- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT In lung disease, persistence of KRT8-expressing aberrant basaloid cells in the alveolar epithelium is associated with impaired tissue regeneration and pathological tissue
remodeling. We analyzed single cell RNA sequencing datasets of human interstitial lung disease and found the profibrotic Interleukin-11 (IL11) cytokine to be highly and specifically
expressed in aberrant KRT8+ basaloid cells. IL11 is similarly expressed by KRT8+ alveolar epithelial cells lining fibrotic lesions in a mouse model of interstitial lung disease. Stimulation
of alveolar epithelial cells with IL11 causes epithelial-to-mesenchymal transition and promotes a KRT8-high state, which stalls the beneficial differentiation of alveolar type 2 (AT2)-to-AT1
cells. Inhibition of IL11-signaling in AT2 cells in vivo prevents the accumulation of KRT8+ cells, enhances AT1 cell differentiation and blocks fibrogenesis, which is replicated by
anti-IL11 therapy. These data show that IL11 inhibits reparative AT2-to-AT1 differentiation in the damaged lung to limit endogenous alveolar regeneration, resulting in fibrotic lung disease.
SIMILAR CONTENT BEING VIEWED BY OTHERS INTERLEUKIN-11 SIGNALING UNDERLIES FIBROSIS, PARENCHYMAL DYSFUNCTION, AND CHRONIC INFLAMMATION OF THE AIRWAY Article Open access 01 December 2020
AUTOCRINE TGF-Β-POSITIVE FEEDBACK IN PROFIBROTIC AT2-LINEAGE CELLS PLAYS A CRUCIAL ROLE IN NON-INFLAMMATORY LUNG FIBROGENESIS Article Open access 31 August 2023 SINGLE-CELL RNA SEQUENCING
REVEALS SPECIAL BASAL CELLS AND FIBROBLASTS IN IDIOPATHIC PULMONARY FIBROSIS Article Open access 09 July 2024 INTRODUCTION The alveolar epithelium plays a pivotal role in lung homeostasis
and protects the lung from inhaled environmental insults and pathogenic infections. In the alveolus, alveolar type 2 cells (AT2 cells) become activated after injury and proliferate and
trans-differentiate into alveolar type-1 cells (AT1 cells) to restore alveolar structure and lung function1,2. A number of human lung pathologies, including idiopathic pulmonary fibrosis
(IPF), chronic obstructive pulmonary disease (COPD), and post-infective lung damage, are characterized by failure of homeostatic AT2-to-AT1 transitions3,4. Recent large-scale single-cell RNA
sequencing (scRNA-seq) studies of human pulmonary fibrosis (PF) have identified transitional cells that exhibit a dysfunctional phenotype and have a reduced capacity to differentiate into
AT1 cells5,6,7,8,9. These disease-associated transitional AT2 cells, coined KRT5−/KRT17+ or aberrant basaloid cells, accumulate in the lungs of patients with IPF6,7 and after severe
SARS-CoV-2 infection10,11,12. An analogous population of transitional cells termed Krt8+ alveolar differentiation intermediate (KRT8 + ADI)/damage-associated transient progenitors
(DATPS)/pre-alveolar type-1 transitional cell state (PATS) are similarly seen in the damaged alveolus in mouse models of lung injury13,14,15. In mice, transitional cells, herein referred to
as Krt8+ transitional cells, can be derived from either AT2 cells or airway stem cells, and possess the capacity to differentiate into mature AT1 cells13,16. Importantly, Krt8+ transitional
cells in the mouse exhibit transcriptional similarities to human disease-associated KRT5-/KRT17+ / aberrant basaloid cells, including signatures of epithelial-mesenchymal transition (EMT),
p53, and cell senescence pathways and expression of KRT8 itself13,15. Krt8+ transitional cells are thought to contribute to fibrosis via the expression of profibrotic and proinflammatory
mediators. Recent studies have shown that elevated TGFβ signaling in AT2 cells and inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α) activity in DATPS maintain the Krt8+
cell state following lung injury in mice17,18,19. Similarly, the persistence of senescent AT2 cells promotes progressive pulmonary fibrosis20. However, it remains unclear whether other
molecular pathways can contribute to the emergence and abnormal maintenance of alveolar transitional cells in this aberrant state. Interleukin-11 (IL11), a member of the IL-6 family of
cytokines, is upregulated in the airways following viral infections and has been associated with a range of respiratory disorders21. We previously reported that IL11 was increased in the
lungs and fibroblasts of patients with IPF, and its expression correlates with disease severity22. A contemporaneous study found that _IL11_ was expressed in a range of cell types in
fibrotic lungs of patients with Hermansky–Pudlak syndrome (HPS) and also in _SFTPC_+ cells in IPF23. More recent pharmacologic studies using siRNA have further confirmed the role of IL11 in
lung fibrosis24. In the current study, we leveraged single-cell RNA sequencing (scRNA-seq) datasets from patients with fibrotic lung disease and analyzed IL11-lineage-labeled cells in a
mouse model to delineate the different lung cell types expressing IL11 in the disease. We examined whether IL11 signaling plays a role in alveolar regeneration via its specific activity in
AT2 cells using conditional _Il11ra1_ deletion in AT2 cells and lineage tracing in mice that were subjected to bleomycin lung injury and also in studies of primary alveolar epithelial cells
and AT2 cells in vitro. We also tested whether a neutralizing anti-IL11 antibody administered to mice with lung injury could promote alveolar regeneration by enhancing AT2-to-AT1
differentiation. In this study, we show that IL11 is uniquely expressed by aberrant basaloid and KRT5-/KRT17+ cells in human lung fibrosis and by Krt8+ transitional cells in the fibrotic
lungs of mice after bleomycin injury. In alveolar epithelial and AT2 cell cultures, IL11 stimulation promotes the expression of ECM proteins and a KRT8+ state that stalls AT2-to-AT1
differentiation. In the bleomycin model of lung fibrosis, the conditional deletion of _Il11ra1_ in AT2 cells prevents the accumulation of profibrotic Krt8+ transitional cells, enhances
alveolar epithelial regeneration, and protects against fibrosis. We further show that therapeutic administration of anti-IL11 antibodies in the bleomycin model similarly prevents the
accumulation of profibrotic Krt8+ transitional cells and enhances regeneration of the injured lung epithelium. These data identify IL11 signaling in AT2 cells for the potentiation of
pathological phenotypes in aberrant transitional epithelial cells in the injured lung and reveal that anti-IL11 may have the potential to enhance alveolar epithelial repair and promote lung
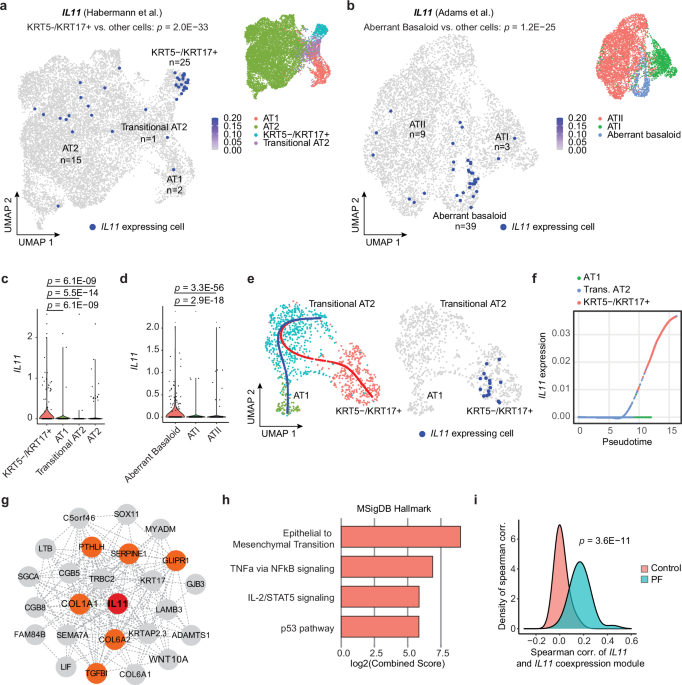
regeneration in severe lung diseases. RESULTS _IL11_ IS EXPRESSED BY KRT5−/KRT17+ CELLS IN HUMAN PF To characterize IL11 and IL11RA expressing cells in human PF, we re-analyzed large-scale
scRNA-seq data of lung cells from patients with PF from two independent studies by Habermann et al. and Adams et al. (GSE135893 and GSE136831, respectively)6,7. Our analysis showed that, in
health, _IL11_ was expressed at very low levels in the lung, and its expression was barely detected across most lung cell types (Supplementary Figs. 1, 2). In contrast, in PF, _IL11_ was
elevated in mesenchymal and epithelial cell populations and rarely detected in immune and endothelial cells (Supplementary Fig. 1). Within mesenchymal cells, _IL11_ was most elevated in
PLIN2+ lipofibroblasts and disease-specific HAS1high fibroblasts (Supplementary Figs. 1, 2), which supports our previous findings22,25 and further associates _IL11_ with pathological
fibroblast activity in PF. _IL11RA_ (which encodes for IL11 receptor subunit alpha), was broadly expressed and more highly enriched in fibroblast and alveolar epithelial cell populations in
the human lung (Supplementary Fig. 2). Amongst the various epithelial cell types identified in the two datasets, we observed particular enrichment of _IL11_ expression in disease-specific
KRT5-/KRT17+ (_P_ = 2.0 × 10−33) and aberrant basaloid cells (_P_ = 1.2 × 10−25) but limited _IL11_ expression in basal, ciliated, MUC5B+, SCGB3A2+, AT2, transitional AT2 or AT1 epithelial
cells (Fig. 1a–d and Supplementary Fig. 3). In contrast, _IL6_, which was recently implicated in airway epithelial dysfunction in fibrotic lung diseases26, was broadly expressed in AT2,
Mesothelial, MUC5AC+ High, MUC5B+ and Goblet epithelial cell types (Supplementary Fig. 4 and Supplementary Data 1) but seen rarely in transitional cells in both control and PF lungs. Since
KRT5−/KRT17+/aberrant basaloid cells may arise from defective AT2-to-AT1 differentiation, we performed trajectory and pseudotime analysis on transitional AT2 cells, KRT5-/KRT17+/aberrant
basaloid and AT1 cells on combined Habermann et al. and Adams et al. datasets. To do this, we first confirmed that the transcriptional profiles between aberrant basaloid and transitional AT2
and KRT5-/KRT17+ cells were highly similar (Supplementary Fig. 5a). The aberrant cells in Adams et al. dataset were then assigned using the classification from the Habermann et al. dataset
(i.e., transitional AT2 or KRT5−/KRT17+) by Seurat’s FindTransfer Algorithm (see Methods) to obtain a consistent nomenclature across these two datasets. Our trajectory analyses showed two
distinct differentiation paths for transitional AT2 cells in PF samples: (1) transitional AT2 to AT1 trajectory and (2) a trajectory from transitional AT2 to KRT5-/KRT17+ cells; with _IL11_
expressed only by KRT5-/KRT17+ cells (Fig. 1e and Supplementary Fig. 5b). Pseudotime analysis revealed that _IL11_ was specifically upregulated along the differentiation trajectory towards
KRT5-/KRT17+ cells but not towards AT1 cells (Fig. 1f and Supplementary Fig. 5c). To delineate a transcriptional program co-expressed with _IL11_ along the KRT5−/KRT17+ cell trajectory, we
performed co-expression analysis to the trajectory using cells assigned to the combined Habermann et al., and Adams et al. datasets and found that the _IL11_ co-expression module was
enriched for genes involved in epithelial-to-mesenchymal transition (EMT) (such as _COL1A1_, _SERPINE1_, _COL6A1_, _PTHLH_, _GLIPR1_, and _TGFBI_), TNFa via NFκB signaling, IL-1/STAT5
signaling and p53 pathway (Fig. 1g, h and Supplementary Data 2, 3). Furthermore, the association between _IL11_ and the _IL11_ co-expression module was highly specific to disease (Fig. 1i
and Supplementary Fig. 5d), suggesting a unique role of IL11 in dysfunctional alveolar epithelial cells in PF. IL11 IS EXPRESSED BY ALVEOLAR KRT8+ CELLS AFTER LUNG INJURY To further
characterize IL11-expressing cells in the injured lung, we used an _IL11__EGFP_ reporter mouse27. We performed single-dose oropharyngeal injections of bleomycin (BLM) (Fig. 2a), a drug that
causes lung epithelial damage and fibrosis, and performed preliminary characterization of lung cells 10 days post-injury by flow cytometry (Supplementary Fig. 6). Using antibodies against a
range of lung cell type markers: CD31 (endothelial cells), CD45 (hematopoietic cells), CD326/EpCAM (epithelial cells), our analysis revealed that IL11EGFP+ cells were rarely observed in the
uninjured lung. However, following BLM injury, we found elevated proportions of IL11EGFP+ cells in hematopoietic (CD45+ CD31−; _P_ = 0.0136), epithelial (CD45− CD31− EpCAM+_; P_ = 0.0002),
and stromal cell populations (CD45− CD31− EpCAM-; _P_ = 0.0200) (Supplementary Fig. 6). IL11EGFP was not detected in endothelial cells (CD45− CD31+) in both injured and uninjured lungs
(Supplementary Fig. 6). Since the low detection/abundance of IL11EGFP+ cells precludes further FACS-based analysis, we next focused on immunohistochemistry to determine the identities of
IL11-expressing cells in the injured lung. To do this, we assessed the lungs of _IL11__EGFP_ reporter mice at 7 or 21 days post-BLM injury by staining for GFP and counterstained for SFTPC
(AT2 marker), PDPN (AT1 marker), PDGFRA (pan-fibroblast marker) or CD45. Consistent with the flow cytometry analysis, IL11EGFP+ cells were very rarely observed in the lungs of uninjured
_IL11__EGFP_ reporter mice. In contrast, in BLM-injured lungs, IL11EGFP expression was notably upregulated in SFTPC+ AT2 cells (Fig. 2b and Supplementary Fig. 7a), PDGFRA+ fibroblasts and a
subset of CD45+ hematopoietic cells (Supplementary Fig. 7b, c) within injured alveolar regions that were marked by areas of dense consolidation of nuclei DAPI staining. IL11EGFP was
localized to numerous SFTPC+ cells adjacent to regions of tissue disruption with enlarged/elongated morphologies suggestive of transitional AT2 cells that have committed towards AT1
differentiation (Fig. 2b, c). Immunostaining for PDPN revealed that IL11EGFP expression was very rarely detected in mature AT1 cells in injured or uninjured lungs (Fig. 2b and Supplementary
Fig. 7d). To investigate if IL11 is expressed by Krt8+ transitional cells, we performed immunostaining for GFP and KRT8 in lung sections from BLM-treated and uninjured _IL11__EGFP_ reporter
mice and excluded airway regions for quantification. In uninjured mice, KRT8 expression was limited to the airways, whereas BLM treatment resulted in the appearance of KRT8+ cells in the
damaged alveolar regions (Fig. 2d and Supplementary Fig. 7e, f). There was an overlap of IL11EGFP expression in a proportion of KRT8 expressing cells in alveolar regions following BLM injury
(Fig. 2e). Additionally, flow cytometry analysis of lung single cell suspension from _IL11__EGFP_ reporter mice for the transitional alveolar epithelial cell marker, Cldn414, showed that
the proportions of GFP expressing Cldn4hi epithelial cells were significantly increased in the lungs after BLM-injury (Fig. 2f–h and Supplementary Fig. 8a–d), with Cldn4hi epithelial cells
being the predominant IL11-expressing epithelial cell subset in the injured lung (Supplementary Fig. 8e–g). We next sought to determine whether IL11-expressing Krt8+ transitional cells are
derived from AT2 cells during lung injury. We utilized _Sftpc-CreER; R26-tdTomato_ (_Sftpc-tdT_) mice to trace AT2 cells and their descendants (AT2-lineage cells) and monitored for the
expression of IL11 specifically in this cell lineage after BLM injury. We exposed _Sftpc-tdT_ mice to tamoxifen prior to BLM treatment and assessed the lungs 14 days post-injury (Fig. 2i).
We performed immunostaining using an anti-IL11 antibody, which showed consistent overlap with anti-GFP in injured _IL11__EGFP_ lungs (Supplementary Fig. 7g), and counterstained for KRT8.
This revealed the emergence of numerous IL11+ KRT8+ _tdT_+ cells with spread out/elongated morphologies 14 days after BLM injury (Fig. 2j and Supplementary Fig. 7h). IL11 and KRT8
immunostaining were not observed in alveolar regions of uninjured _Sftpc-tdT_ mice, as expected. These findings revealed that IL11-expressing Krt8+ transitional cells are derived from
activated AT2 cells after lung injury. IL11 INDUCES ECM PRODUCTION IN ALVEOLAR EPITHELIAL CELLS To investigate the functional importance of IL11 in alveolar epithelial cells, we performed
two-dimensional (2D) cultures of primary human pulmonary alveolar epithelial cells (HPAEpiC). By immunostaining, we first confirmed that HPAEpiC expressed high levels of SFTPC and did not
stain positive for AGER (Supplementary Fig. 9a). HPAEpiC expressed high levels of IL11RA and its co-receptor IL6ST (gp130) but lacked detectable IL6R expression (Supplementary Fig. 9a). To
test whether IL11 directly induces profibrotic EMT-like processes in alveolar epithelial cells, we stimulated HPAEpiC with IL11 (5 ng/ml, 24 h) and monitored for the expression of pathologic
ECM components (Collagen I, fibronectin)7 along with KRT8 using immunostaining and immunofluorescence quantification (Fig. 3a). In parallel, we treated HPAEpiC with TGFβ1 (5 ng/ml; 24 h), a
potent inducer of both EMT and KRT8 expression in AT2 cells28,29,30,31, and simultaneously added a neutralizing IL11 antibody (X203) or an IgG control antibody to investigate the effect of
IL11 signaling downstream of TGFβ stimulation (Fig. 3a). This revealed that IL11 and TGFβ1 treatment led to upregulation of Collagen I, fibronectin, and KRT8 expression, as compared to
untreated epithelial cells (Fig. 3b–d). By ELISA, we found that TGFβ1 stimulation significantly induced IL11 secretion by HPAEpiC (Supplementary Fig. 9b). The effects of TGFβ1 on the
expression of ECM proteins and KRT8 were significantly blunted by X203 (Fig. 3b–d). AT2 cell proliferation is crucial for alveolar repair after injury1,32,33 and we tested the effects of
IL11 or TGFβ1 on human alveolar epithelial cell proliferation. By EdU staining, we found that exposure of cells to either IL11 or TGFβ1 (24 h) impaired HPAEpiC proliferation (Supplementary
Fig. 9c–e). Furthermore, the anti-proliferative effects of TGFβ1 on HPAEpiC could be reversed by X203. These data shows that IL11 directly induces KRT8 expression and EMT processes while
impairing proliferation of human alveolar epithelial cells. Next, we performed bulk RNA sequencing (RNA-seq) of IL11- or TGFβ1-stimulated HPAEpiC (5 ng/ml, 24 h) to evaluate the
transcriptional effects of these cytokines on alveolar epithelial cells. RNA-seq analysis revealed that TGFβ1 induced transcriptomic features characteristic of KRT5−/KRT17+ cells from human
fibrotic lungs (such as the elevated expression of _CDKN2A_, _CDKN2B_, _CDH2_, _COL1A1_, _FN1_, _SOX9_, _SOX4_, _KRT8_, _KRT17_, _KRT18_, and reduced expression of _NKX2-1)_ as compared to
untreated cells (Supplementary Fig. 10a, b). Along with these changes, _IL11_ was amongst the top upregulated genes in TGFβ1 treated HPAEpiC (4.65-fold, _Padj_ = 1.28e-62) (Supplementary
Data 4). In contrast to TGFβ1 treatment and consistent with data from other cell types22,34, IL11 (5 ng/ml, 24 h) did not result in significant changes in global transcription levels in
HPAEpiC (Supplementary Fig. 10a,b), despite inducing the expression of several ECM-related and KRT8 proteins (Fig. 2b–d). In human cardiac and lung fibroblasts, IL11-stimulated ERK
activation induces profibrotic protein expression and myofibroblast differentiation22,34. Correspondingly, the effects of IL11 on the expression of KRT8 and ECM proteins Collagen I and
fibronectin expression in HPAEpiC were blocked by the ERK inhibitor U0126 (Supplementary Fig. 10c, d), supportive of an important role for IL11-ERK post-transcriptional gene regulation in
human alveolar epithelial cells. In keeping with this, we observed numerous p-ERK+ IL11+ _tdT_+ cells within injured regions of lungs from BLM-injured _Sftpc-tdT_ mice (Supplementary Fig.
10e) and a similar increase in p-ERK+ GFP+ KRT8+ cells in the injured lungs of IL11EGFP reporter mice (Supplementary Fig. 10f), indicating the activation of IL11-ERK signaling in
transitional alveolar epithelial cells after lung injury that was not apparent in uninjured lungs. To provide additional evidence to support the role of IL11 in driving pathologic ECM
protein expression by lung epithelial cells, we performed similar in vitro experiments on human small airway epithelial cells (HSAEC) and on primary mouse AT2 cells that were isolated from
tamoxifen-exposed _Sftpc-tdT_ mice by FACS sorting for constitutive _tdT_+-expressing cells and cultured these primary cells under 2D conditions (Fig. 3f–j and Supplementary Fig. 11). By
immunostaining, we observed that IL11 and TGFβ1 treatment significantly increased the expression of Collagen I and fibronectin and secreted collagen by HSAEC and Collagen I expression in
mouse AT2 cells (Fig. 3g–j and Supplementary Fig. 11c, e, g). Furthermore, in these cells, the effects of TGFβ on the expression of these ECM proteins were largely dependent on downstream
IL11-signaling and could be blocked by X203-treatment (Fig. 3g–j and Supplementary Fig. 11d, e, g). Similarly, the effects of IL11 on the expression of these ECM proteins could be
significantly blunted by ERK inhibition (Fig. 3k, l and Supplementary Fig. 11d, e, h). Taken together, our data suggests that IL11-ERK signaling induces EMT-like features in lung epithelial
cell dysfunction across species. IL11 STALLS AT2-TO-AT1 CELL DIFFERENTIATION IN VITRO AT2 cells largely increase their cell area and spontaneously undergo differentiation towards AT1-like
cells when cultured under prolonged 2D culture conditions. Under these conditions, AT2 cells upregulate KRT8 during early differentiation, followed by a decline of KRT8 and the subsequent
upregulation of mature AT1 markers (such as PDPN) during late differentiation13,29. To test if IL11 stalls the transition of AT2 cells into mature AT1 cells, we isolated mouse AT2 cells from
tamoxifen-exposed _Sftpc-tdT_ mice and cultured these primary AT2 cells under 2D culture conditions followed by treatment with IL11 (5 ng/ml) from day 1 to day 5 (Fig. 3m). By
immunostaining and cell surface area analysis of _tdT_+ cells, we found that numerous cells expressed PDPN and greatly increased their surface area by 5 days of culture in untreated cells
(Fig. 3n, o and Supplementary Fig. 12). In contrast, exposure to IL11 from days 1 to 5 stalled AT1 differentiation with cells expressing higher levels of KRT8, lower levels of PDPN and with
reduced cell surface area, as compared to controls (Fig. 3n, o and Supplementary Fig. 12). Since prolonged TGFβ signaling impairs terminal AT1 maturation17,29,35, we further hypothesized
that the maladaptive effects of prolonged TGFβ-exposure on AT1 maturation might be mediated, in part, by IL11. We tested for this by first priming AT2 cells with TGFβ1 for 2 consecutive
days, followed by subsequent TGFβ1 treatment with X203 or IgG antibodies for an additional 2 days (Fig. 3m). Similar to the effects of sustained IL11 treatment, we found that cells treated
with TGFβ1 followed by coincubation with IgG resulted in stalled AT1 differentiation, with cells that were less enlarged and expressed higher levels of KRT8 as compared to controls (Fig. 3n,
o and Supplementary Fig. 12). On the other hand, coincubation with X203 partially-relieved the stalled AT1 differentiation phenotype and significantly increased cell surface area and PDPN
expression as compared to IgG-treated cells (Fig. 3n, o and Supplementary Fig. 12). These data show that IL11 directly promotes AT2 cell dysfunction by causing the accumulation of Krt8+
transitional cells and delaying the terminal differentiation of AT1 cells. IL11 SIGNALING IN AT2 CELLS PROMOTES LUNG FIBROSIS IN VIVO Having established that IL11 stimulation triggered
EMT-related features in primary human alveolar and distal airway epithelial cells and mouse AT2 cells, we next surveyed mouse single-cell sequencing datasets and profiled the RNA expression
of IL11 receptor (_Il11ra1_) in the adult mouse lung. Consistent with our human scRNA-seq analysis (Supplementary Fig. 3), _Il11ra1_ was found to be highly expressed in mouse lung stromal
populations (fibroblasts, smooth muscle cells), mesothelial cells and moderately expressed by macrophages and alveolar epithelial cells (Supplementary Fig. 13a–c). Notably, _Il11ra1_ was
consistently expressed in AT2 cells and injury emergent AT2-derived cells such as activated AT2 and Krt8 ADI, further illustrating the potential for auto/paracrine IL11-signaling across
AT2-lineage cells in the injured mouse lung. We next employed a genetic loss-of-function approach to test the importance of IL11 signaling specifically in AT2 cells for lung fibrogenesis. We
utilized _Sftpc-CreER; Il11ra1__fl/fl_ mice in which _Il11ra1_ could be temporally and conditionally deleted in AT2 and AT2-derived cells upon tamoxifen treatment. Mice were injected with
tamoxifen 14 days prior to BLM-treatment and the lungs were assessed 12 and 21 days post-injury (Fig. 4a). Tamoxifen-exposed _Sftpc-CreER; Il11ra1__+/+_ mice were used as controls. The
deletion of _Il11ra1_ in AT2 cells from tamoxifen-exposed _Sftpc-CreER; Il11ra1__fl/fl_ mice was verified by qPCR of FACS-sorted CD31- CD45- EpCAM+ MHCII+ cells (Supplementary Fig. 13d, e).
At baseline, the lungs of mice with AT2 cell-specific _Il11ra1_ deletion appeared histologically normal (Supplementary Fig. 13f). Further histology assessment of lungs from BLM-injured
_Sftpc-CreER; Il11ra1__+/+_ control mice at both 12 and 21 day time points indicated severe disruption to the lung architecture, increased collagen deposition and higher histopathological
fibrosis scores, as compared to uninjured mice (Fig. 4b and Supplementary Fig. 14a–d). These pathologies were significantly reduced in mice where _Il11ra1_ was deleted in AT2 cells. Lung
hydroxyproline content was also significantly reduced in mice lacking _Il11ra1,_ specifically in AT2 cells, as compared to controls (Fig. 4c and Supplementary Fig. 14c). There was a
non-statistical trend of improved survival, body weights, and decreased lung weights in AT2-specific _Il11ra1_-deleted mice by the end of the 21 day study period (Supplementary Fig. 14e–g).
Serum surfactant protein D (SFTPD) levels, a marker of lung inflammation and epithelial injury36 was elevated in BLM-injured control mice but was significantly reduced in injured mice with
AT2 cell-specific _Il11ra1_ deletion (Supplementary Fig. 14h). Immunostaining for KRT8 revealed that KRT8-expressing cells were rarely observed in the alveolar compartment of AT2
cell-specific _Il11ra1_ deleted mice post-BLM injury, as compared to controls (Fig. 4d). Furthermore, western blot analysis of lung lysates further confirmed the overall reduction in KRT8
expression and a corresponding increase in AGER protein in the lungs of _Sftpc-tdT_; _Il11ra1__fl/fl_ mice as compared to controls (Supplementary Fig. 14i). Taken together, these data
indicate that the loss of IL11-signaling in AT2 cells prevents the development of lung fibrosis. IL11 SIGNALING POTENTIATES FIBROTIC KRT8+ CELL STATE IN VIVO Recent studies have revealed
that several pathological pathways (such as EMT, TGF-beta signaling, and p53 pathway) are highly enriched in aberrant transitional epithelial cells in human PF and in mouse Krt8+
transitional cells, and that these aberrant cells may secrete profibrotic factors and express pathologic ECM in the fibrotic lung6,7,37. We asked whether IL11-signaling in AT2 cells promotes
fibrosis by regulating the profibrotic phenotype of aberrant transitional cells. To this end, we performed scRNA-seq on sorted epithelial cells (CD31− CD45− EpCAM+) from the lungs of
_Sftpc-CreER; Il11ra1__fl/fl_ and _Il11ra1__+/+_ mice 12 days post-BLM challenge (_n_ = 1 mouse/uninjured groups and _n_ = 2 mice/BLM-injured groups) (Fig. 4e). Our analysis recapitulated
known homeostatic epithelial cell types in the lungs, such as AT2 (_Bex2, Lpl_), proliferating AT2 (_Mki67, Birc5,_ and _Ube2c_), club (_Scgb1a1_ and _Cyp2f2_), ciliated cells (_Tppp3_ and
_Dynlrb2_), and mature AT1 cells (_Igfbp2 and Cped1_). We also captured injury-emergent cell populations, including activated AT2 cells (_Lcn2, Il33_), Krt8+ transitional cells (_Cldn4,
Krt8_) and immature AT1 cells (_Rtkn2, Krt8_) (Supplementary Fig. 15a, b)13,38. _Il11ra1_ expression was reduced across AT2-lineage cells (AT2, activated AT2 and Krt8+ transitional cells) in
_Sftpc-CreER; Il11ra1__fl/fl_ mice, indicating the loss of IL11-signaling across various AT2-lineage cells in this model (Supplementary Fig. 15c). Amongst the various AT2 cells and
injury-emergent populations (Fig. 4f and Supplementary Fig. 15d), we identified a marked reduction in the proportion of Krt8+ transitional cells in _Sftpc-CreER; Il11ra1__fl/fl_ mice
post-BLM challenge (11.8 vs 1.95 %) (Fig. 4g and Supplementary Fig. 15e) which was consistent with our earlier histological analysis. We then focused on Krt8+ transitional cells. Gene set
enrichment analysis (GSEA) of differentially expressed genes between _Sftpc-CreER; Il11ra1__fl/fl_ and control Krt8+ transitional cells reveals significant downregulation of Hallmark
pathways of Krt8+ ADI (FDR <0.05) such as “EMT”, “TGF-beta signaling”, and “p53 pathway”13 in _Sftpc-CreER; Il11ra1__fl/fl_ cells (Fig. 4h, Supplementary Fig. 16a, b, and Supplementary
Data 5). From the leading-edge analysis of the GSEA result, we constructed a de-novo gene set of EMT, specific to Krt8+ ADI (see Methods). Notably, the transcriptomic signatures of this
Krt8+ ADI EMT program, along with the expression of several ECM and profibrotic genes such as _Col1a1_ (_P_adj = _1.9E-3_), _Fn1_ (_P_adj = _3.3E-5_), and _Ccn2_ (_P_adj = _3.6E-4_) were
significantly downregulated in Krt8+ transitional cells from _Sftpc-CreER; Il11ra1__fl/fl_ mice as compared to controls (Fig. 4i and Supplementary Fig. 16d and Supplementary Data 6). Taken
together, abrogated IL11 signaling may attenuate the profibrotic phenotype of Krt8+ transitional cells. We further investigated if IL11-signaling mediates the dysregulation of the
differentiation from AT2 to AT1 cells. Therefore, we performed separate Slingshot39 trajectory analysis of AT2 and injury emergent cells (activated AT2, AT1, and Krt8+ transitional cells)
from BLM-injured _Sftpc-CreER; Il11ra1__fl/fl_ and _Il11ra1__+/+_ control mice, with origin of differentiation set at AT2 cells (Supplementary Fig. 16e). In injured controls, we found two
distinct trajectories from (1) AT2 to Krt8+ transitional cells and (2) AT2 to AT1 cells. In the first trajectory predicted, the destination at Krt8+ cells implies that Krt8+ may be a
terminally differentiated state, likely potentiated by disease-causing cues and reminiscent of the differentiation trajectories of AT2 to aberrant basaloid or KRT5-/KRT17+ cells in our
earlier human scRNA-seq analysis (Fig. 1). In contrast, only a single trajectory from AT2 to AT1 cells was observed for cells from _Sftpc-CreER; Il11ra1__fl/fl_ mice which suggests that AT2
cells lacking IL11 signaling may undergo effective terminal differentiation to AT1 cells. To specifically test the hypothesis that IL11 signaling in AT2 cells promotes the accumulation of
AT2-cell derived Krt8+ transitional cells and delays AT2-to-AT1 differentiation after lung injury in vivo, we crossed _Sftpc-CreER; Il11ra1__fl/fl_ mice with R26-tdTomato (_tdT)_ mice
(_Sftpc-tdT; Il11ra1__fl/fl_) to allow the simultaneous deletion of _Il11ra1_ and the constitutive expression of _tdT_ specifically in AT2 and AT2-derived cells upon tamoxifen
administration. _Sftpc-tdT; Il11ra1__+/+_ mice were used as controls. We similarly injected tamoxifen 14 days prior to BLM injury and assessed the lungs of mice 12 days post-BLM treatment
(Fig. 4j). Following BLM injury, immunostaining revealed numerous KRT8+ _tdT_+ cells and few newly differentiated AT1 cells (PDPN+ _tdT_+ cells) in _Sftpc-tdT; Il11ra1__+/+_ control mice
(Fig. 4k, l and Supplementary Fig. 17b). In contrast, parenchymal damage in BLM-treated _Sftpc-tdT; Il11ra1__fl/fl_ mice was markedly reduced and KRT8+ _tdT_+ cells were rarely observed in
alveolar regions from these mice, which mirrored the phenotypes observed earlier with _Sftpc-CreER; Il11ra1__fl/fl_ mice. Instead, we found numerous newly differentiated AT1 cells including
regions of completely formed alveoli that were PDPN+ _tdT_+ and AGER+ _tdT_+ in BLM-treated _Sftpc-tdT; Il11ra1__fl/fl_ mice (Fig. 4k, m and Supplementary Fig. 17b, c). These results show
that the deletion of _Il11ra1_ in AT2 cells only reduces KRT8+ cell accumulation and greatly enhances AT2-to-AT1 differentiation after BLM-injury. We next validated the importance of
IL11-signaling in AT2 cells for the acquisition of profibrotic transitional epithelial cell phenotypes identified from the earlier scRNA-seq findings. We performed immunostaining of Collagen
I or CTGF and KRT8 in the lungs of _Sftpc-tdT; Il11ra1__fl/fl_ mice after BLM injury. In _Sftpc-tdT; Il11ra1__+/+_ controls, we observed the presence of numerous Collagen I- and
CTGF-expressing AT2-derived transitional cells (Collagen I+ KRT8+ _tdT_+ cells and CTGF+ KRT8+ _tdT_+ cells) in fibrotic regions of the lung (Fig. 4n and Supplementary Fig. 17d). In
contrast, the expression of Collagen I and CTGF were markedly diminished in the lungs of BLM-challenged _Sftpc-tdT; Il11ra1__fl/fl_ mice along with the lack of observable Collagen I- and
CTGF-expressing lineage-traced cells (Fig. 4n and Supplementary Fig. 17d). Taken together, these findings further support the concept that IL11-signaling in AT2 cells impairs epithelial
regeneration by promoting the differentiation of ECM-producing profibrotic KRT8+ cells that may contribute directly to aberrant lung remodeling. _IL11_ DELETION IN AT2 CELLS DOES NOT PREVENT
LUNG FIBROSIS Having determined earlier that IL11 expression is elevated in AT2 and AT2-derived Krt8+ transitional cells following lung injury (Fig. 2), we next sought to investigate
whether the autocrine and/or paracrine activity of _Il11_, secreted by AT2 and AT2-derived cells is important for lung fibrogenesis. We utilized _Sftpc-CreER; Il11__fl/fl_ mice in which
_Il11_ could be temporally and conditionally deleted in AT2 cells upon tamoxifen treatment. _Sftpc-CreER; Il11__+/+_ mice were used as controls. Mice were injected with tamoxifen 14 days
prior to BLM-treatment and the lungs were assessed for fibrosis 21 days post-injury (Supplementary Fig. 18). However, lung histopathological assessment and hydroxyproline content analysis
revealed that BLM-injured _Sftpc-CreER; Il11__fl/fl_ mice had comparable levels of lung collagen content and fibrosis to injured controls (Supplementary Fig. 18). There were also no apparent
benefits of _Il11_-deletion in AT2 cells for survival at the end of the 21 day study period (Supplementary Fig. 18). These findings indicate that the deletion of _Il11_ specifically in AT2
and AT2-lineage cells does not prevent aberrant remodeling after lung injury and further suggests that IL11 expression from non AT2-lineage cells, such as fibroblasts or airway
progenitor-derived transitional cells may be of greater importance for lung fibrogenesis. IL11 INHIBITION PROMOTES ALVEOLAR REGENERATION IN VIVO In our previous therapeutic studies, we
showed that X203-treatment significantly diminished lung inflammation and reversed established lung fibrosis in BLM-injured mice22. We next investigated whether anti-IL11 antibodies could
promote AT2-to-AT1 differentiation and enhance alveolar regeneration when administered after lung injury. To test this, we performed BLM-induced injury to tamoxifen-exposed _Sftpc-tdT_ mice
followed by X203 or IgG control antibody administration starting from day 4 after injury, at a time point where alveolar KRT8+ cells begin to accumulate, and assessed the lungs on day 12
(Fig. 5a and Supplementary Fig. 19a). As compared to uninjured lungs, we observed widespread architectural disruption in IgG-treated mice, with a large increase in KRT8+ cells that adopted
elongated morphologies, along with a decline in the number of _tdT__+_ cells (Fig. 5b, c). Additionally, in IgG-treated mice, we found an increase in non-lineage-labeled KRT8_+_ cells
(KRT8_+_ _tdT_−) that stained weakly for the AT1 marker PDPN (Fig. 5b and Supplementary Fig. 19b), likely reflecting an influx of airway/progenitor cells that have committed to alveolar
fates in regions of severe lung injury13,16,40,41. As compared to IgG-treated mice, BLM-induced parenchymal damage and fibrosis, as assessed by histopathological scoring of Masson’s
trichrome staining and lung hydroxyproline content was significantly attenuated by X203-treatment (Supplementary Fig. 19c–e). These changes coincided with reduced numbers of alveolar KRT8_+_
cells and proportions of lineage-labeled transitional cells (KRT8_+_ _tdT__+_ cells) and lineage-negative cells (KRT8_+_ _tdT_− cells) (Fig. 5b–d and Supplementary Fig. 19b). Flow cytometry
analysis of lung Cldn4hi _tdT__+_ epithelial cells further confirmed the reduction in the proportion of AT2-derived transitional cells following X203-treatment (Fig. 5e and Supplementary
Fig. 20a–d). Furthermore, X203-treatment partially restored _tdT__+_ cell numbers after injury to levels similar to those seen in uninjured lungs (Fig. 5b, c and Supplementary Fig 20b),
which was associated with increased proliferation of surviving _tdT_+ AT2 cells as determined by immunostaining for Ki67 (Supplementary Fig. 20e). Consistent with the role of IL11-ERK
signaling in promoting a KRT8+ cell state, as seen in vitro, we found numerous p-ERK+ KRT8+ cells in the lungs after BLM-injury in IgG-treated mice (Supplementary Fig. 21a). The occurrence
of p-ERK+ KRT8+ cells were reduced in the lungs of X203-treated mice (Supplementary Fig. 21a). Immunostaining for AT1 markers PDPN or AGER revealed that X203-treatment led to significantly
enhanced differentiation of lineage-labeled cells into AT1 cells (PDPN_+_ _tdT__+_ or AGER_+_ _tdT__+_ cells) as compared to IgG (Fig. 5b, d and Supplementary Fig. 21b, c). Flow
cytometry-based quantification of PDPN_+_ _tdT__+_ epithelial lung cells and western blot analysis of KRT8 and AGER expression in lung lysates from X203 or IgG-treated mice further supported
these observations (Fig. 5f and Supplementary Fig. 21d). Lastly, to uncover additional mechanisms by which X203 prevents fibrosis, we performed similar scRNA-seq analysis on lung single
cells suspensions of Cd45− Cd31− EpCAM+ epithelial cells from uninjured and X203 or IgG-treated mice 12 days post-BLM (_n_ = 1 mouse/group) and focused our analysis on AT2-derived
injury-emergent cell populations (Fig. 5g and Supplementary Fig. 22a). Consistent with our histological and flow cytometry findings, the scRNA-seq analysis revealed that X203-treatment
reduced the proportion of Krt8+ transitional cells as compared to IgG (32.5 vs. 15.0%) (Fig. 5h). Pathway analysis of differentially expressed genes in Krt8+ transitional cells revealed that
the expression levels of genes related to unfolded protein response, TGF-beta signaling and EMT were modestly reduced following X203-treatment (Fig. 5i, Supplementary Fig. 22b, and
Supplementary Data 7). Similar to scRNA-seq data on epithelial cells from AT2-specific _Il11ra1_-deleted mice (Fig. 4), the transcriptomic signatures of EMT-related genes in Krt8+
transitional cells were significantly reduced by X203-treatment (Supplementary Fig. 22c). Immunostaining of lungs sections for Collagen I, CTGF and for an ER-stress marker XBP1, further
confirmed that the pharmacological inhibition of IL11 diminished the expression of pathologic ECM and profibrotic proteins by Krt8+ transitional cells (Collagen I+ KRT8+ _tdT_+ cells, CTGF+
KRT8+ _tdT_+ cells and XBP1+ KRT8+ _tdT_+ cells) after BLM injury (Fig. 5j and Supplementary Fig. 22d, e). DISCUSSION Severe respiratory diseases such as IPF and SARS-COV-2 pneumonia are
associated with defects in alveolar epithelial repair and irreversible loss of alveolar epithelial cells, which ultimately leads to fibrosis and lung function decline. We previously
discovered an important role for IL11 in lung fibrosis, mediated via its profibrotic activity in lung fibroblasts and _IL11_ expression was confirmed in diseased fibroblasts in the current
study22,25,42. Here, we show that _IL11_ is specifically upregulated in aberrant alveolar epithelial cells in human PF, and its expression is associated with pathological pro-EMT and
inflammatory gene signatures in diseased epithelial cells. In complementary studies of mice with severe lung injury, we found that IL11 is expressed by activated AT2 cells, Cldn4hi, and
Krt8+ transitional cells. Due to the complex signaling milieu that occurs following severe lung injury, multiple pathways likely contribute to the emergence and maintenance of Krt8+
transitional cells, among which TGFβ, which shows IL11 dependency for its effects, is of particular importance17,35. While inflammatory cytokines such as IL-1β and TNFα induce AT2 cell
proliferation, IL-1β also primes a subset of _Il1r1_-expressing AT2 cells for differentiation into DATPS14,43. Intriguingly, while IL6 is a therapeutic target in some forms of PF26,44, we
show that the cell types expressing IL6 in the fibrotic lung differ from those expressing IL11 and IL11, but not IL6, expression is enriched in aberrant epithelial cells. Our data identify
an IL11-stimulated ERK-dependent signaling pathway that promotes and maintains AT2 cells in a profibrotic KRT8+ state and induces the protein expression of pathologic ECM by alveolar
epithelial cells in vitro and in vivo. These findings further highlight the underappreciated potential of aberrant lung epithelial cells for the direct contribution of ECM components and
profibrotic factors that drive pathological lung remodeling. Furthermore, we found that the effects of TGFβ on the induction of ECM proteins and KRT8 expression in human alveolar and distal
airway epithelial cells and mouse AT2 cells was, in part, mediated by IL11 signaling. Our data are consistent with a previous report that showed the importance of IL11-dependent ERK
signaling in promoting EMT and senescence of AT2 cells in a _Bmi-1_ deficient model of premature senescence45. Additionally, emerging evidence from a recent study utilizing lung epithelial
cell organoids showed that IL11 negatively impacted the formation of SFTPC-expressing organoids, suggesting additional roles for IL11 in causing alveolar epithelial progenitor dysfunction46.
These findings may have implications for other airway/lung disorders such as Hermansky–Pudlak syndrome-associated pulmonary fibrosis, severe asthma, and severe viral pneumonitis, including
SARS-COV-2 infection, where IL11 levels are elevated and implicated in disease pathogenesis11,23,47,48,49. Specialized lung mesenchymal cells form a supportive niche that maintains the
progenitor properties of AT2 cells under homeostatic conditions50,51. In disease, impaired alveolar repair may arise due to disruption of this supportive niche and the development instead of
a profibrotic niche composed of pathological fibroblasts and dysfunctional alveolar epithelial cells52. Given the elevated expression of IL11 in aberrant mesenchymal and epithelial cell
types in PF and its roles in both fibroblast activation and AT2 cell dysfunction, we propose that IL11 may cause multiple aspects of pathobiology in different cell types in the diseased
niche (Supplementary Fig. 23). In support of this concept, a recent study demonstrated that the expression of IL11 by pathological lung fibroblasts from ILD-patients can potentially initiate
aberrant epithelial differentiation signatures in iPSC-derived alveolar organoid systems53. There are limitations to our study. Although several recent studies have shown that IL11 is
upregulated in the lungs and _SFTPC_+ cells from patients with IPF22,23,24, in situ studies of IL11 expression in diseased human lung tissue are required to further validate these findings.
Although our data on _Sftpc-CreER;Il11__fl/fl_ mice demonstrated that the specific deletion of _Il11_ in AT2 cells only was not sufficient to protect mice against lung fibrosis, we did not
dissect other cell type(s) expressing IL11 that can impact AT2-to-AT1 differentiation and fibrosis, despite our earlier studies suggesting a dominant role for IL11 secretion from fibroblasts
for fibrosis phenotypes25. Moreover, conventional signaling studies on primary AT2 cells pose significant challenges due to the lack of proliferative capacity and rapid loss of primary
phenotypes in vitro. Hence, the downstream molecular mechanisms of IL11 signaling in AT2 cells remain to be elucidated. In light of recent evidence highlighting the importance of distal
airway secretory/basal cells in aberrant alveolar repair and fibrosis52,54,55,56, the effects of IL11 on the recruitment and differentiation of airway/ progenitor cells towards KRT8+ and AT1
cells require study. In conclusion, we suggest that IL11 causes lung pathology in severe lung disease through at least two pathological processes. First, causing AT2 dysfunction and
maintenance of a profibrotic KRT8+ cell state, thus limiting terminal AT1 differentiation and impairing alveolar regeneration. And second, stimulating fibroblast-to-myofibroblast
transformation and the expression of pathologic ECM proteins by profibrotic KRT8+ cells that leads to lung fibrosis and inflammation25. Hence, anti-IL11 therapeutics, which are advancing
towards clinical trials in patients with PF, may promote alveolar regeneration and mitigate lung fibrosis that would differentiate anti-IL11 therapy from anti-fibrotics currently used in the
clinic. METHODS ETHICS All experiments and animal procedures were approved and performed in accordance with guidelines set by the Institutional Animal Care and Use Committee at SingHealth
(Singapore) and the SingHealth Institutional Biosafety Committee. COMPUTATIONAL ANALYSIS OF SCRNA-SEQ DATASETS OF HUMAN PULMONARY FIBROSIS Processed human PF scRNA-seq datasets by Habermann
et al. and Adams et al., were downloaded from GEO with the accession number GSE135893 and GSE136831, respectively. Cell-type annotations and Uniform Manifold Approximation and Projection
(UMAP) coordinates provided by the authors were used in subsequent analyses. TRAJECTORY ANALYSIS We re-classified alveolar epithelial cells in the Adams et al., dataset with cell-type
annotations defined by Habermann et al., using Seurat’s default label transfer pipeline. The quality of label transfer was evaluated by the Jaccard Index (See Assessment of transcriptomic
similarities between epithelial cell-types below). Transitional AT2, KRT5-/KRT17+, and AT1 cells were extracted from the Habermann and Adams et al. dataset for Slingshot trajectory analysis
(Slingshot 1.8.0)39, and the analysis was performed separately for each dataset. Briefly, Slingshot derives differentiation paths from a specified origin and calculates for each cell a
pseudotime, which approximates the differentiation progression of a cell toward the destination of the trajectory. In this analysis, transitional AT2 cells were specified as the origin, and
two differentiation trajectories were derived, one to KRT5−/KRT17+ cells and the other to AT1 cells. Change in IL11 expression was evaluated along the two trajectories by fitting a
generalized additive model (GAM) with the expression of IL11 against pseudotime. ASSESSMENT OF TRANSCRIPTOMIC SIMILARITIES BETWEEN EPITHELIAL CELL-TYPES We examined transcriptional
similarities of different epithelial clusters using the Jaccard index (a cluster here refers to cells of the same cell-type from a specific study, e.g., AT2 cells from Habermann et al.
dataset). First, we performed differentially expressed gene (DEGs) analysis in epithelial cells from the same study, and for each cell-type retained upregulated DE genes with log2 fold
change (log2FC) above the 85th percentile of the FC distribution and discarded genes with expression proportion in a cluster less than 40% compared to other cell-types. We refer to these
genes as “markers” of a cluster, and a Jaccard index value was derived for all possible cluster pairing (of all epithelial clusters pooling together both datasets). A Jaccard index between
cluster A and cluster B was calculated by dividing the size of the intersection of their markers over the size of the union of their markers. NETWORK ANALYSIS Cells assigned to the
differentiation trajectory from transitional AT2 to KRT5−/KRT17+ cells by Slingshot analysis were selected for IL11 co-expression analysis, done individually in Habermann et al., and Adams
et al., dataset. Briefly, spearman correlations were calculated between the expression of IL11 and genes expressed in the selected cells. Genes with Spearman correlation with FDR <0.2
were kept. In summary, 103 genes were found to be significantly correlated with IL11 in Adams et al dataset, 378 genes in Habermann et al. dataset, and 32 genes in both datasets. Using the R
package EnrichR (enrichR 3.1.0)39,57, functional pathway enrichment analysis was performed on genes significantly correlated with IL11 (in individual datasets and combined) querying several
annotation databases including KEGG 2019 and MSigDB Hallmark 2020. Pathway terms with FDR <0.1 were retained. De-novo network construction was performed on the 32 genes significantly
correlated with IL11 in both datasets. Each node in the network represents a gene and each edge (connecting a pair of genes) the Spearman correlation between the expression of the two genes
in transitional AT2 and KRT5-/KRT17+ cells from Habermann et al., dataset. A graphical representation of the network was constructed in Cytoscape (Cytoscape 3.8.2)58, and genes overlapping
with the MSigDB Hallmark EMT process were colored. COMPUTATIONAL ANALYSIS OF SCRNA-SEQ DATASETS OF MURINE PULMONARY FIBROSIS Raw murine PF scRNA-seq datasets were downloaded by GEO with the
following accession numbers: GSE141259, GSE184854, and GSE12703. Cell-type classifications from GSE141259 were used to annotate cell clusters in the other two datasets. MOUSE STUDIES Animals
were maintained in a specific pathogen-free environment and had ad libitum access to food and water, with a 12-h light/dark cycle, at an ambient temperature of 21–24 °C and humidity of
40–70%. The following mice strains were maintained on a C57BL/6 background and used for the study: Sftpctm1(cre/ERT2)Blh (_Sftpc-CreER_)59, B6.Cg-_Gt(ROSA)26Sor__tm9(CAG-tdTomato)Hze_/J
(_R26-tdTomato_ mice), C57BL/6-_Il11ra1__em1Cook_/J (_Il11ra1__fl/fl_ mice)25, C57BL/6-_Il11__tm1.1Cook_/J (_IL11__EGFP_ reporter mice)27_, Il11__fl/fl_ mice60_. Sftpc-CreER_ mice were
crossed with _R26-tdTomato_ mice to generate _Sftpc-CreER; R26-tdTomato_ (_Sftpc-tdT_) mice for lineage tracing experiments. To model the deletion of _Il11ra1_ in AT2 cells, _Sftpc-CreER_
mice were crossed with _Il11ra1__fl/fl_ mice to generate _Sftpc-CreER; Il11ra1__fl/fl_ mice. Similarly, to model the deletion of _Il11_ in AT2 cells, _Sftpc-CreER_ mice were crossed with
_Il11__fl/fl_ mice to generate _Sftpc-CreER; Il11__fl/fl_ mice. _Sftpc-CreER; Il11ra1__fl/fl_ mice were further crossed with _R26-tdTomato_ mice to generate S_ftpc-tdT; Il11ra1__fl/fl_ mice.
_Sftpc-tdT_ mice were injected intraperitoneally with three consecutive doses of 100 mg/kg tamoxifen (Sigma-Aldrich) starting 14 days prior to bleomycin administration. _Sftpc-CreER;
Il11ra1__fl/fl_ mice, _Sftpc-CreER; Il11__fl/fl_ mice, and _Sftpc-tdT; Il11ra1__fl/fl_ mice were injected intraperitoneally with four doses of 75 mg/kg tamoxifen (Sigma-Aldrich) starting
from 14 days prior to bleomycin administration. Therapeutic doses of monoclonal anti-IL11 (X203, Genovac) were established previously22. X203 or IgG control antibodies were injected
intraperitoneally at 20 mg/kg starting from day 4 and subsequently on day 7 and day 10 post-bleomycin administration in the 12-day model of lung fibrosis. BLEOMYCIN MODEL OF LUNG INJURY The
bleomycin model of lung fibrosis was performed as previously described22. Briefly, male mice at 10–14 weeks of age were anesthetized by isoflurane inhalation and subsequently administered a
single dose of bleomycin (Sigma-Aldrich) oropharyngeally at 0.75 U/kg body weight (for _IL11__EGFP_ reporter mice) or 1.5 U/kg body weight (for all other mouse strains) in a volume of saline
not exceeding 50 µl per mouse. Uninjured control mice received equal volumes of saline oropharyngeally. Mice were sacrificed at indicated time points post-bleomycin administration and the
lungs were collected for downstream analysis. MOUSE LUNG DISSOCIATION, FLOW CYTOMETRY, AND FACS ANALYSIS Mice lung dissociation was performed as previously described with slight
adjustments61. Briefly, the lungs were perfused with cold sterile saline through the right ventricle. The lungs were then intratracheally inflated with 1.5 ml of Dispase 50 U/ml (Corning)
followed by installation of 0.5 ml of 1% low melting agarose (Bio-Rad) via the trachea. The lungs were excised and incubated on an orbital shaker for 45 min at room temperature. Each lobe
was then minced into small pieces in DMEM (GIBCO) supplemented with 10% FBS (GIBCO) and 0.33 U/ml DNase I (Roche) and placed on the orbital shaker for an additional 10 min. The cells were
then filtered through a 100 µm cell strainer and centrifuged at 400×_g_ for 5 min at 4 °C. The cell pellet was resuspended in ACK-buffer (GIBCO), incubated for 2 min on ice, and then
filtered through a 40 µm cell strainer. The cells were centrifuged at 400×_g_ for 5 min at 4 °C and resuspended in DPBS (GIBCO) supplemented with 5% FBS, and then stained with the following
antibodies: EpCAM-BV785 (BioLegend #118245), CD45-APC (BioLegend, 103112), CD31-APC/Cy7 (BioLegend, 102534), I-A/I-E - AlexaFluor488 (MHC-II) (BioLegend, 107616) and 4’,
6-diamidino-2-phenylindole (DAPI) (Life Technologies, 62248) was used to eliminate dead cells. The cells were then sorted on the BD FACS Aria III system (BD Bioscience). For flow cytometry
analysis, lung single-cell suspensions were obtained as described above. The cells were then stained for the following markers (CD45-APC, CD31-APC/Cy7, EpCAM-BV785, PDPN-FITC; all antibodies
at 1:200 dilution), fixed in 4% paraformaldehyde, permeabilized with 0.1% triton-X in DPBS, and stained for intracellular proteins in this order: Firstly, cells were stained with
anti-Claudin 4 primary (Invitrogen, 36-4800, 1:100) and anti-rabbit Alexa Fluor 488 secondary (Invitrogen, A32731, 1:200) antibody, followed sequentially by PE-conjugated anti-GFP primary
antibody (Abcam, ab303588) staining to prevent potential binding and overlap of Alexa Fluor 488 secondary and anti-GFP signals. The cells were then analyzed on the BD LSR Fortessa system (BD
Biosciences) and data was analyzed using FlowJo software (Tree Star). HUMAN AND MOUSE CELL CULTURES Human pulmonary alveolar epithelial cells (HPAEpiC) (ScienCell Research Laboratories,
3200) were supplied at passage 1, cultured in complete AEpiCM (ScienCell Research Laboratories), and were directly used for experiments after 24 h of acclimatization. Human small airway
epithelial cells (HSAEC) (Lonza Bioscience, CC-2547) were cultured in SAGMTM small airway epithelial cell growth medium kit (Lonza Bioscience, CC-3118) and used for experiments at passage 3.
Briefly, HPAEpiC or HSAEC were seeded at a density of 1.5e4 cells per well in 96-well CellCarrier plates (PerkinElmer) or 3e5 cells per well in six-well tissue culture plates (Corning).
HPAEpiC and HSAEC were synchronized in AEpiCM basal medium or SABMTM basal medium, respectively, for 16 h prior to cytokine or antibody treatment. For the assessment of cell proliferation,
cells were pulsed with EdU for 22 h prior to cell fixation and stained using the Click-iT EdU Labeling kit (Thermo Fisher Scientific, C10350) according to the manufacturer’s protocol. For
mouse AT2 cell cultures, _tdTomato_ positive (_tdT_+) cells were isolated from _Sftpc-tdT_ mice lungs by FACS sorting for live CD45- CD31- EpCAM+ _tdT_+ cells. The FACS-sorted cells were
then seeded at a density of 2e4 cells per well in rat tail collagen (Invitrogen, A1048301) coated 96-well CellCarrier plates (PerkinElmer) and cultured in DMEM supplemented with 10% FBS.
Mouse AT2 cells were allowed to adhere for 24 h prior to cytokine or antibody treatment. The various cytokines and antibodies used for in vitro experiments are as follows: Recombinant human
IL11 (UniProtKB:P20809, GenScript), recombinant human TGFβ1 (PHP143B, Bio-Rad), anti-IL11 antibody (X203, Genovac), IgG antibody (IIE10, Genovac), U0126 (Cell Signaling Technology, 9903),
recombinant mouse IL11 (UniProtKB: P47873, GenScript), recombinant mouse TGFβ1 (R&D Systems, 7666-MB). IN VITRO IMMUNOFLUORESCENCE IMAGING AND ANALYSIS Immunofluorescence imaging and
quantification of HPAEpiC, HSAEC, and mouse AT2 cells were performed on the Operetta High-Content Imaging System (PerkinElmer) as previously described in ref. 22. The cells were first fixed
in 4% paraformaldehyde (Thermo Fisher Scientific) and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS). The cells were then incubated with the following primary
antibodies against: KRT8 (Merck Millipore, MABT329, 1:100), Collagen I (Abcam, ab34710, 1:100), fibronectin (Abcam, ab2413, 1:100), IL11RA (Abcam, ab125015, 1:100), gp130 (Thermo Fisher
Scientific, PA5-28932, 1:100), IL6RA (Thermo Fisher Scientific, MA1-80456, 1:100), SFTPC (Santa Cruz, sc-518029; 1:100), or AGER (R&D Systems, MAB1179, 1:100) and visualized using Alexa
Flour 488-conjugated secondary antibodies. Cellular morphology was assessed by counterstaining with Phalloidin-iFluor 555 reagent (Abcam, ab176756). The permeabilization step was omitted for
membrane staining of gp130, IL6RA, and IL11RA. Plates were scanned and images were collected with the Operetta high-content imaging system (PerkinElmer). The percentages of proliferating
cells (EdU+ve cells) were quantified using the Harmony software version 3.5.2 (PerkinElmer). Each treatment condition was run in duplicate wells, and 7 to 14 fixed non-overlapping fields
were imaged and analyzed per treatment group. Quantification of immunofluorescence of protein markers in HPAEpiC and HSAEC experiments was performed using the built-in cell analysis tool on
the Columbus software (version 2.7.2, PerkinElmer). To determine the fluorescence intensities for each cell, individual cells were denoted based on the DAPI nuclei staining, and cell areas
were established based on the total cytoplasmic Alexa Fluor 488 signal. Fluorescence intensities of cytoplasmic Alexa Fluor 488 signals within each demarcated cell area were concurrently
measured, and fluorescence intensities for each cell were then further normalized to their respective area. Mean intensity/area per analyzed field are presented as one datapoint.
Quantification of immunostaining intensity of KRT8, PDPN, and Collagen I in mouse AT2 cells were analyzed by Fiji software and fluorescence intensities were normalized to cell area. RNA-SEQ
Total RNA was isolated from HPAEpiC with or without TGFβ1 or IL11 stimulation using RNeasy columns (Qiagen). RNA was quantified using Qubit™ RNA Broad Range Assay Kit (Life Technologies) and
assessed for degradation based on RNA Quality Score (RQS) using the RNA Assay and DNA 5 K/RNA/CZE HT Chip on a LabChip GX Touch HT Nucleic Acid Analyzer (PerkinElmer). TruSeq Stranded mRNA
Library Prep kit (Illumina) was used to assess transcript abundance following standard instructions from the manufacturer. Briefly, poly(A) + RNA was purified from 1 µg of total RNA with RQS
>9, fragmented, and used for cDNA conversion, followed by 3′ adenylation, adapter ligation, and PCR amplification. The final libraries were quantified using Qubit™ DNA Broad Range Assay
Kit (Life Technologies) according to the manufacturer’s guide. The quality and average fragment size of the final libraries were determined using DNA 1 K/12 K/Hi Sensitivity Assay LabChip
and DNA High Sensitivity Reagent Kit (PerkinElmer). Libraries with 16 unique dual indexes were pooled and sequenced on a NextSeq 500 benchtop sequencer (Illumina) using the NextSeq 500 High
Output v2 kit and 75-bp paired-end sequencing chemistry. RNA-SEQ ANALYSIS Libraries were demultiplexed using bcl2fastq v2.19.0.316 with the --no-lane-splitting option. Adapter sequences were
then trimmed using trimmomatic v0.3662 in paired end mode with the options MAXINFO:35:0.5 MINLEN:35. Trimmed reads were aligned to the _Homo sapiens_ GRCh38 using STAR v.2.2.163 with the
options --outFilterType BySJout --outFilterMultimapNmax 20 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --alignIntronMin 20 --alignIntronMax 1000000 retained
for counting. Counts were calculated at the gene level using the FeatureCounts module from subread v.1.5.164, with the options -O -s 2 -J -T 8 -p -R -G. The combined transcript model
annotation file was constructed using Ensembl hg38 and FANTOM5 hg38 as previously described65 and used as an annotation to prepare STAR indexes and for FeatureCounts. Differential expression
analyses were performed in R v4.2.0 using the Bioconductor package DESeq2 v1.36.066, using the Wald test for comparisons. For sample groups, the design for the model was specified as ~
stimulus (IL11/TGFβ1/baseline) + source (commercial tube 1–4), to account for the confounding effect of different batches of cells. SAMPLE PREPARATION FOR MOUSE LUNG SCRNA-SEQ Mouse lung
single-cell suspensions were generated and stained with antibodies as described above for FACS analysis. Lung epithelial cells were enriched by sorting for live CD45− CD31− EpCAM+ cells and
the cells from different mice were then labeled with unique sample oligo barcodes (sample-tag) using BDTM Ms Single Cell Sample Multiplexing Kit (BD Biosciences) and evaluated for cell
concentration and viability using C-Chip disposable hemocytometer (NanoEnTek) on a BD Rhapsody Scanner (BD Biosciences). SINGLE-CELL CAPTURE, CDNA LIBRARY CONSTRUCTION, AND SEQUENCING
Single-cell capturing was performed using the BD Rhapsody™ Express Single-Cell Analysis System (BD Biosciences). Libraries were generated using BD Rhapsody Whole-Transcriptome Analysis (WTA)
Amplification kit according to the manufacturer’s protocols. Briefly, 15,000 cells from each uniquely tagged sample were loaded together with beads with oligonucleotide barcodes onto the
cartridge containing microwells. Individual cells were lysed allowing the hybridization of mRNA-sample-tag molecules with beads before pooling and cDNA conversion. Sample-tags with barcode
information were denatured off of the beads, PCR-amplified, and indexed to generate sample-tag libraries. Then, random primers were hybridized to the cDNA on the remaining beads without
sample-tags, followed by an extension with an enzyme. Second-strand complementary DNA were then synthesized and ligated with adapters for PCR amplification to generate the
whole-transcriptome libraries. Next, the sample-tag and whole-transcriptome libraries were combined and spiked with 5% PhiX genome to increase the library complexity. The final libraries
were subsequently sequenced on a NovaSeq sequencer (Illumina) using a 150-bp paired-end run. SINGLE-CELL DATA PRE-PROCESSING AND ANALYSIS Raw sequencing data were demultiplexed using
bcl2fastq v2.19.0.316 with the --no-lane-splitting option. FASTQ files were demultiplexed using the unique sample-tags, trimmed, mapped, and annotated using the BD Rhapsody™ Sequence
Analysis Pipeline (Revision 2.0) on the Seven Bridges Genomics platform (accessed on June 2023). Low-quality read pairs were removed based on read length, average base quality score, and
highest single-base frequency. Filtered reads were aligned to the _Mus musculus_ genome (GRCm39 assembly) using STAR v2.7.4a embedded in the pipeline and annotated using the mouse GENCODE
release M31 GTF. Reads with identical cell labels, identical unique molecular identifier (UMI) sequences, and identical genes were collapsed into a single raw molecule, followed by removing
artifacts using recursive substitution error correction (RSEC) developed by BD Biosciences. Cells that had been identified as doublets or labeled as “undetermined” and genes that were
expressed in <3 cells were removed. Next, cells with expression of ≥200 genes, ≥500 unique molecular identifier (UMI) counts, >0.8 log10GenesPerUMI, and mitochondrial gene fraction
<30% were further processed. Seurat v5.0.1 was used to perform anchor-based CCA integration on the datasets. The top eight integrated components were used for uniform manifold
approximation and projection (UMAP) dimensional reduction before rejoining layers and data visualization in two dimensions. Unsupervised clustering was performed by the FindClusters function
using a shared nearest neighbor (SNN) modularity optimization-based clustering algorithm. The resulting clusters were manually labeled based on the top differentially expressed genes in
each cluster against all other clusters and conserved among the groups. PSEUDOBULK DIFFERENTIAL EXPRESSION, GSEA, AND ADDMODULESCORE ANALYSES Krt8+ transitional epithelial cells labeled as
Krt8ADI (_Krt8_+, _Cldn4_+) were subset from the entire epithelial cell dataset. Raw counts in all Krt8ADI cells were extracted from the Seurat object and subsequently aggregated to form
count matrices for each sample. Differential expression analyses were performed in R v4.2.0 using the Bioconductor package DESeq2 v1.38.3, using the Wald test for comparisons. Gene set
enrichment analyses (GSEA) were run using the fgsea v.1.22.0, pre-ranking the gene list by the “stat” column of the DESeq2 results output, and 105 permutations against mouse MSigDB (msigdbr
v.7.5.1) “Hallmark”67 mouse gene sets. Enriched gene set activities were calculated using the AddModuleScore Seurat function to compare the expression of the genes of interest as defined by
the GSEA leading-edge analysis. Wilcoxon test was then performed to study the difference in gene set activities between genotypes or treatment groups. Visualizations were generated with
Seurat, Nebulosa, scCustomize, and ggplot2 R packages68,69. For trajectory analysis, AT2, Activated AT2, Krt8ADI, and immature and mature AT1 were subset from the alveolar epithelial cell
dataset and split into two different Seurat objects based on the genotypes for Slingshot trajectory analyses (v2.6.0). Pseudotime estimate, lineage(s) assignment, and simultaneous principal
curve(s) were constructed by Slingshot _getLineages()_ and _getCurves()_ functions for different genotypes to infer cell transitional stages. LUNG HISTOLOGY AND IMMUNOHISTOCHEMISTRY Mouse
lung tissue (left lobes) were fixed in 10% formalin for 16–20 h, dehydrated and embedded in paraffin, and sectioned (7 µm) for Masson’s trichrome staining as described previously22.
Histological analysis for fibrosis was performed blinded to genotype and treatment exposure as previously described22. For immunostaining, the lungs were fixed in 4% paraformaldehyde at 4 °C
for 16 h, followed by serial 15 to 30% sucrose in PBS dehydration for 48 h. The tissues were then embedded in an OCT compound prior to sectioning (10 µm). The sections were incubated
overnight at 4 °C with the following primary antibodies: KRT8 (Merck Millipore, MABT329, 1:100), p-ERK (Cell Signaling Technology, 4370 or 5726, 1:100), Ki67 (Abcam, ab16667, 1:100), SFTPC
(Abcam, ab211326, 1:100), GFP (Abcam, ab290/ab6673, 1:100), AGER (R&D Systems, MAB1179, 1:200), Podoplanin (R&D Systems, AF3244, 1:200), IL11 (Invitrogen, PAS-95982, 1:100), CD45
(Proteintech, 20103-1-AP, 1:50), PDGFRA (R&D Systems, AF1062, 1:100), Collagen I (Abcam, ab21286, 1:100), CTGF (Abcam, ab6992, 1:100), XBP1 (Abcam, ab37152, 1:100). Alexa
Fluor-conjugated secondary antibodies (Invitrogen, 1:500) were incubated at room temperature for 60 min. Nuclei were stained with DAPI (Invitrogen, 1:1000). Images were captured using the
Leica DMi8 microscope (Leica Microsystems) with a 20X or 40X objective. The cells were counted based on positive staining for immunohistochemistry markers and DAPI using Fiji software. Five
to ten non-overlapping images for each unique marker were analyzed per mouse lung, and the mean values per mouse were presented. COLORIMETRIC ASSAYS Detection of secreted IL11 into the
supernatant of HPAEpiC and HSAEC cultures were performed using the human IL-11 ELISA kit (R&D systems, D1100) according to manufacturers’ instructions. Detection of SFTPD in mouse serum
was performed using the mouse SP-D ELISA kit (ab240683) according to the manufacturer’s instructions. Total lung hydroxyproline content of the right lung of _Sftpc-CreER; Il11__fl/fl_ mice
or the right caudal lung lobes of _Sftpc-tdT and Sftpc-CreER; Il11ra1__fl/fl_ mice were measured using the Quickzyme Total Collagen assay kit (Quickzyme Biosciences) as previously
described22. Soluble collagen in HSAEC culture supernatants were quantified using the Sirius Red collagen detection kit (9062, Chondrex) following the manufacturer’s protocol. WESTERN BLOT
Total proteins were extracted from mouse right lung tissues using RIPA lysis buffer (Thermo Fisher Scientific) containing protease and phosphatase inhibitors (Thermo Fisher Scientific).
Protein concentrations were determined by a BCA protein assay kit (Thermo Fisher Scientific). Protein lysates were separated by SDS-PAGE before being transferred onto PVDF membranes and
stained with the following primary antibodies against: RAGE/AGER (Proteintech, 16346-1-AP), KRT8 (Merck Millipore, MABT329) or GAPDH (Cell Signaling, 2118). Blots were then incubated with
the appropriate secondary antibodies before being visualized using ECL Western Blotting substrate (Thermo Fisher Scientific). STATISTICAL ANALYSIS Statistical analyses for in vitro and in
vivo data were performed using GraphPad Prism (v9). Analyses of experimental data were performed using two-tailed Student’s _t_-test or one-way ANOVA, as indicated in the figure legends. For
comparisons between multiple treatment groups, _P_ values were corrected for multiple testing using Tukey’s test. _P_ values <0.05 were considered statistically significant. REPORTING
SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY All data associated with this study are
presented in the paper or in the Supplementary Materials. Raw RNA sequencing data generated for this study have been uploaded onto Gene Expression Omnibus under the accession GSE261794.
Source data are provided in the Supplementary Information/Source Data file. Source data are provided with this paper. CODE AVAILABILITY All codes generated for this study are available on
Zenodo repository: [https://doi.org/10.5281/zenodo.13315637] and the GitHub link: [https://github.com/henryhuang12345/IL11_AEC_2024]. REFERENCES * Barkauskas, C. E. et al. Type 2 alveolar
cells are stem cells in adult lung. _J. Clin. Invest._ 123, 3025–3036 (2013). Article CAS PubMed PubMed Central Google Scholar * Desai, T. J., Brownfield, D. G. & Krasnow, M. A.
Alveolar progenitor and stem cells in lung development, renewal and cancer. _Nature_ 507, 190–194 (2014). Article ADS CAS PubMed PubMed Central Google Scholar * Basil, M. C. et al. The
cellular and physiological basis for lung repair and regeneration: past, present, and future. _Cell Stem Cell_ 26, 482–502 (2020). Article CAS PubMed PubMed Central Google Scholar *
Zepp, J. A. & Morrisey, E. E. Cellular crosstalk in the development and regeneration of the respiratory system. _Nat. Rev. Mol. Cell Biol._ 20, 551–566 (2019). Article CAS PubMed
PubMed Central Google Scholar * Xu, Y. et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. _JCI Insight_ 1, e90558 (2016).
Article PubMed PubMed Central Google Scholar * Adams, T. S. et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. _Sci.
Adv._ 6, eaba1983 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Habermann, A. C. et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial
and mesenchymal lineages in pulmonary fibrosis. _Sci. Adv._ 6, eaba1972 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Reyfman, P. A. et al. Single-cell transcriptomic
analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. _Am. J. Respir. Crit. Care Med._ 199, 1517–1536 (2019). Article CAS PubMed PubMed Central Google
Scholar * Morse, C. et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. _Eur. Respir. J._ 54, 1802441 (2019). Article CAS PubMed PubMed Central
Google Scholar * Delorey, T. M. et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. _Nature_ 595, 107–113 (2021). Article ADS CAS PubMed PubMed Central
Google Scholar * Sinha, S. et al. COVID-19 lung disease shares driver AT2 cytopathic features with Idiopathic pulmonary fibrosis. _EBioMedicine_ 82, 104185 (2022). Article CAS PubMed
PubMed Central Google Scholar * Melms, J. C. et al. A molecular single-cell lung atlas of lethal COVID-19. _Nature_ 595, 114–119 (2021). Article ADS CAS PubMed PubMed Central Google
Scholar * Strunz, M. et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. _Nat. Commun._ 11, 3559 (2020). Article ADS CAS
PubMed PubMed Central Google Scholar * Choi, J. et al. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. _Cell Stem
Cell_ 27, 366–382.e7 (2020). Article CAS PubMed PubMed Central Google Scholar * Kobayashi, Y. et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell
state in pulmonary fibrosis. _Nat. Cell Biol._ 22, 934–946 (2020). Article CAS PubMed PubMed Central Google Scholar * Choi, J. et al. Release of Notch activity coordinated by IL-1β
signalling confers differentiation plasticity of airway progenitors via Fosl2 during alveolar regeneration. _Nat. Cell Biol._ 23, 953–966 (2021). Article CAS PubMed PubMed Central Google
Scholar * Wu, H. et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. _Cell_ 184, 845–846 (2021). Article CAS PubMed Google Scholar *
Auyeung, V. C. et al. IRE1α drives lung epithelial progenitor dysfunction to establish a niche for pulmonary fibrosis. _Am. J. Physiol. Lung Cell. Mol. Physiol._ 322, L564–L580 (2022).
Article CAS PubMed PubMed Central Google Scholar * Katzen, J. et al. Disruption of proteostasis causes IRE1 mediated reprogramming of alveolar epithelial cells. _Proc. Natl Acad. Sci.
USA_ 119, e2123187119 (2022). Article CAS PubMed PubMed Central Google Scholar * Yao, C. et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. _Am. J.
Respir. Crit. Care Med._ 203, 707–717 (2021). Article CAS PubMed PubMed Central Google Scholar * Ng, B., Cook, S. A. & Schafer, S. Interleukin-11 signaling underlies fibrosis,
parenchymal dysfunction, and chronic inflammation of the airway. _Exp. Mol. Med._ 52, 1871–1878 (2020). Article CAS PubMed PubMed Central Google Scholar * Ng, B. et al. Interleukin-11
is a therapeutic target in idiopathic pulmonary fibrosis. _Sci. Transl. Med._ 11, eaaw1237 (2019). Article CAS PubMed Google Scholar * Strikoudis, A. et al. Modeling of fibrotic lung
disease using 3D organoids derived from human pluripotent stem cells. _Cell Rep._ 27, 3709–3723.e5 (2019). Article CAS PubMed PubMed Central Google Scholar * Bai, X. et al. Inhaled
siRNA nanoparticles targeting inhibit lung fibrosis and improve pulmonary function post-bleomycin challenge. _Sci. Adv._ 8, eabn7162 (2022). Article ADS CAS PubMed PubMed Central Google
Scholar * Ng, B. et al. Fibroblast‐specific IL11 signaling drives chronic inflammation in murine fibrotic lung disease. _FASEB J._ 34, 11802–11815 (2020). Article CAS PubMed Google
Scholar * Stancil, I. T. et al. Interleukin-6–dependent epithelial fluidization initiates fibrotic lung remodeling. _Sci. Transl. Med._ 14, eabo5254 (2022). Article CAS PubMed PubMed
Central Google Scholar * Widjaja, A. A. et al. Redefining IL11 as a regeneration-limiting hepatotoxin and therapeutic target in acetaminophen-induced liver injury. _Sci. Transl. Med._ 13,
eaba8146 (2021). Article CAS PubMed Google Scholar * Willis, B. C. & Borok, Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. _Am. J. Physiol. Lung
Cell. Mol. Physiol._ 293, L525–L534 (2007). Article ADS CAS PubMed Google Scholar * Jiang, P. et al. Ineffectual type 2–to–type 1 alveolar epithelial cell differentiation in idiopathic
pulmonary fibrosis: persistence of the KRT8hi transitional state. _Am. J. Respir. Crit. Care Med._ 201, 1443–1447 (2020). Article PubMed PubMed Central Google Scholar * Zhao, L., Yee, M.
& O’Reilly, M. A. Transdifferentiation of alveolar epithelial type II to type I cells is controlled by opposing TGF-β and BMP signaling. _Am. J. Physiol. Lung Cell. Mol. Physiol._ 305,
L409–L418 (2013). Article CAS PubMed PubMed Central Google Scholar * Enomoto, Y. et al. Autocrine TGF-β-positive feedback in profibrotic AT2-lineage cells plays a crucial role in
non-inflammatory lung fibrogenesis. _Nat. Commun._ 14, 4956 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * Liberti, D. C. et al. Alveolar epithelial cell fate is
maintained in a spatially restricted manner to promote lung regeneration after acute injury. _Cell Rep._ 35, 109092 (2021). Article CAS PubMed PubMed Central Google Scholar * Khalil,
N., O’Connor, R. N., Flanders, K. C., Shing, W. & Whitman, C. I. Regulation of type II alveolar epithelial cell proliferation by TGF-beta during bleomycin-induced lung injury in rats.
_Am. J. Physiol._ 267, L498–L507 (1994). CAS PubMed Google Scholar * Schafer, S. et al. IL-11 is a crucial determinant of cardiovascular fibrosis. _Nature_ 552, 110–115 (2017). Article
ADS CAS PubMed PubMed Central Google Scholar * Riemondy, K. A. et al. Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. _JCI
Insight_ 5, e123637 (2019). Article PubMed Google Scholar * Gaunsbaek, M. Q., Rasmussen, K. J., Beers, M. F., Atochina-Vasserman, E. N. & Hansen, S. Lung surfactant protein D (SP-D)
response and regulation during acute and chronic lung injury. _Lung_ 191, 295–303 (2013). Article CAS PubMed PubMed Central Google Scholar * Wang, F. et al. Regulation of epithelial
transitional states in murine and human pulmonary fibrosis. _J. Clin. Invest._ 133, e165612 (2023). Article CAS PubMed PubMed Central Google Scholar * DiGiovanni, G. T. et al.
Epithelial Yap/Taz are required for functional alveolar regeneration following acute lung injury. _JCI Insight_ 8, e173374 (2023). Article PubMed PubMed Central Google Scholar * Street,
K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. _BMC Genomics_ 19, 477 (2018). Article PubMed PubMed Central Google Scholar * Vaughan, A. E.
et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. _Nature_ 517, 621–625 (2015). Article ADS CAS PubMed Google Scholar * Liu, Q. et al. Lung
regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. _Nat. Genet._ 51, 728–738 (2019). Article CAS PubMed Google Scholar * Ng, B. et al. Similarities
and differences between IL11 and IL11RA1 knockout mice for lung fibro-inflammation, fertility and craniosynostosis. _Sci. Rep._ 11, 14088 (2021). Article ADS CAS PubMed PubMed Central
Google Scholar * Katsura, H., Kobayashi, Y., Tata, P. R. & Hogan, B. L. M. IL-1 and TNFα contribute to the inflammatory Niche to enhance alveolar regeneration. _Stem Cell Rep._ 12,
657–666 (2019). Article CAS Google Scholar * Khanna, D. et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. _Lancet Respir. Med._ 8,
963–974 (2020). Article CAS PubMed Google Scholar * Chen, H. et al. TGF-β1/IL-11/MEK/ERK signaling mediates senescence-associated pulmonary fibrosis in a stress-induced premature
senescence model of Bmi-1 deficiency. _Exp. Mol. Med._ 52, 130–151 (2020). Article CAS PubMed PubMed Central Google Scholar * Kortekaas, R. K. et al. Interleukin-11 disrupts alveolar
epithelial progenitor function. _ERJ Open Res._ 9, 00679–02022 (2023). Article PubMed PubMed Central Google Scholar * Minshall, E. et al. IL-11 expression is increased in severe asthma:
association with epithelial cells and eosinophils. _J. Allergy Clin. Immunol._ 105, 232–238 (2000). Article CAS PubMed Google Scholar * Gamage, A. M. et al. Infection of human nasal
epithelial cells with SARS-CoV-2 and a 382-nt deletion isolate lacking ORF8 reveals similar viral kinetics and host transcriptional profiles. _PLoS Pathog._ 16, e1009130 (2020). Article CAS
PubMed PubMed Central Google Scholar * Winkler, E. S. et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. _Nat. Immunol._
21, 1327–1335 (2020). Article CAS PubMed PubMed Central Google Scholar * Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A. & Desai, T. J. Single-cell Wnt signaling
niches maintain stemness of alveolar type 2 cells. _Science_ 359, 1118–1123 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Zepp, J. A. et al. Distinct mesenchymal
lineages and Niches promote epithelial self-renewal and myofibrogenesis in the lung. _Cell_ 170, 1134–1148.e10 (2017). Article CAS PubMed PubMed Central Google Scholar * Kathiriya, J.
J. et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5 basal cells. _Nat. Cell Biol._ 24, 10–23 (2022). Article CAS PubMed Google Scholar * Kastlmeier, M.
T. et al. Cytokine signaling converging on in ILD fibroblasts provokes aberrant epithelial differentiation signatures. _Front. Immunol._ 14, 1128239 (2023). Article CAS PubMed PubMed
Central Google Scholar * Jaeger, B. et al. Airway basal cells show a dedifferentiated KRT17Phenotype and promote fibrosis in idiopathic pulmonary fibrosis. _Nat. Commun._ 13, 5637 (2022).
Article ADS CAS PubMed PubMed Central Google Scholar * Kadur Lakshminarasimha Murthy, P. et al. Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. _Nature_
604, 111–119 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Basil, M. C. et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli.
_Nature_ 604, 120–126 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool.
_BMC Bioinformatics_ 14, 128 (2013). Article PubMed PubMed Central Google Scholar * Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular
interaction networks. _Genome Res._ 13, 2498–2504 (2003). Article CAS PubMed PubMed Central Google Scholar * Rock, J. R. et al. Multiple stromal populations contribute to pulmonary
fibrosis without evidence for epithelial to mesenchymal transition. _Proc. Natl Acad. Sci. USA_ 108, E1475–E1483 (2011). Article CAS PubMed PubMed Central Google Scholar * Dong, J. et
al. Hepatocyte specific gp130 signalling underlies APAP induced liver injury. _Int. J. Mol. Sci._ 23, 7089 (2022). Article CAS PubMed PubMed Central Google Scholar * Chen, Q. & Liu,
Y. Isolation and culture of mouse alveolar type II cells to study type II to type I cell differentiation. _STAR Protoc._ 2, 100241 (2021). Article CAS PubMed Google Scholar * Bolger, A.
M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. _Bioinformatics_ 30, 2114–2120 (2014). Article CAS PubMed PubMed Central Google Scholar *
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS PubMed Google Scholar * Liao, Y., Smyth, G. K. & Shi, W. The Subread
aligner: fast, accurate and scalable read mapping by seed-and-vote. _Nucleic Acids Res._ 41, e108 (2013). Article PubMed PubMed Central Google Scholar * Chothani, S. P. et al. A
high-resolution map of human RNA translation. _Mol. Cell_ 82, 2885–2899.e8 (2022). Article CAS PubMed Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of
fold change and dispersion for RNA-seq data with DESeq2. _Genome Biol._ 15, 550 (2014). Article PubMed PubMed Central Google Scholar * Liberzon, A. et al. The molecular signatures
database (MSigDB) hallmark gene set collection. _Cell Syst._ 1, 417–425 (2015). Article CAS PubMed PubMed Central Google Scholar * Alquicira-Hernandez, J. & Powell, J. E. Nebulosa
recovers single-cell gene expression signals by kernel density estimation. _Bioinformatics_ 37, 2485–2487 (2021). Article CAS PubMed Google Scholar * Hao, Y. et al. Integrated analysis
of multimodal single-cell data. _Cell_ 184, 3573–3587.e29 (2021). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the
National Medical Research Council (NMRC) Singapore (NMRC/STaR/0029/2017 to S.A.C., MOH-OFIRG21nov-0006 to A.A.W., and NMRC-OFYIRG21jun-0022 to B.N. and S.V.), NMRC Central Grant to NHCS
(MOH-CIRG18nov-0002 to S.A.C.), Goh Cardiovascular Research Award (Duke-NUS-GCR/2015/0014 to S.A.C.), Tanoto Foundation (to S.A.C.), Advanced Manufacturing and Engineering Young Individual
Research Grant (AME YIRG) of Agency for Science, Technology and Research (A*STAR) award (A2084c0157 to W.-W.L. and B.N.); and a research grant from Boehringer Ingelheim. We thank Dr. Jinrui
Dong for her support with in vivo experiments and Mr. Daryl Yeong for his support for histology. We thank Laura and Charles Lou at AMC research core services for their technical support for
FACS analysis. AUTHOR INFORMATION Author notes * These authors contributed equally: Kevin Y. Huang, Chee Jian Pua. AUTHORS AND AFFILIATIONS * National Heart Research Institute Singapore,
National Heart Center Singapore, Singapore, Singapore Benjamin Ng, Chee Jian Pua, Wei-Wen Lim, An An Hii & Stuart A. Cook * Cardiovascular and Metabolic Disorders Program, Duke-National
University of Singapore Medical School, Singapore, Singapore Benjamin Ng, Kevin Y. Huang, Sivakumar Viswanathan, Wei-Wen Lim, Fathima F. Kuthubudeen, Yu-Ning Liu, Benjamin L. George, Anissa
A. Widjaja, Enrico Petretto & Stuart A. Cook * Center for Computational Biology, Duke-National University of Singapore Medical School, Singapore, Singapore Enrico Petretto * MRC-London
Institute of Medical Sciences, Hammersmith Hospital Campus, London, United Kingdom Stuart A. Cook Authors * Benjamin Ng View author publications You can also search for this author inPubMed
Google Scholar * Kevin Y. Huang View author publications You can also search for this author inPubMed Google Scholar * Chee Jian Pua View author publications You can also search for this
author inPubMed Google Scholar * Sivakumar Viswanathan View author publications You can also search for this author inPubMed Google Scholar * Wei-Wen Lim View author publications You can
also search for this author inPubMed Google Scholar * Fathima F. Kuthubudeen View author publications You can also search for this author inPubMed Google Scholar * Yu-Ning Liu View author
publications You can also search for this author inPubMed Google Scholar * An An Hii View author publications You can also search for this author inPubMed Google Scholar * Benjamin L. George
View author publications You can also search for this author inPubMed Google Scholar * Anissa A. Widjaja View author publications You can also search for this author inPubMed Google Scholar
* Enrico Petretto View author publications You can also search for this author inPubMed Google Scholar * Stuart A. Cook View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS B.N. and S.A.C. conceptualized the study. K.Y.H. and C.J.P. performed computational analyses. B.N., F.F.K., B.L.G., and S.V. performed in vitro and ex vivo
experiments. B.N., W.-W.L., F.F.K., and Y.-N.L. performed in vivo studies and histological evaluations. C.J.P. and A.A.H. performed scRNA-seq and bulk RNA-seq experiments. B.L.G. performed
animal genotyping and provided administrative support. B.N., E.P., and S.A.C. supervised the study. B.N., A.A.W., and S.A.C acquired funding for the study. Data were curated, processed, and
visualized by B.N., K.Y.H., C.J.P., and W.-W.L. B.N. and S.A.C. drafted, reviewed, and edited the manuscript with input from all other authors. CORRESPONDING AUTHORS Correspondence to
Benjamin Ng or Stuart A. Cook. ETHICS DECLARATIONS COMPETING INTERESTS S.A.C. is a co-inventor of the patent applications (WO/2017/103108) and (WO/2018/109170). S.A.C., W.-W.L., and B.N. are
co-inventors of the patent application (WO/2019/073057). S.A.C. is a co-founder and shareholder of Enleofen Bio PTE LTD, a company that develops anti-IL11 therapeutics, which were acquired
for further development by Boehringer Ingelheim. The remaining authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Andrew Gelman,
Hiroyasu Nakano and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE
DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5 SUPPLEMENTARY DATA 6 SUPPLEMENTARY DATA
7 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use,
sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative
Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ng, B., Huang, K.Y., Pua, C.J. _et al._ Interleukin-11 causes alveolar type 2 cell dysfunction and prevents alveolar regeneration. _Nat
Commun_ 15, 8530 (2024). https://doi.org/10.1038/s41467-024-52810-8 Download citation * Received: 13 December 2022 * Accepted: 19 September 2024 * Published: 02 October 2024 * DOI:
https://doi.org/10.1038/s41467-024-52810-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative





