immobilion Western Detection System Kit (Millipore) and Amersham Imager 600 (GE
Healthcare Life Sciences). Afterwards blots were stripped (Thermo Scientific, Restore™ PLUS Western Blot Stripping Buffer), re-saturated, stained again using anti-γTubulin primary antibody
(Sigma-Aldrich, 1:2000, T6557) and anti-mouse secondary antibody HRP-conjugated (Promega, 1:2500, W4028). QPCR AND RNA EXTRACTION IN ZEBRAFISH For each condition 50 4.5dpf zebrafish embryos
were anesthetized with tricaine, heads were cut off and placed in 300 μl of Trizol (Invitrogen, #15596026). Samples were sonicated (3x5s) with Bandelin Electronic Sonoplus HD 2070
Homogenisator (Bandelin) and lysates were cleared by centrifugation. After the addition of 60 μl of chloroform, samples were vortexed and centrifuged at 13,000 x _g_ for 15 min in 4 °C.
Supernatants were transferred to new tubes, and RNA extraction was continued using PureLink RNA mini Kit (Invitrogen, #12183018 A) with on-column PureLink DNase treatment (Invitrogen,
#12185-010). The RNA template from three independent experiments for each condition was used for cDNA synthesis using the SuperScript IV Reverse Transcriptase (Invitrogen, #18090010). Gene
expression levels were analyzed using the CFX96 Real-Time System (Bio-Rad) and SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, #1725270). The data were analyzed based on ref. 63 using
three reference genes (β-Actin (_ACTB_)64, Ribosomal Protein L13a (_RPL13A_)65, TATA-Box Binding Protein (_TBP_)). For inflammation analysis three markers were used: Interleukin-6 (_IL6_)40,
Nuclear factor NF-kappa-B subunit 2 (_NFΚB2_) and Interleukin-1 (_IL1_)38. Table 1 contains primers used in the analysis. LIPIDOMIC ANALYSIS Lipid extraction, lipidomics, and data analysis
were adapted from ref. 66 and performed at the Functional Genomic Center Zurich, ETH Zurich and University of Zurich. Briefly, each sample (control RAW, NPC1 inhibitor treated RAW, HeLa-fed
RAW, HeLa-fed and NPC1 inhibitor treated RAW) was re-suspended in 100 μl of isopropanol / methanol (1:1) by shaking 20 min, 20 °C, 100 x g. Samples were centrifuged (16,000 x g, 10 min, 20
°C) and 70 μL of the supernatant was transferred into glass vial for LC-MS. The QCpool consisted of 10 μL of each sample. For the LC method a Waters Premier BEH C18 column (50 mm × 2.1 mm)
was employed at 60 °C, paired with a Thermo Vanquish Horizon Binary Pump. A 2 µL sample was used, with a flow rate of 1 mL for 7.5 min. The gradient transitioned from 15% Buffer B to 99%
Buffer B (Buffer A: 60% acetonitrile, 40% H2O, 5 mm NH4acetate; Buffer B: 90% isopropanol, 10% acetonitrile, 5 mm NH4acetate). For the MS method a Thermo Q Exactive HF DDA top5 with two
different resolutions (MS1 resolution 60‘000; MS2 resolution 15‘000) was used. The stepped normalized collision energy was 10, 20, and 30. Data analysis was conducted using the Thermo
Compound Discoverer software 3.3. Lipid identification was achieved by matching to lipidblast library, followed by visual inspection of the mirror plots (library vs data) to confirm the
match and the presence of diagnostic fragments according to the lipids standard initiative guidelines. Only lipids that matched the expected fragmentation spectra were included in the
analysis. For identification of cholesterol and its derivatives see Supplementary Table 1. LIGHT MICROSCOPY OF FIXED CELLS Confocal analysis of fixed cells was performed with an Andor
Dragonfly 200 Sona spinning-disc microscope with a Nikon 40x/NA 1.25 silicon oil objective and 40 μm spinning disc to capture 15–20 μm thick stacks with a z-step of 0.2 μm. HMC3 and human
patients’ fibroblasts in vitro feeding screens were imaged on a GE IN Cell Analyzer 2500HS microscope with Nikon 40x/NA 0.95 air objective to capture 300 randomized areas per sample. All
images were analyzed in Fiji67. LIGHT MICROSCOPY OF LIVE AND FIXED ZEBRAFISH Zebrafish embryos were anaesthetized with tricaine and pre-selected based on the expression of the desired
fluorophore, using a Nikon ZMZ18 fluorescent stereoscope. Embryos were embedded in 1–1.5% low-melting (LM) agarose (PeqGOLD Low Melt Agarose, PeqLab Biotechnologie GmbH), dissolved in E3
medium with tricaine. Embryos were mounted on glass-bottom dishes (Greiner Bio-One, #627871) for confocal microscopy or pulled together with agar, into glass capillaries (Brand, #701904)
with a rod (Brand, #701932), and then pushed halfway out, into the microscopy chamber for light-sheet microscopy. Both the imaging dishes and the microscopy chamber were filled with E3
medium containing PTU and tricaine during the entire imaging period. For confocal microscopy, an Andor Dragonfly 200 Sona spinning-disc microscope with a Nikon 20x/NA 0.95 WI objective or
Nikon 40x/NA 1.25 silicon oil objective and 40 μm spinning disc were used to capture 70–90 μm thick stacks with a z-step of 0.5 μm. The same conditions were used for fixed zebrafish embryos.
For light-sheet microscopy, a Zeiss Z.1 microscope with a W Plan-APO 20x/NA 1.0 WI imaging objective and 10x/NA 0.2 air illumination objectives were used to capture 40–75 μm thick stacks
with a z-step of 0.4–0.5 μm, every 15–30 s. ELECTRON MICROSCOPY Transmission Electron Microscopy on RAW 264.7 mammalian macrophages fed with apoptotic HeLa and treated with U18666a, an NPC1
inhibitor, was performed at the Center for Microscopy and Image Analysis, University of Zurich. Cells were grown on 12 mm cover glasses treated with L-polylysine and containing gold
reference markers (20 nm thickness), which were sputter coated onto the discs using an appropriate mask (Leica Microsystems, Vienna, Austria). Cells were fixed with 2.5% glutaraldehyde in
0.1 M sodium cacodylate buffer (pH 7.35, pre-warmed to 37 °C) for 1 h, followed by 1% OsO4 for 1 h in 0.1 M cacodylate buffer, and 1% aqueous uranyl acetate for 1 hour both at RT. Samples
then were dehydrated in an ethanol series and embedded in Epon/Araldite (Sigma-Aldrich). To target specific cells from in vitro feeding assays that presented the clear phenotype of the
enlarged gastrosome, we imaged the resin block with a fluorescent microscope (Thunder, Leica Microsystems, Vienna, Austria) and identified the cells of interest based on the gold marks and
the weak fluorescence of the sample compared with the resin. Ultrathin (70 nm) sections were post-stained with lead citrate and examined with a Talos 120 transmission electron microscope at
an acceleration voltage of 120KV using a Ceta digital camera and the MAPS software package (Thermo Fisher Scientific, Eindhoven, The Netherlands). LIGHT-SHEET IMAGE PRE-PROCESSING For 3D
analysis of light-sheet microscopy images, 3D volumes as czi files were converted to H5 files using Fiji67 and BigDataProcessor68. Cells were tracked and individual frames were saved using
BigDataTools and BigDataTracker for further analysis. For 2D analysis, maximum projections of hyperstacks were used. IMAGE ANALYSIS MICROGLIAL MORPHOLOGY Maximum projections of individually
cropped cells were used to create a Gaussian blurred image for further thresholding and mask generation in Fiji67. Shape descriptors, including circularity, were measured using the created
mask. VESICLE AND PHAGOSOME DETECTION Vesicles were detected as empty spaces in microglia labelled with cytoplasmic fluorescent marker (_TgBAC(csf1ra:GAL4-VP16)_, _Tg(UAS-E1B:NTR-mCherry)_).
Phagosomes were defined as vesicles that form at the plasma membrane upon the closure of a phagocytic cup. MICROGLIA AND VESICLE SEGMENTATION 3D microglia segmentations were generated in
Imaris using surface detection function. The largest vesicle was segmented manually using the same software. MICROGLIAL PHAGOCYTOSIS Quantification of phagocytosis rate was performed as
previously described in ref. 42. Maximum projections of individual cells were created to count successful phagocytic events in Fiji67. The total number of counts from one cell was divided by
the duration of the time-lapse to estimate the number of events per hour ratio. UNCOLLECTED APOPTOTIC CELLS QUANTIFICATION 3D volumes from acridine orange staining experiments were
processed in Imaris using the spot detection function. Automated selection was corrected manually to consider only spots in the optic tectum of zebrafish and to estimate the number of
uncollected apoptotic neurons in each sample. DIAMETER OF THE VESICLE The area of vesicles was measured in Fiji67 using a polygonal tool on the chosen frame from hyperstacks representing the
middle of the vesicle, or in the case of data acquired with widefield microscopy, on the only frame available. Diameter was calculated based on the following formula: $$2r=2\cdot
\sqrt{\frac{{area}}{\pi }}$$ VESICLE TRACKING Volumes of single cell crops were used to track individual vesicles using the MTrackJ plug-in69 in Fiji67. For each time point a tracker was
placed in the center of the vesicle in the chosen frame representing the middle of the vesicle. The center z-plane was used for measuring the diameter of the vesicle in the subsequent time
points. STATISTICAL TESTING _N_ refers to the number of biological replicates (e.g., fish embryos) and _n_ to the number of items (e.g., cells) analyzed, unless otherwise specified. For
statistical analysis, Python 3.8 and statannot packages were used. Conditions were compared using an unpaired, two-tailed, nonparametric Mann-Whitney U test with Bonferroni correction.
REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The data and materials supporting
the findings of this study are available from the corresponding author upon request. Source data are provided with this paper. The imaging data generated in this study have been deposited in
the EBI BioStudies database under accession number S-BSST1537. Source data are provided with this paper. REFERENCES * Sierra, A. et al. Microglia shape adult hippocampal neurogenesis
through apoptosis-coupled phagocytosis. _Cell Stem Cell_ 7, 483–495 (2010). Article PubMed CAS PubMed Central Google Scholar * Villani, A. et al. Clearance by Microglia Depends on
Packaging of Phagosomes into a Unique Cellular Compartment. _Dev. Cell_ 49, 77–88 (2019). Article PubMed CAS Google Scholar * Keren-Shaul, H. et al. A Unique Microglia Type Associated
with Restricting Development of Alzheimer’s Disease. _Cell_ 169, 1276–1290 (2017). Article PubMed CAS Google Scholar * Li, Q. et al. Developmental Heterogeneity of Microglia and Brain
Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. _Neuron_ 101, 207–223.e10 (2019). Article PubMed CAS Google Scholar * Zareba, J. & Peri, F. Microglial ‘fat shaming’ in
development and disease. _Curr. Opin. Cell Biol._ 73, 105–109 (2021). Article PubMed CAS Google Scholar * Cantuti-Castelvetri, L. et al. Defective cholesterol clearance limits
remyelination in the aged central nervous system. _Science_ 359, 684–688 (2018). Article ADS PubMed CAS Google Scholar * Brekk, O. R., Honey, J. R., Lee, S., Hallett, P. J. &
Isacson, O. Cell type-specific lipid storage changes in Parkinson’s disease patient brains are recapitulated by experimental glycolipid disturbance. _Proc. Natl Acad. Sci. USA_ 117,
27646–27654 (2020). Article ADS PubMed CAS PubMed Central Google Scholar * Marschallinger, J. et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory
state in the aging brain. _Nat. Neurosci._ 23, 194–208 (2020). Article PubMed CAS PubMed Central Google Scholar * Arbaizar‐Rovirosa, M. et al. Aged lipid‐laden microglia display
impaired responses to stroke. _EMBO Mol. Med._ 15, 1–21 (2023). Article Google Scholar * Polese-Bonatto, M. et al. Niemann-Pick Disease Type C: Mutation Spectrum and Novel Sequence
Variations in the Human NPC1 Gene. _Mol. Neurobiol._ 56, 6426–6435 (2019). Article PubMed CAS Google Scholar * Kwon, H. J. et al. Structure of N-Terminal Domain of NPC1 Reveals Distinct
Subdomains for Binding and Transfer of Cholesterol. _Cell_ 137, 1213–1224 (2009). Article PubMed PubMed Central Google Scholar * Li, X., Saha, P., Lib, J., Blobel, G. & Pfeffer, S.
R. Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. _Proc. Natl. Acad. Sci. USA_ 113, 10079–10084 (2016). Article ADS PubMed
CAS PubMed Central Google Scholar * Trinh, M. N., Brown, M. S., Seemann, J., Goldstein, J. L. & Lu, F. Lysosomal cholesterol export reconstituted from fragments of Niemann-Pick C1.
_ELife_ 7, 1–14 (2018). Article Google Scholar * Wang, N. L. et al. The Presence of Vacuolated Kupffer Cells Raises a Clinical Suspicion of Niemann-Pick Disease Type C in Neonatal
Cholestasis. _Front. Genet._ 13, 1–5 (2022). Google Scholar * Appelqvist, H. et al. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. _Am. J. Pathol._ 178,
629–639 (2011). Article PubMed CAS PubMed Central Google Scholar * Appelqvist, H. et al. Sensitivity to Lysosome-Dependent Cell Death Is Directly Regulated by Lysosomal Cholesterol
Content. _PLoS ONE_ 7, 1–11 (2012). Article Google Scholar * Chung, C., Puthanveetil, P., Ory, D. S. & Lieberman, A. P. Genetic and pharmacological evidence implicates cathepsins in
Niemann-Pick C cerebellar degeneration. _Hum. Mol. Genet._ 25, 1434–1446 (2016). Article PubMed CAS PubMed Central Google Scholar * Higashi, Y., Murayama, S., Pentchev, P. G. &
Suzuki, K. Cerebellar degeneration in the Niemann-Pick type C mouse. _Acta Neuropathologica_ 85, 175–184 (1993). Article PubMed CAS Google Scholar * Erickson, R. P. & Bernard, O.
Studies on neuronal death in the mouse model of Niemann-Pick C disease. _J. Neurosci. Res._ 68, 738–744 (2002). Article PubMed CAS Google Scholar * Wu, Y. P. et al. Apoptosis accompanied
by up-regulation of TNF-α death pathway genes in the brain of Niemann-Pick type C disease. _Mol. Genet. Metab._ 84, 9–17 (2005). Article PubMed CAS Google Scholar * Cougnoux, A., et al.
Necroptosis in Niemann–Pick disease, type C1: a potential therapeutic target. _Cell Death Disease_ 7 https://doi.org/10.1038/cddis.2016.16 (2016). * Funakoshi, T., Aki, T., Tajiri, M.,
Unuma, K. & Uemura, K. Necroptosis-like neuronal cell death caused by cellular cholesterol accumulation. _J. Biol. Chem._ 291, 25050–25065 (2016). Article PubMed CAS PubMed Central
Google Scholar * Prasad, A., Fischer, W. A., Maue, R. A. & Henderson, L. P. Regional and developmental expression of the Npc1 mRNA in the mouse brain. _J. Neurochem._75, 1250–1257
(2000). Article PubMed CAS Google Scholar * Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. _J.
Neurosci._ 34, 11929–11947 (2014). Article PubMed CAS PubMed Central Google Scholar * Colombo, A. et al. Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in
microglia. _Nat. Commun._ 12, 1158 (2021). Article ADS PubMed CAS PubMed Central Google Scholar * Cologna, S. M. et al. Human and mouse neuroinflammation markers in Niemann-Pick
disease, type C1. _J. Inherit. Metab. Dis._ 37, 83–92 (2014). Article PubMed CAS Google Scholar * Cougnoux, A. et al. Microglia activation in Niemann-Pick disease, type C1 is amendable
to therapeutic intervention. _Hum. Mol. Genet._ 27, 2076–2089 (2018). Article PubMed CAS PubMed Central Google Scholar * German, D. C. et al. Neurodegeneration in the Niemann-Pick C
mouse: Glial involvement. _Neuroscience_ 109, 437–450 (2002). Article PubMed CAS Google Scholar * Baudry, M., Yao, Y., Simmons, D., Liu, J. & Bi, X. Postnatal development of
inflammation in a murine model of Niemann-Pick type C disease: Immunohistochemical observations of microglia and astroglia. _Exp. Neurol._ 184, 887–903 (2003). Article PubMed CAS Google
Scholar * Kavetsky, L. et al. Increased interactions and engulfment of dendrites by microglia precede Purkinje cell degeneration in a mouse model of Niemann Pick Type-C. _Sci. Rep._ 9,
14722 (2019). Article ADS PubMed PubMed Central Google Scholar * Kroll, F. et al. A simple and effective f0 knockout method for rapid screening of behaviour and other complex
phenotypes. _eLife_ 10, e59683 (2021). Article PubMed CAS PubMed Central Google Scholar * Gray, C. et al. Simultaneous intravital imaging of macrophage and neutrophil behaviour during
inflammation using a novel transgenic zebrafish. _Thrombosis Haemost._ 105, 811–819 (2011). Article CAS Google Scholar * Casano, A. M., Albert, M. & Peri, F. Developmental Apoptosis
Mediates Entry and Positioning of Microglia in the Zebrafish Brain. _Cell Rep._ 16, 897–906 (2016). Article PubMed CAS Google Scholar * Lu, F. et al. Identification of NPC1 as the target
of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. _eLife_ 4, 1–16 (2015). Article Google Scholar * Naureckiene, S. et al. Identification of HE1 as the second
gene of Niemann-Pick C disease. _Science_ 290, 2298–2301 (2000). Article ADS PubMed CAS Google Scholar * Subramanian, K. & Balch, W. E. NPC1/NPC2 function as a tag team duo to
mobilize cholesterol. _Proc. Natl Acad. Sci. USA_ 105, 15223–15224 (2008). Article ADS PubMed CAS PubMed Central Google Scholar * Tseng, W. C. et al. Modeling Niemann-Pick disease type
C1 in zebrafish: A robust platform for in vivo screening of candidate therapeutic compounds. _DMM Dis. Models Mechanisms_ 11, dmm034165 (2018). Article Google Scholar * Wiweger, M.,
Majewski, L., Adamek-Urbanska, D., Wasilewska, I. & Kuznicki, J. npc2-Deficient Zebrafish Reproduce Neurological and Inflammatory Symptoms of Niemann-Pick Type C Disease. _Front. Cell.
Neurosci._ 15, 1–17 (2021). Article Google Scholar * Suzuki, M. et al. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal
transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: A potential basis for glial cell activation in the NPC brain. _J. Neurosci._ 27, 1879–1891
(2007). Article PubMed CAS PubMed Central Google Scholar * Varela, M., Dios, S., Novoa, B. & Figueras, A. Characterisation, expression and ontogeny of interleukin-6 and its
receptors in zebrafish (_Danio rerio_). _Dev. Comp. Immunol._ 37, 97–106 (2012). Article PubMed CAS Google Scholar * Shiau, C. E., Kaufman, Z., Meireles, A. M. & Talbot, W. S.
Differential Requirement for irf8 in Formation of Embryonic and Adult Macrophages in Zebrafish. _PLoS ONE_ 10, e0117513 (2015). Article PubMed PubMed Central Google Scholar * Möller, K.
et al. A role for the centrosome in regulating the rate of neuronal efferocytosis by microglia in vivo. _eLife_ 11, e82094 (2022). Article PubMed PubMed Central Google Scholar * Silva,
N. J., Dorman, L. C., Vainchtein, I. D., Horneck, N. C. & Molofsky, A. V. In situ and transcriptomic identification of microglia in synapse-rich regions of the developing zebrafish
brain. _Nat. Commun._ 12, 1–12 (2021). Article Google Scholar * Pommier, Y. Topoisomerase I inhibitors: camptothecins and beyond. _Nat. Rev. Cancer_ 6, 789–802 (2006). Article PubMed CAS
Google Scholar * Curado, S. et al. Conditional targeted cell ablation in zebrafish: A new tool for regeneration studies. _Dev. Dyn._ 236, 1025–1035 (2007). Article PubMed CAS Google
Scholar * Mazaheri, F. et al. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. _Nat. Commun._ 5, 4046 (2014). Article ADS PubMed CAS Google Scholar *
Varsano, N. et al. Two polymorphic cholesterol monohydrate crystal structures form in macrophage culture models of atherosclerosis. _Proc. Natl Acad. Sci. USA_ 115, 7662–7669 (2018). Article
ADS PubMed CAS PubMed Central Google Scholar * Maxfield, F. R. & Wüstner, D. Analysis of Cholesterol Trafficking with Fluorescent Probes. _Methods Cell Biol._ 108, 367–393 (2012).
Article PubMed CAS PubMed Central Google Scholar * Chen, J. W., Murphy, T. L., Wlllingham, M. C., Pastan, I. & August, J. T. Identification of two lysosomal membrane glycoproteins.
_J. Cell Biol._ 101, 85–95 (1985). Article PubMed CAS Google Scholar * Janabi, N., Peudenier, S., Heron, B., Ng, K. H. & Tardieu, M. Establishment of human microglial cell lines
after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. _Neurosci. Lett._ 195, 105–108 (1995). Article PubMed CAS Google Scholar * Soga, M. et
al. Erratum: HPGCD Outperforms HPBCD as a Potential Treatment for Niemann-Pick Disease Type C during Disease Modeling with iPS Cells. _Stem Cells_ 33, 2885–2886 (2015). Article Google
Scholar * Davidson, C. D. et al. Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. _Ann. Clin. Transl. Neurol._ 3, 366–380 (2016). Article PubMed CAS PubMed
Central Google Scholar * Singhal, A., Krystofiak, E. S., Jerome, W. G. & Song, B. 2-Hydroxypropyl-gamma-cyclodextrin overcomes NPC1 deficiency by enhancing lysosome-ER association and
autophagy. _Sci. Rep._ 10, 1–14 (2020). Article Google Scholar * Ferreira, C. R. & Gahl, W. A. Lysosomal storage disease. _Transl. Sci. Rare Dis._ 2, 1–71 (2017). PubMed PubMed
Central Google Scholar * Yang, S. T., Kreutzberger, A. J. B., Lee, J., Kiessling, V. & Tamm, L. K. The role of cholesterol in membrane fusion. _Chem. Phys. Lipids_ 199, 136–143 (2016).
Article PubMed CAS PubMed Central Google Scholar * Kellner-Weibel, G. et al. Crystallization of free cholesterol in model macrophage foam cells. _Arteriosclerosis, Thrombosis, Vasc.
Biol._ 19, 1891–1898 (1999). Article CAS Google Scholar * Shu, F. et al. Cholesterol crystal-mediated inflammation is driven by plasma membrane destabilization. _Front. Immunol._ 9, 1–13
(2018). Article Google Scholar * He, W. T. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. _Cell Res._ 25, 1285–1298 (2015). Article PubMed CAS
PubMed Central Google Scholar * Cheng, X. T. et al. Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. _J. Cell Biol._ 217, 3127–3139
(2018). Article PubMed CAS PubMed Central Google Scholar * Hartenstein, V. & Martinez, P. Phagocytosis in cellular defense and nutrition: a food-centered approach to the evolution
of macrophages. _Cell Tissue Res._ 377, 527–547 (2019). Article PubMed CAS PubMed Central Google Scholar * Matz, J. M. Plasmodium’s bottomless pit: properties and functions of the
malaria parasite’s digestive vacuole. _Trends Parasitol._ 38, 525–543 (2022). Article PubMed Google Scholar * Rovira, M. et al. Zebrafish Galectin 3 binding protein is the target antigen
of the microglial 4C4 monoclonal antibody. _Dev. Dyn._ 252, 400–414 (2022). Article PubMed Google Scholar * Taylor, S. C. et al. The Ultimate qPCR Experiment: Producing Publication
Quality, Reproducible Data the First Time. _Trends Biotechnol._ 37, 761–774 (2019). Article PubMed CAS Google Scholar * Tsarouchas, T. M. et al. Dynamic control of proinflammatory
cytokines Il-1β and Tnf-α by macrophages in zebrafish spinal cord regeneration. _Nat. Commun._ 9, 4670 (2018). Article ADS PubMed PubMed Central Google Scholar * Lin, C., Spikings, E.,
Zhang, T. & Rawson, D. Housekeeping genes for cryopreservation studies on zebrafish embryos and blastomeres. _Theriogenology_ 71, 1147–1155 (2009). Article PubMed CAS Google Scholar
* Ganguin, A. A., Skorup, I., Streb, S., Othman, A. & Luciani, P. Formation and Investigation of Cell-Derived Nanovesicles as Potential Therapeutics against Chronic Liver Disease. _Adv.
Healthc. Mater._ 12, e2300811 (2023). Article PubMed Google Scholar * Schindelin, J. et al. Fiji: An open-source platform for biological-image analysis. _Nat. Methods_ 9, 676–682 (2012).
Article PubMed CAS Google Scholar * Tischer, C. et al. BigDataProcessor2: a free and open-source Fiji plugin for inspection and processing of TB sized image data. _Bioinformatics_ 37,
3079–3081 (2021). Article PubMed CAS PubMed Central Google Scholar * Meijering, E., Dzyubachyk, O., & Smal, I. Methods for cell and particle tracking. In _Methods in Enzymology_
(1st ed., Vol. 504) (2012). https://doi.org/10.1016/B978-0-12-391857-4.00009-4. Download references ACKNOWLEDGEMENTS We are grateful to Cornelia Henkel for helping with fish work, Max
Brambach for his assistance with data analysis and Darren Gilmour for providing feedback on the manuscript. We would like to thank Nicolas Rieser for helping with qPCR experiments. The
authors acknowledge the support of the Center for Microscopy and Image Analysis, University of Zurich for light and electron microscopy experiments. We are thankful to Forbes D. Porter for
sharing the _npc1__hg37_ mutant fishline and to Valérie Wittamer for sharing the 4C4 antibody. This project was supported by Swiss National Science Foundation Grants 310030_212794 and
31003A_182733 to F.P., J.Z. and A.V. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Molecular Life Sciences, University of Zurich, Zurich, Switzerland Joanna Zareba, Elena F.
Cattaneo, Ambra Villani & Francesca Peri * Functional Genomic Center Zurich, ETH Zurich and University of Zurich, Zurich, Switzerland Alaa Othman & Sebastian Streb Authors * Joanna
Zareba View author publications You can also search for this author inPubMed Google Scholar * Elena F. Cattaneo View author publications You can also search for this author inPubMed Google
Scholar * Ambra Villani View author publications You can also search for this author inPubMed Google Scholar * Alaa Othman View author publications You can also search for this author
inPubMed Google Scholar * Sebastian Streb View author publications You can also search for this author inPubMed Google Scholar * Francesca Peri View author publications You can also search
for this author inPubMed Google Scholar CONTRIBUTIONS J.Z. and F.P. designed the study and wrote the manuscript. J.Z. conducted the majority of experiments and analyzed the data. E.F.C.
participated in the initial characterization of NPC1-deficient embryos. A.V. contributed to EM experiments and cholesterol detection in BAI1 knockdowns. A.O. and S.S. performed lipidomic
analysis. CORRESPONDING AUTHOR Correspondence to Francesca Peri. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION
_Nature Communications_ thanks Ryuta Koyama, Cody Smith and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION
SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY MOVIE 1 SUPPLEMENTARY MOVIE 2 SUPPLEMENTARY MOVIE 3 SUPPLEMENTARY MOVIE 4 SUPPLEMENTARY
MOVIE 5 SUPPLEMENTARY MOVIE 6 SUPPLEMENTARY MOVIE 7 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Zareba, J., Cattaneo, E.F., Villani, A. _et al._ NPC1 links cholesterol
trafficking to microglial morphology via the gastrosome. _Nat Commun_ 15, 8638 (2024). https://doi.org/10.1038/s41467-024-52874-6 Download citation * Received: 11 March 2024 * Accepted: 19
September 2024 * Published: 05 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52874-6 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative