- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Domestication – the artificial selection of wild species to obtain variants with traits of human interest – was integral to the rise of complex societies. The oversupply of food was
probably associated with the formalization of food preservation strategies through microbial fermentation. While considerable literature exists on the antiquity of fermented food, only few
eukaryotic microbes have been studied so far for signs of domestication, less is known for bacteria. Here, we tested if cheese starter cultures harbour typical hallmarks of domestication by
characterising over 100 community samples and over 100 individual strains isolated from historical and modern traditional Swiss cheese starter cultures. We find that cheese starter cultures
have low genetic diversity both at the species and strain-level and maintained stable phenotypic traits. Molecular clock dating further suggests that the evolutionary origin of the bacteria
approximately coincided with the first archaeological records of cheese making. Finally, we find evidence for ongoing genome decay and pseudogenization via transposon insertion related to a
reduction of their niche breadth. Future work documenting the prevalence of these hallmarks across diverse fermented food systems and geographic regions will be key to unveiling the joint
history of humanity with fermented food microbes. SIMILAR CONTENT BEING VIEWED BY OTHERS FUNCTIONAL STRAIN REDUNDANCY AND PERSISTENT PHAGE INFECTION IN SWISS HARD CHEESE STARTER CULTURES
Article Open access 06 August 2021 META-ANALYSIS OF CHEESE MICROBIOMES HIGHLIGHTS CONTRIBUTIONS TO MULTIPLE ASPECTS OF QUALITY Article 13 August 2020 LARGE-SCALE GENOME-WIDE ANALYSIS LINKS
LACTIC ACID BACTERIA FROM FOOD WITH THE GUT MICROBIOME Article Open access 25 May 2020 INTRODUCTION Domestication is the process of modifying wild species through artificial selection to the
benefit of a “domesticator”, which is usually human1,2. This process was integral to the rise of complex human societies3,4,5. In particular, the domestication of crop plants6 and
livestock7 over the past 12,000 years4 enabled the emergence of social complexity and initiated large-scale anthropogenic changes in the earth’s biosphere8. Plant and animal domestication is
characterised by phenotypic and genetic changes from the wild ancestor, such as adaptation of body mass9 or change in nutrient content10, which overall contributed to the production of food
surplus. This oversupply of food was associated with the formalisation of food preservation strategies by microbial fermentation – a metabolic process that converts sugars into acids – to
produce, for example, fermented vegetables, wine or cheese11,12. This leads to a decrease in pH, which reduces undesired microbial growth and prevents spoilage of stored food. These
processes were likely adopted by humans many millennia ago13. For cheese, the earliest archaeological evidence can be found within the Neolithic (ca. 12th-5th millennium B.C.)11,13 and
likely became more common and important during the Copper, Bronze and Iron Age (ca. 5th-1st millennium B.C.)14. Fermented food products have diversified in a myriad of forms all over the
globe15,16, constituting healthy and tasty components of the human diet, which are key in many cultures and sustainable opportunities for the future of food in others17. This raises the
fundamental question of whether, as for plants and animals, fermented food microbes have also been domesticated by humans, and if so, when and how this has happened18,19. It is commonly
accepted that some fermented food microbes have been maintained through continuous passaging20,21 and artificial selection for specific traits (e.g., shelf life or taste). Accordingly, we
expect them to present typical “hallmarks” of domestication, i.e., genomic and phenotypic signatures associated with (microbial) domestication that distinguish them from their wild
counterparts. In fact, a handful of microbial domestication cases have been documented empirically22,23, mostly for eukaryotic microbes. For example, _Saccharomyces cerevisiae_ used for
bread making24 and alcoholic fermentation25,26, _Aspergillus oryzae_ used for sake, soy sauce, and miso production27 and _Penicillium camemberti_ found on camembert cheese rind28 show
genomic and phenotypic characteristics that distinguish them from their wild counterparts18,29,30. Thus, while the genomic signatures of domestication are well-defined for eukaryotic
microbes, we know less about the collective genomic and phenotypic consequences of bacterial domestication used for food fermentation29,31. For example, _Oenococcus oeni_, which is
responsible for the malolactic conversion in winemaking, is thought to have rapidly diverged from its ancestor due to the emergence of hypermutator strains, but it remains unclear to what
extent subsequent evolution was influenced by human domestication32. Similarly, strains of _Lacticaseibacillus paracasei_ isolated from ripening cheese show signs of adaptation to milk but
lack other hallmarks of domestication33. Altogether, this suggests that most suspected cases of domestication in fermented food bacteria lack a complete characterisation of domestication
hallmarks previously observed in microbial eukaryotes like yeast. To explain this discrepancy, we posit that, unlike yeasts (i.e., in beer, bread and wine), the previously studied bacteria
have not undergone continuous passaging with rapid and iterative bursts of growth solely on the fermented foods but rather persisted in food-associated environments (e.g., grape skin for
_Oenococcus oeni_). This reduces the strength with which artificial selection can act on evolution. In contrast, cheese starter cultures (starter cultures) are a promising candidate to test
for domestication in fermentation. These cultures have been passaged and selected for thousands of years via backslopping (i.e., continuous re-inoculation of previous-day whey) and have been
extensively selected for flavour and rapid acidification purposes34. Starter cultures are categorised by their usage temperature: mesophilic (below 42 °C) and thermophilic (above 42 °C).
Mesophilic cultures, dominated by _Lactococcus lactis_ or _L. cremoris_, show taxonomic and genetic adaptations to the dairy environment, namely the loss of genes encoding functions needed
in animal and plant environments29. However, these species maintain comparatively large genomes30 and occur in multiple environments31 partially due to the presence of diverse plasmids32,33.
Thermophilic starter cultures are dominated by three thermophilic bacteria, namely _Streptococcus thermophilus_ and _Lactobacillus delbrueckii_ subsp. _lactis_ (hereafter only _L.
delbrueckii_) and _Lactobacillus helveticus_35. Although previous genomic analyses have shown signs of genome decay in these species22,36,37,38,39,40, systematic screening for domestication
hallmarks in these microbial communities is still lacking20,21. In this study, we aimed to detect signs of domestication in thermophilic starter cultures and to date the potential
domestication events. To this end, we collected both modern and historic (1970s) samples of 11 undefined starter cultures that are continuously passaged as undefined starter cultures
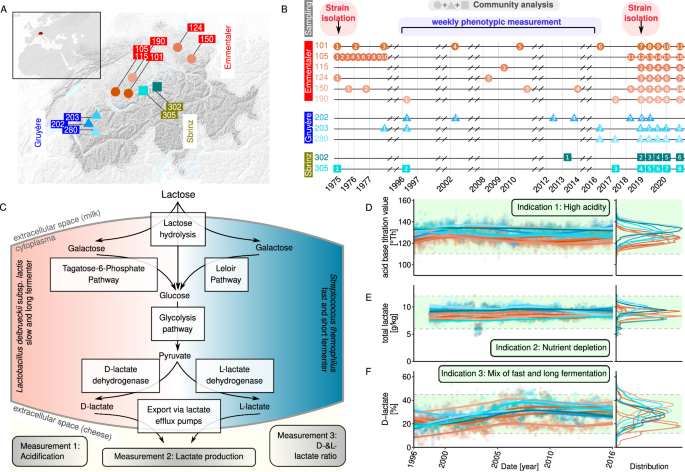
(described in refs. 41,42,43,44) and used to make three cheese varieties in different regions in Switzerland (Fig. 1A). We characterised over 1000 samples phenotypically, and about 100
metagenomes and more than 100 bacterial isolates from historical and modern Swiss starter cultures were genetically characterised (Fig. 1B). By conducting species dating and genomic analysis
of additional isolates from public databases, sampled in different geographic locations and dairy fermentations, we expanded on previously proposed hallmarks of domestication for eukaryotes
to define five specific hallmarks of microbial community domestication for bacteria29: (i) phenotypic reliability over time, as a result of the selection for food preservation, (ii) simple
and stable microbial diversity both at the species and strain-level, as a result of continuous passaging, (iii) evolutionary origin of focal species coinciding with the start of food
preservation, (iv) gradual genome decay and (v) adaptation to the food environment by the gradual reduction of niche breadth. Collectively, our results suggest that thermophilic starter
cultures have been domesticated by humans for millennia. RESULTS FOOD PRESERVATION BY RAPID AND RELIABLE ACIDIFICATION AS A RESULT OF LACTOSE FERMENTATION BY THERMOPHILIC CHEESE STARTER
CULTURES The first hallmark of domestication predicts phenotypic stability over time as a result of continuous passaging in a stable environment (i.e., milk) and selection for a specific
trait, especially food preservation but also ripening and flavouring, as determined by a characteristic aroma and favourable process parameters. The underlying phenotypic properties that
give rise to food preservation (i.e., preventing the growth of undesired microbes) are the rapid and reliable (i) acidification of the environment and (ii) reduction of the amount of easily
accessible nutrients (Fig. 1C). We analysed over 1000 weekly routine quality-control measurements for three key phenotypes of food preservation via lactose fermentation, namely
acidification, lactate production, and D- to L-lactate ratio (Fig. 1B, C). In routine starter culture quality control, acidification values above 110 acid base titration value (°Th), amount
of detected lactate between 6 and 12 g/kg and D- to L-lactate ratio between 12 and 45 are generally regarded as efficient milk fermentation. Firstly, we titrated 10 ml of > 1000 samples
and found that acidification potential was consistently (> 110 acid base titration value (°Th)) and reproducibly high over time with no significant temporal trend (Fig. 1D, mixed effect
linear model time slope estimates with _p_ < .05). Secondly, the amount of detected lactate and the ratio of the corresponding enantiomers, D- and L-lactate, slightly but not dramatically
increased over time (Fig. 1E, F, mixed effect linear model time slope estimates with _p_ < .05). The former suggests a stable nutrient depletion by accumulation of lactate as a final
product. The latter suggests that the relative metabolic contribution of the two dominant community members is similar across time. This is essential as _S. thermophilus_ ferments rapidly
but only until pH 5, whereas _L. delbrueckii_ commonly starts significantly fermenting from pH 5 downwards for a longer time. In summary, these results show that starter cultures are
phenotypically stable, measured by rapidly (within the quality control boundaries) and reliably (no significant trend over time, and the distribution of phenotypes within the range of what
is considered efficient in the industry, in Fig. 1d) acidify milk, as expected when communities are selected for specific traits. CHEESE STARTER CULTURES ARE SIMPLE AND STABLE MICROBIAL
COMMUNITIES The second hallmark of domestication predicts that the microbial diversity, both at the species and strain level are simple and stable as a result of the passaging in a contained
and highly stable nutrient-rich environment. We tested this by selecting a total of 98 starter cultures for shotgun sequencing (6–11 Mio. reads per sample; see circles, triangles, and
squares in Fig. 1B) and determined their taxonomic composition using a short read taxonomic profiler (mOTU245). As expected, we found that most samples were dominated by only two species,
_Streptococcus thermophilus_ and _Lactobacillus delbrueckii_ subsp. _lactis_ (Fig. 2A). Yet, we noted two apparent signs of instability over time: (i) _L. helveticus_ was only present in
early samples of some cheese starters, and (ii) the relative abundance of _S. thermophilus_ and _L. delbrueckii_ varied across samples of the same starter culture (Fig. 2A). The former
observation suggests that _L. helveticus_ may have been lost over time without changing the phenotypic properties of the starter cultures. The latter observation may be due to the fact that
the precise sampling time points during the acidification process by the starter cultures were not controlled for and that _L. delbrueckii_ growth is delayed relative to _S. thermophilus_
during acidification41 by their mutualistic interaction termed protocooperation46,47. Also, we cannot exclude that sample degradation through time may have biased the community composition
(the older samples are typically not viable anymore). The stable D- to L-lactate ratio (Fig. 1F) corroborates that the integrated metabolic activity of the two species at the sampling
timepoint remains stable over time. Altogether, this suggests that the species-level composition of the starter culture was remarkably stable over nearly 50 years of sampling, with no
additional species identified in the metagenomic sequences. To assess the within-species diversity, we mapped the metagenomic reads against a reference database containing isolate genomes of
each of the three species and quantified the number of polymorphic sites detected in core genes (i.e., genes identified in all strains of a given species, see methods) in each sample. The
proportion of polymorphic sites was similar over the samples, with around 0.11% (SD = 0.72 %) and 0.02 % (SD = 0.03 %) for _S. thermophilus_ and _L. delbrueckii_, respectively (SFig. 1).
This is comparatively low with respect to bacteria found in non-food fermentation systems like in the gut of animals (3% and 2–10% polymorphic sites within species in the gut microbiota of
human48 and honey bees49, respectively, even for _Lactobacillus_ species). To determine the actual number of strains the detected within-species diversity corresponds to, we genotyped >
2000 colonies from the 11 starter cultures (Fig. 1B) and sequenced the genomes of 112 isolates. Using an all-against-all genomic distance analysis implemented in poppunk50, we found that the
sequenced genomes separate into 12 _S. thermophilus_, two _L. delbrueckii_ and two _L. helveticus_ sub-species clades (see “Methods”). Overall, the sub-species clades within the different
species are very similar (min ANI: Sterm = 98.6 %, Ldel = 98.9 %, Supplementary Fig. 251). These sub-species clades accounted for most of the SNPs detected by metagenomic sequencing (93% and
78% of the metagenomic SNPs from _S. thermophilus_ and _L. delbrueckii_, respectively, Fig. 2B and Supplementary Fig. 3), suggesting that we have isolated and sequenced most of the
sub-species diversity present in the analysed starter cultures. In the case of _L. delbrueckii_ and _L. helveticus_, a single sub-species clade dominated in all analysed samples, while for
_S. thermophilus_, one to four sub-species clades per sample were detected. Finally, to test if the milk environment in which these bacteria have been passaged cannot carry a larger amount
of diversity at the sub-species level, we inoculated sterile milk with a diverse synthetic community containing all 11 dominant _L. delbrueckii_ isolates (all from the same sub-species
clade) and all 11 dominant _S. thermophilus_ isolates (from seven different sub-species clades) each from one of the 11 starter cultures. We inoculated them at equal amounts and co-cultured
them in five replicates for 27 passages (~ 123 generations) (Fig. 2C). After 21 generations, we found that five of the 11 original _S. thermophilus_ strains were still present. However,
after 123 generations, only one strain could be detected in the community (Fig. 2C), the most dominant strain in RMK 101 and 150). This experiment suggests that only a limited number of
strains can co-exist due to passaging, as observed in natural settings, but that other local factors (not mimicked in our experiment) might contribute to the dominance of alternative strains
in different starter cultures. In summary, these results show that undefined starter cultures have a simple community composition at the species and strain levels that shows a high degree
of stability, in particular at the species level, likely due to the continuous passaging in a stable, closed and nutrient-rich environment. THE ONSET OF CHEESE STARTER STRAIN DIVERSIFICATION
COINCIDES WITH THE ORIGIN OF DAIRY FERMENTATION IN HUMANS The third hallmark of domestication predicts that the evolutionary origin of starter bacteria should coincide with or postdate the
start and diversification of cheese making or dairy fermentation in prehistoric times, as referenced by the archaeological record. The actual timing of the origin of cheese making is
difficult to estimate from archaeological evidence because cheese making tools (wooden vessels, cloth strainers, leather sacks) and residues are usually organic and unlikely to be well
preserved over millennia52,53. The first archaeological evidence of cheese making based on organic matter found in ceramic stainers places the latest estimate of cheese-making origin around
7000 BC in the Near East13 and around 6000 BC in Europe11. We thus hypothesise that the origin of the starter bacteria should similarly fall within the Neolithic, and possibly even into the
Copper, Bronze, and Iron Ages, when important innovations in cheese making occurred11. To test this, we sought to date the evolutionary transition(s) from a non-dairy environment to a dairy
environment using phylogenetic comparative methods. This approach first maps strain or species habitat preference (dairy/non-dairy) onto a phylogeny to identify the evolutionary
transition(s) between habitats and then, in a second step, uses the molecular clock and the dates of historical samples as calibration points to estimate the age of this transition. We
assembled a genomic database of 234 strains from the three cheese starter bacteria as well as closely related species by combining our own dataset with publicly available data (complete list
of genomes provided in Supplementary Data 1). We mapped the isolation source on whole genome phylogenies (for within-species phylogenetic relationships) and on rRNA phylogenies (for
between-species relationships). Most strains from _S. thermophilus, L. delbrueckii_ and _L. helveticus_ were isolated from cheese starter cultures or other milk products, with a few isolates
from faecal samples (Supplementary Fig. 4). Corroborating previous findings54, ancestral niche reconstruction suggests that with a high likelihood of 99.99%, 99.99% and 76% the niche of the
ancestors of all known strains of _S. thermophilus, L. delbrueckii_ subsp. _lactis_, and _L. helveticus_, respectively, were already associated with dairy products (Maximum likelihood
model, Supplementary Fig. 5). In addition, although the three closest related sister species to our focal species are of animal rather than dairy origin, they all encode enzymes to degrade
milk (LacG or LacZ galactosidases, Fig. 3A–C). This suggests that these milk-adapted sister clades probably evolved with the appearance of milk in mammals roughly 200 million years ago55,
but only our focal species are found in the dairy environment. Therefore, we propose that the origin of cheese starter bacteria is located somewhere between the split from the sister species
(stem age) and the most recent common ancestor (MRCA) of the strains within the species (crown age) of the three starter culture species (branches highlighted in black in Fig. 3A–C). To
estimate the age of the non-dairy-to-dairy transition, we took advantage of two independent molecular clocks of the stem and crown age. Stem ages were determined using the divergence time
from the rRNA phylogeny. Assuming an rRNA substitution rate of 1 substitution per site per 100 million years56, we estimated the divergence of _S. thermophilus_, _L. delbrueckii_ subsp_.
lactis_ and _L. helveticus_ from their sister taxon at around 432,000, 264,000 and 1,044,000 years ago, respectively (Fig. 3A–C). While this is more recent than most other species within the
two genera (Fig. 3G), it is still three orders of magnitude older than the first report of dairy fermentation. Crown ages were determined using dated core-genome phylogenies of our focal
species (Fig. 3D–F and Supplementary Fig. 6). Using a molecular clock based on the core genome phylogeny and historical samples as calibration points57, we estimated a substitution rate of
1.1 SNPs per clonal core genome per year, which falls in the average range typically observed for other bacterial species58,59. We extrapolate that the crown age of _S. thermophilus_, _L.
delbrueckii_ subsp. _lactis_ and _L. helveticus_ were around 3221, 7798 and 4304 years ago, respectively (Fig. 3G). This method provides an approximate age range for the different clades
rather than precise dating represented by the large estimated confidence interval errors, ranging from 2163 to 12,708 years ago. Nevertheless, it indicates that the origin of known
strain-level diversity within species is roughly similar to the expected origin of dairy fermentation from archaeological records11. CHEESE STARTER BACTERIA SHOW GENOME DECAY BY TRANSPOSON
EXPANSION The fourth hallmark of domestication predicts that cheese starter bacteria show signs of gene loss and genome decay60. This is expected when bacteria thrive in stable environments
with extensive nutrient availability and relatively small population sizes61. This hallmark additionally predicts that the onset of genome decay must have started after the onset of
domestication itself. To test this hallmark, we compared the genome size and the extent of genome decay (number of pseudogenes) of our focal cheese starter strains to their closest relative.
To put these estimates into a broader context, we also included data on insect-associated bacterial endosymbionts that show extensive genome decay due to pseudogenization62. All three
starter culture species had smaller genome sizes (Wilcoxon test, _p_ < 0.05, Fig. 4A and Supplementary Fig. 7) and a higher number of pseudogenes (Wilcoxon, _p_ < 0.05, Fig. 4A and
Supplementary Fig. 8) than closely related species. We found that many of the detected pseudogenes in the three starter culture species were the result of insertion events of transposons
(mean = 45%, std = 9%; Supplementary Fig. 9) belonging to 15 different IS element families (Supplementary Fig. 10). Three other _Lactobacillus_ species had similar signs of genome decay, all
of which were also food fermentation-associated species, namely _L. delbrueckii_ subsp. _bulgaricus_, _L. kefiranofaciens_ and _L. amylolyticus_. The observed genome decay of our focal
genomes is in the range of host-restricted symbionts such as _Rickettsia_ spp. and _Wolbachia spp_., but not as extreme as for obligate endosymbionts of plant-sap feeding insects that have
co-evolved with their host for millions of years. Overall, our findings suggest that all three starter culture species have experienced pseudogenization via transposon insertion during their
evolutionary history. If genome decay started at the onset of domestication as predicted from the hallmark, the timing of pseudogenization events should be more recent than the onset of
domestication itself. To estimate the onset of genome decay, we reconstructed the evolutionary history of pseudogenization events by mapping the presence/absence of modern pseudogenes onto
the strain phylogenies. For each pseudogene, we identified the most recent common ancestor (MRCA) of the genomes which contained the pseudogene or in which the gene was completely missing
(assuming the pseudogene was lost, Supplementary Fig. 11). While we observed a few pseudogenes originating around the crown age (oldest peak, Fig. 4B), we found that most pseudogenes likely
originated in the last millennia (intermediate peak, Fig. 4B) or even as recent as in the last century (most recent peak, Fig. 4B). The lag between the crown age (prehistoric) and the
increase in pseudogenization in the last millennia, could arise if cheese making remained a spontaneous fermentation-like process for millennia before being more tightly controlled by
backslopping. We conclude that the overall age similarity between the loss of functional genes (i.e., pseudogenization) and the cheese making development (within the last couple of
millennia) for all three focal species suggests that the persistent bottleneck and selection pressures of cheese-making are associated with recent and ongoing genome decay. REDUCTION OF THE
NICHE BREADTH AND ADAPTATION TO THE CHEESE MAKING ENVIRONMENT The fifth hallmark of domestication predicts a reduction of the niche breadth associated with the loss of non-essential
functions due to the adaptation to the stable and nutrient-rich cheese environment. We first tested the ability of starter culture bacteria to grow on a wide range of carbon sources that are
representative of diverse non-dairy environments. We found that _S. thermophilus_ and _L. delbrueckii_ can metabolise only 5 and 9, respectively, of the 92 tested carbon sources, while
their closest non-dairy relatives could metabolise 12 (58% drop), 61 (85% drop), respectively, (Supplementary Figs. 12, 13). We then sought to explore whether the pseudogenization of genes
observed in these species could explain the loss of these metabolic capabilities. We tested whether cellular functions - in particular carbon metabolism - were enriched in pseudogenes.
Pseudogenes were spread across many orthologous gene families (OGs). Of a total of 19,728 OGs in all three species, we identified 5639 (29%) containing at least one pseudogene. Three COG
categories were overrepresented among the OGs containing pseudogenes, independently in each of the three starter culture species: carbohydrate (G), amino acid (E) and inorganic ion transport
and metabolism (I) (χ2 > 0.3, _p_-value < 0.05, Fig. 5, Supplementary Fig. 14). More specifically, the three KEGG metabolic modules (i) pentose phosphate pathway (ko00030), (ii)
fructose and mannose metabolism (ko00051) and (iii) starch and sucrose metabolism (ko00500) were commonly pseudogenized (Supplementary Fig. 15). To confirm that the identified pseudogenes
are indeed non-functional, we carried out RNA-seq of cheese during the first 24 h of cheese making to look at their expression levels. We found that pseudogenized genes were generally less
expressed than non-pseudogenized genes throughout the first 24 hours of fermentation (RNA-seq experiment, Supplementary Fig. 17). In summary, these results show that recent pseudogenization
affected genes involved in the degradation of carbohydrates that occur in plants but not in milk, suggesting that cheese making strains have lost genes that appear non-essential today in a
stable and predictable dairy environment but were likely more important in their ancestral niche. DISCUSSION Overall, fermented foods represent a stable, microbe-rich environment where
human-controlled continuous passaging and selection results in the establishment of defined and stable microbial communities with specific phenotypic properties. So far, apart from the
domestication of some eukaryotic microbes (e.g., Saccharomyces cerevisiae used in beer, wine or bread), we know surprisingly little about the history of fermented food microbes, and whether
they have been domesticated as extensively as cattle and crop plants remains an open question29,31. We can conclude from our study that thermophilic starter cultures show clear signs of
domestication. (i) They are highly reliable in their acidification and lactose utilisation, likely because of the ongoing selection of the preservation properties (hallmark 1). (ii) They
contain simple and stable microbial communities perhaps because of continuous passaging (hallmark 2). (iii) The timing of the origin of the strains is roughly similar to the emergence of
dairy fermentation and the resulting anthropogenic selection pressure (hallmark 3). (iv) They show clear signs of recent and ongoing genome decay that can be expected from stable and
nutrient-rich environments with relatively small population sizes and continuous passaging (hallmark 4). (v) They show a reduction of the niche breadth and adaptation to the cheese making
environment, suggesting that their current niche is restricted to dairy fermentation batches (hallmark 5). We acknowledge that the domestication hallmarks highlighted here could vary
substantially across species and systems depending on the processes and routes of domestication63. We suggest that comparing and classifying various fermented food systems into distinct
routes of domestication represents an exciting avenue for future research (as previously considered for eukaryotes64). In addition, the list of hallmarks we provide here is not necessarily
complete. For example, recent studies have looked at the evolution of the pangenome of the different clades of _L. delbrueckii_40,65 and _S. thermophilus_39. While these studies similarly
conclude that these species have a distinct evolutionary history tied to dairy fermentation, it currently remains unclear if the pangenome is open and expanding39. Notably, the accessory
genome contains numerous carbohydrate utilisation genes suggesting a broad nutrient range. Here, we suggest that the size of the pangenome is likely also a consequence of extensive and
ongoing pseudogenization because of a substantial fraction of highly mobile genes in the accessory genome, namely active transposases, phages or phage defence genes40,41,66,67.
Interestingly, phages targeting _S. thermophilus_ have adapted to the specific dairy conditions similarly to their host, potentially playing an important role in shaping the remaining
genetic diversity of the bacterial species68. In any case, the addition of more high-quality genomes and a broader sampling in dairy and non-dairy environments will enable us to understand
the pangenome diversity better and explore how it relates to domestication history. Finally, including a wider diversity of cheese starter bacteria from all over the world will probably help
push back the date of the onset of the diversification of cheese bacteria and represent an important research avenue. Collectively, we suggest that thermophilic starter cultures have been
domesticated by humans for millennia. Domestication probably started during the Neolithic era when cheese making emerged but likely gradually continued for millennia14. In our data, this is
evidenced by the large difference in timing of origin between cheese starter members as well as the delay between the origin of the current strain diversity and the pseudogenization. While
the exact timing of the transition from a spontaneous dairy fermentation to backslopping is impossible to pinpoint precisely, our estimates of starter culture origin using molecular data
roughly agree with the archaeological record. Overall, we suggest that the bacteria have likely been in repeated cycles of selection for rapid and reliable acidification and also genetic
drift through the repeat subsampling by backslopping. Our study fills an important knowledge gap by addressing if, when and how microbes have evolved to anthropogenic usage and provide a
conceptual framework that can be applied to other fermented food products. For example, from the pseudogenization analysis (Fig. 4), we pinpoint candidate species that might have been
domesticated, in particular, _L. delbrueckii_ subsp. _bulgaricus_ which has previously been associated with a distinct evolutionary history69 and genomic repertoire40 tightly linked to
yogurt production. Moreover, it will be instrumental in guiding decision makers in the cheese community to decide on optimal community and genomic diversity guidelines, as, for example, has
already been done in beer brewing70. While rapid and reliable acidification is the primary role of starter cultures, many secondary roles, most notably phage defence and flavour production,
play key roles in the selection of starter cultures today. Key questions in domestication research ask whether, for a given domesticate, there was a single domestication event that was
restricted to a particular geographic area that spread across regions or if there were multiple independent domestication events12. For cheese starter bacteria, this remains unknown. In the
case of crop plants and animals, the most likely scenario seems to depend on the domesticate and the continent1,64. The origin of _S. cerevisiae_ is likely Asian, but it is still unclear
whether the domestication happened initially in Asia and the domesticate then spread or whether the wild ancestor spread first and then got domesticated several times in different
places24,71, as reviewed in ref. 29. In the case of cheese starter bacteria, data from across a larger diversity of continents and traditional cheese varieties will be key to differentiating
between a single or a multiple-origin scenario and will probably help push back the date of the onset of diversification of cheese bacteria. In addition, the data we present here do not
encompass closely related “wild” strains that might share a very recent common ancestor with starter culture strains. Discovering and characterising these wild relatives will be instrumental
to reconstructing the evolutionary history of starter culture and testing the single vs. multiple origin scenarios as well as the degree of hybridisation between domesticated and wild
strains. Previous studies have already suggested that these species likely do not have an original niche in the human gut54,72,73,74 potentially having a plant origin. However, we speculate
here that the ancestral niche of cheese starter bacteria might be the gut of milk-feeding (i.e., juvenile) dairy animals that were originally used as a source for rennet75. METHODS COMMUNITY
SAMPLING The starter cultures were continuously passaged at Agroscope (Liebefeld, Switzerland) as undefined starter cultures. The process of maintaining an undefined starter culture is
explained extensively in ref. 41. In short, a stock culture is aliquoted into several samples, which are used on a weekly basis to fulfil the demand of cheese makers. The initial stock is
maintained as a freeze-dried ampule at 4 °C and is only passaged and aliquoted if necessary. The number of passages is irregular and not documented. In general, the stock cultures were
passaged more frequently from 1970 to approximately 2000, as frequently as every month since we have numerous samples dated one month apart. Today, the frequency of passaging is reduced to a
minimum, which can be as rare as every 5 years. The historic samples were stored at 4 °C throughout the years, and DNA isolation was done collectively in 2019. The DNA isolation was done as
previously described76. In short, the DNA was isolated with the EZ1 DNA Tissue kit on the BioRobot EZ1 robot (Qiagen, Hombrechtikon, Switzerland). STRAIN ISOLATION AND BACTERIAL COUNTS
Strains were isolated in 1975 and 2019 (Fig. 1B). The isolates from 1975 were picked from MR11 plates (for both species) and stored in the strain collection of the Agroscope. The isolates
from 2019 were isolated, as previously explained in Somerville et al. 202241. In short, all 11 starter cultures were plated on two selective media SPY9.377 and MR11 (MRS adjusted to pH 5.4
according to ISO7889) for _S. thermophilus_ and _L. delbrueckii_, respectively. Ninety-six colonies per species were randomly picked and cultured in liquid media for 24 h at 37 °C. For
genotyping, DNA from 100 μL of culture was extracted using the EtNa DNA isolation method78, and mini-satellite PCR for strain identification of _S. thermophilus_ and _L. delbrueckii_ was
done as explained previously41. In short, the length of a mini-satellite region in the two species was evaluated by quantifying the PCR length (all primers in Supplementary Data 2) on a
Fragment Analyser™ (Advanced Analytical Technologies, Ankeny, IA, USA). Moreover, colony-forming units (CFU/ml) were determined by serial dilution and plate counting with an Eddy Jet Spiral
Plater and SphereFlash Automatic Colony Counter (both from IUL, Barcelona, Spain). A full description, including all primers used, can be found in the supplemental methods. METAGENOME AND
GENOME SAMPLE PREPARATION AND SEQUENCING Ninety-eight samples, including historic freeze-dried ampules, present working stocks and starter cultures, were prepared for shotgun metagenome
sequencing. The DNA was isolated as previously explained41, and Nextera flex libraries were prepared and subjected to HiSeq4000 150PE (Illumina) sequencing at the Genomic Technologies
Facility in Lausanne, Switzerland. Further, a subset of genomic samples was sequenced on a minION (Nanopore) with a rapid barcoding kit. RAW READ ANALYSIS The raw reads for both metagenomic
and genomic samples were handled similarly. All adaptors and barcodes were removed with trim galore (v0.6.10)79. Reads mapping to the cow genome from the milk was removed with KneadData
(v0.7.3) (https://bitbucket.org/biobakery/kneaddata). The reads were mapped with bwa mem (bwa-0.7.18-r1243)80. The genomes and metagenomes were assembled with SPAdes (v3.13.1)81 for short
reads and Flye for long reads (v2.9.3-b1797)82. Extensive genome polishing was done with Racon (v1.3.3)83 and freebayes (v1.3.7)84. GENOME ANALYSIS The genome assemblies were submitted to
NCBI and annotated with PGAP85. In addition, eggnog mapper was used to identify annotations of pseudogenes86. The pairwise ANI values were calculated with fastANI (v1.33)87. Additional
genomes and their respective PGAP of the three focal species were downloaded from NCBI RefSeq (12.01.2020). Only completely assembled genomes were used as we have previously observed a
substantial number of genes (and pseudogenes) not being assembled with Illumina only assemblies due to the repetitive nature of the tranposase-rich genomes67. Orthofinder (v2.3.1) was used
to identify single-copy core genes within the species88. METAGENOME ANALYSIS The metagenomic raw reads were profiled for species abundance with mOTU2 (v0.3.2)45. In addition, the metagenomic
reads were mapped against reference genomes of the three focal species with bwa mem (bwa-0.7.18-r1243)89. We performed a single nucleotide analysis (SNV) with freebayes-parallel (v1.3.7) on
the genomes and metagenomes in comparison to random reference genomes84. The observed SNVs were filtered with vcftools (v0.1.16)90 and SNPeffect (v4.3t)91 to include only SNVs with a
minimum allele frequency of 0.05, read coverage of 5 and in single-copy-genes (identified with OrthoFinder). The metagenomic SNVs were compared to the previously identified SNVs from the
reference genomes. Sub-species clade frequency was calculated by averaging the allele frequency of all sub-species clade-specific SNVs (for details, see script). PROPAGATION EXPERIMENT AND
MEASUREMENTS For the propagation experiment, we created a pooled starting sample consisting of one random isolate of _L. delbrueckii_ and one of _S.thermophilus_ per starter culture (in
total 22 isolates) (see selection in Supplementary Data 1). The starting sample was propagated in five replicates as described in Somerville et al. 202241. In short, the samples were
propagated to simulate the production of starter cultures. We conducted two passages per week. On the first day, 100 µl of the freeze-dried sample was inoculated into 10 ml autoclaved
organic milk media (BM) and incubated for 18 h at 37 °C. For the second passage on the next day, the pre-culture was inoculated into 10 ml autoclaved BM and incubated for another 18 h at 37
°C. For the final step, 100 µl of the incubated samples were transferred into a freeze-dry ampule and stored at − 30 °C for at least 1 h. Thereafter, the samples were freeze-dried for 7
hours until dry. For pH measurements, we used the hydroplate system (PreSens, Germany). The pH was normalised with pH standards of pH 4 and 7. The measurements were done in four replicates
for 30 h at 37 °C. SPECIES-LEVEL LACTIC ACID BACTERIA PHYLOGENY All representative genomes from lactic acid bacteria species were downloaded from NCBI RefSeq. Moreover, from the genus
_Streptococcus_ and _Lactobacillus_, all genomes deposited on NCBI before 12.01.2020 were included. The annotations were screened for Galactosidase (lacZ and lacG). The phylogeny was
reconstructed by concatenating the 16S, 23S and 5S rRNA sequence of the genomes, making a multi-sequence-alignment file with mafft-linsi (v7.526)92 and calculating the phylogeny with RaxML
(v8.2.12) and the “GTRCAT” model93. The plot was created in R with ggtree94. STRAIN-LEVEL DATED WHOLE GENOME PHYLOGENY The preliminary species tree was calculated with OrthoFinder
(v2.3.1)88. Therefore, we back-translated the single copy core genes into the nucleotide space and created a core genome species tree and gene trees as described in ref. 95 using MAFFT
(v7.526)96 and RAxML (v8.2.12) with 100 bootstrap rounds and the GTRCAT93. Moreover, by including the gene tree and the species tree we calculated the clonal species tree with ClonalFrameML
(v1.12)97. Therefore a dated phylogeny was calculated with BactDating (v1.1.2) and 107 Bayes repetitions using our historical samples as time calibration points57. From the subsequent
phylogeny, we predicted sub-species clades for our genome isolates with poppunk (v1.2.0)50. In order to quantify the presence of sub-species clades in the metagenomes, we identified all
sub-species clade-specific SNVs in the core genes. The dated phylogeny and the information from the isolation source (Supplementary Data 1) were used to reconstruct ancestral habitat
reconstruction with the ace function from the ape package98. TRANSCRIPTOME ANALYSIS Samples for transcriptome analysis were collected throughout the first 24 hours of a regular gruyere-type
cheese making process at the cheese pilot plant at Agroscope (Liebefeld, Switzerland). The samples were immediately stored in liquid nitrogen and the RNA extraction was carried out with the
Qiagen EZ1 extraction robot and the RNA Tissue kit. Illumina libraries were prepared with the TruSeq Str-RNA Zero, and sequencing was performed on a 150 PE HiSeq 4000 (Illumina) at the
Genomic Technologies Facility in Lausanne, Switzerland. The Illumina sequences were cleaned with trim galore (v0.6.10)79 and sortmeRNA (v2.1)99. Mapping of reads to isolate reference genomes
was performed with bwa mem (bwa-0.7.18-r1243)89 and gene counting with HT-seq (v0.11.2)100. Further, sample and gene normalisation was performed with DESeq2101. PHENOTYPIC ASSAYS OF
ISOLATES The carbohydrate utilisation profiles were determined using Biolog™ phenotypic microarray plates PM01 and PM02A. Samples were prepared according to the manufacturer’s protocols.
Plates were incubated in Omnilog™ for 48 h at 37 °C. The equipment records the contrast difference every 15 min to generate growth curves. This data was evaluated using Biologs data
acquisition software™ and the opm package in ref. 102. PHENOTYPIC ASSAYS OF STARTER CULTURES The starter cultures were regularly monitored as part of the regular in-house quality control at
Agroscope starting in 1996 (acidity, lactate ratio) and 2000 (total lactate) until 2020 (last measurement included in the manuscript). Specifically, (1) the sample acidity was measured
weekly, (2) the total lactate and (3) the lactate ratio was measured monthly. The acidity of the cultures was determined as follows: 10 mL reconstituted skim milk was inoculated using 0.1 ‰
(10 µl) of the corresponding culture and incubated for 18 h at 38 °C. 1 drop of phenolphthalein was added to the sample and then titrated with 0.1 M NaOH till a visible colour change was
detected. The recorded volume of 0.1 M NaOH in mL was multiplied by 10 and rounded to the whole number resulting in the determined °Th (or Clark degree). Total lactate (D- and L-Lactate) was
analysed enzymatically according to the instruction protocol of the kit manufacturer (Boehringer, Manheim, Germany) using an automated spectrophotometric analyser (Gallery, Thermo,
Switzerland). The proportion of L-lactic acid to total lactic acid was calculated as a percentage. This method has previously been published103. The linear mixed effects model was fitted
into the phenotypic data with the lmer function in the lme4 package104. STATISTICS AND REPRODUCIBILITY No statistical methods were used to predetermine sample size, but rather all available
samples were used. The experiments and lab work were, wherever possible, randomised and blinded. If not mentioned otherwise, the data analyses and statistics were done in R (R Core Team,
2020) and plotting was done using ggplot2105. The wilcox.test function from the rstatix package and all other tests (_t_ test and Pearson chi-square) from the base package was used. All
details and specific parameters can be retrieved in the available code (see “Code availability” section). Moreover, the version numbers for all tools used in this study are provided in the
supplemental methods. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Raw sequence
data and, if applicable, assembled genomes that support the findings of this study have been deposited in on NCBI with the BioProject codes: PRJNA717134 (Illumina only genomes), PRJNA717134,
PRJNA1083966 (ONT and Illumina genomes), PRJNA1048529 (shotgun metagenomics) and PRJNA1157897 (RNA-seq). In addition, all data frames, intermediate figures and outputs are available on
Zenodo (https://doi.org/10.5281/zenodo.10783580). CODE AVAILABILITY The complete code is available on Git Hub (https://github.com/Freevini/Starter-Culture-diversity) or as a permanent
version on Zenodo (https://doi.org/10.5281/zenodo.10783580). REFERENCES * Diamond, J. Evolution, consequences and future of plant and animal domestication. _Nature_ 418, 700–707 (2002).
Article ADS CAS PubMed Google Scholar * Purugganan, M. D. What is domestication? _Trends Ecol. Evol._ 37, 663–671 (2022). Article PubMed Google Scholar * Driscoll, C. A. &
Macdonald, D. W. O’Brien SJ. From wild animals to domestic pets, an evolutionary view of domestication. _Proc. Natl. Acad. Sci. USA_ 106, 9971–9978 (2009). Article ADS CAS PubMed PubMed
Central Google Scholar * Lazaridis, I. et al. Genomic insights into the origin of farming in the ancient Near East. _Nature_ 536, 419–424 (2016). Article ADS CAS PubMed PubMed Central
Google Scholar * Weisdorf, J. L. From foraging to farming: Explaining the Neolithic Revolution. _J. Econ. Surv._ 19, 561–586 (2005). Article Google Scholar * Yousef, E. A. A., Müller,
T., Börner, A. & Schmid, K. J. Comparative analysis of genetic diversity and differentiation of cauliflower (Brassica oleracea var. botrytis) accessions from two ex situ genebanks. _PLoS
ONE_ 13, e0192062 (2018). Article PubMed PubMed Central Google Scholar * Pitt, D. et al. Domestication of cattle: Two or three events? _Evol. Appl._ 12, 123 (2019). Article PubMed
Google Scholar * Ellis, E. C. Anthropogenic transformation of the terrestrial biosphere. _Philos. Trans. A Math. Phys. Eng. Sci._ 369, 1010–1035 (2011). ADS PubMed Google Scholar *
Milla, R. et al. Phylogenetic patterns and phenotypic profiles of the species of plants and mammals farmed for food. _Nat. Ecol. Evol._ 2, 1808–1817 (2018). Article PubMed Google Scholar
* Meyer, R. S., DuVal, A. E. & Jensen, H. R. Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. _N. Phytol._ 196,
29–48 (2012). Article Google Scholar * Salque, M. et al. Earliest evidence for cheese making in the sixth millennium BC in northern Europe. _Nature_ 493, 522–525 (2013). Article ADS CAS
PubMed Google Scholar * Wilkin, S. et al. Dairy pastoralism sustained eastern Eurasian steppe populations for 5000 years. _Nat. Ecol. Evol._ 4, 346–355 (2020). Article PubMed PubMed
Central Google Scholar * Evershed, R. P. et al. Earliest date for milk use in the Near East and southeastern Europe linked to cattle herding. _Nature_ 455, 528–531 (2008). Article ADS
CAS PubMed Google Scholar * Greenfield, H. J. & Arnold, E. R. Go(a)t milk? New perspectives on the zooarchaeological evidence for the earliest intensification of dairying in south
eastern Europe. _World Archaeol._ 47, 792–818 (2015). Article Google Scholar * Marco, M. L. et al. The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus
statement on fermented foods. _Nat. Rev. Gastroenterol. Hepatol._ 18, 196–208 (2021). Article PubMed PubMed Central Google Scholar * Gänzle, M. The periodic table of fermented foods:
limitations and opportunities. _Appl. Microbiol. Biotechnol._ 106, 2815–2826 (2022). * Castro, G. R., Nadda, A. K., Nguyen, T. A., Sharma, S. & Bilal, M. _Nanomaterials for Bioreactors
and Bioprocessing Applications_. (Elsevier, 2023). * Gibbons, J. G. & Rinker, D. C. The genomics of microbial domestication in the fermented food environment. _Curr. Opin. Genet. Dev._
35, 1–8 (2015). Article CAS PubMed PubMed Central Google Scholar * Warinner, C. An archaeology of microbes. _J. Anthropol. Res._ 78, 420–458 (2022). Article Google Scholar * Swanson,
K. S. et al. The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of synbiotics. _Nat. Rev. Gastroenterol. Hepatol._
17, 687–701 (2020). Article PubMed PubMed Central Google Scholar * Arias-Sánchez, F. I., Vessman, B. & Mitri, S. Artificially selecting microbial communities: If we can breed dogs,
why not microbiomes? _PLoS Biol._ 17, e3000356 (2019). Article PubMed PubMed Central Google Scholar * Douglas, G. L. & Klaenhammer, T. R. Genomic evolution of domesticated
microorganisms. _Annu Rev. Food Sci. Technol._ 1, 397–414 (2010). Article CAS PubMed Google Scholar * Dunn, R. R., Wilson, J., Nichols, L. M. & Gavin, M. C. Toward a global ecology
of fermented foods. _Curr. Anthropol._ 62, S220–S232 (2021). Article Google Scholar * Peter, J. et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. _Nature_ 556,
339–344 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Gonçalves, M. et al. Distinct domestication trajectories in top-fermenting beer yeasts and wine yeasts. _Curr.
Biol._ 26, 2750–2761 (2016). Article PubMed Google Scholar * Gallone, B. et al. Domestication and divergence of saccharomyces cerevisiae beer yeasts. _Cell_ 166, 1397–410 (2016). Article
CAS PubMed PubMed Central Google Scholar * Gibbons, J. G. et al. The evolutionary imprint of domestication on genome variation and function of the filamentous fungus Aspergillus
oryzae. _Curr. Biol._ 22, 1403–1409 (2012). Article CAS PubMed PubMed Central Google Scholar * Ropars, J. et al. Domestication of the emblematic white cheese-making fungus Penicillium
camemberti and its diversification into two varieties. _Curr. Biol._ 30, 4441–53 (2020). Article CAS PubMed Google Scholar * Steensels, J., Gallone, B., Voordeckers, K. & Verstrepen,
K. J. Domestication of industrial microbes. _Curr. Biol._ 29, R381–R393 (2019). Article CAS PubMed Google Scholar * Landis, E. A. et al. The diversity and function of sourdough starter
microbiomes. _Elife_ 10, e61644 (2021). * Kelly, W. J., Ward, L. J. H. & Leahy, S. C. Chromosomal diversity in Lactococcus lactis and the origin of dairy starter cultures. _Genome Biol.
Evol._ 2, 729–744 (2010). PubMed PubMed Central Google Scholar * Marcobal, A. M., Sela, D. A., Wolf, Y. I., Makarova, K. S. & Mills, D. A. Role of hypermutability in the evolution of
the genus Oenococcus. _J. Bacteriol._ 190, 564–570 (2008). Article CAS PubMed Google Scholar * Smokvina, T. et al. Lactobacillus paracasei comparative genomics: towards species
pan-genome definition and exploitation of diversity. _PLoS ONE_ 8, e68731 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Roux, E. et al. The genomic basis of the
Streptococcus thermophilus health-promoting properties. _BMC Genom._ 23, 210 (2022). Article CAS Google Scholar * Hill, C. & Paul Ross R. _Genetic Modification in the Food Industry._
(1998). * Bolotin, A. et al. Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. _Nat. Biotechnol._ 22, 1554–1558 (2004). Article CAS
PubMed PubMed Central Google Scholar * van de Guchte, M. et al. The complete genome sequence of Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution. _Proc. Natl.
Acad. Sci. USA_ 103, 9274–9279 (2006). Article ADS PubMed PubMed Central Google Scholar * Schmid, M. et al. Comparative genomics of completely sequenced lactobacillus helveticus genomes
provides insights into strain-specific genes and resolves metagenomics data down to the strain level. _Front. Microbiol._ 9, 63 (2018). Article PubMed PubMed Central Google Scholar *
Alexandraki, V. et al. Comparative genomics of streptococcus thermophilus support important traits concerning the evolution, biology and technological properties of the species. _Front.
Microbiol._ 10, 2916 (2019). Article PubMed PubMed Central Google Scholar * Baek, M. G., Kim, K. W. & Yi, H. Subspecies-level genome comparison of Lactobacillus delbrueckii. _Sci.
Rep._ 13, 3171 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * Somerville, V. et al. Functional strain redundancy and persistent phage infection in Swiss hard cheese
starter cultures. _ISME J._ 16, 388–399 (2022). Article CAS PubMed Google Scholar * Sieuwerts, S., de Bok, F. A. M., Hugenholtz, J. & van Hylckama Vlieg, J. E. T. Unraveling
microbial interactions in food fermentations: from classical to genomics approaches. _Appl. Environ. Microbiol._ 74, 4997–5007 (2008). Article ADS CAS PubMed PubMed Central Google
Scholar * Sieuwerts, S. et al. Mixed-culture transcriptome analysis reveals the molecular basis of mixed-culture growth in Streptococcus thermophilus and Lactobacillus bulgaricus. _Appl
Environ. Microbiol._ 76, 7775–7784 (2010). Article ADS CAS PubMed PubMed Central Google Scholar * Iskandar, C. F., Cailliez-Grimal, C., Borges, F. & Revol-Junelles, A. M. Review of
lactose and galactose metabolism in Lactic Acid Bacteria dedicated to expert genomic annotation. _Trends Food Sci. Technol._ 88, 121–132 (2019). Article CAS Google Scholar * Milanese, A.
et al. Microbial abundance, activity and population genomic profiling with mOTUs2. _Nat. Commun._ 10, 1014 (2019). Article ADS PubMed PubMed Central Google Scholar * Smid, E. J. &
Lacroix, C. Microbe–microbe interactions in mixed culture food fermentations. _Curr. Opin. Biotechnol._ 24, 148–154 (2013). Article CAS PubMed Google Scholar * Sieuwerts, S. Microbial
interactions in the yoghurt consortium: Current status and product implications. _SOJ Microbiol. Infect. Dis._ 4, 01–05 (2016). Article Google Scholar * Fontana, F. et al. Multifactorial
microvariability of the Italian raw milk cheese microbiota and implication for current regulatory scheme. _mSystems_ 8, e0106822 (2023). Article PubMed Google Scholar * Kamilari, E.,
Tsaltas, D., Stanton, C. & Ross, R. P. Metataxonomic mapping of the microbial diversity of Irish and Eastern mediterranean cheeses. _Foods_ 11, 2483 (2022). * Lees, J. A. et al. Fast and
flexible bacterial genomic epidemiology with PopPUNK. _Genome Res._ 29, 304–316 (2019). Article CAS PubMed PubMed Central Google Scholar * Rodriguez-R, L. M. et al. An ANI gap within
bacterial species that advances the definitions of intra-species units. _MBio_ 15, e0269623 (2024). Article PubMed Google Scholar * McGovern, P. E. & Hall, G. R. Charting a future
course for organic residue analysis in archaeology. _J. Archaeol. Method Theory_ 23, 592–622 (2016). Article Google Scholar * Stott, A. W. et al. Direct dating of archaeological pottery by
compound-specific 14C analysis of preserved lipids. _Anal. Chem._ 75, 5037–5045 (2003). Article CAS PubMed Google Scholar * Pasolli, E. et al. Large-scale genome-wide analysis links
lactic acid bacteria from food with the gut microbiome. _Nat. Commun._ 11, 2610 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Pickrell, J. How the earliest mammals
thrived alongside dinosaurs. _Nature_ 574, 468–472 (2019). Article ADS CAS PubMed Google Scholar * Kuo, C. H. & Ochman, H. Inferring clocks when lacking rocks: the variable rates of
molecular evolution in bacteria. _Biol. Direct_ 4, 35 (2009). Article PubMed PubMed Central Google Scholar * Didelot, X., Croucher, N. J., Bentley, S. D., Harris, S. R. & Wilson, D.
J. Bayesian inference of ancestral dates on bacterial phylogenetic trees. _Nucleic Acids Res._ 46, e134 (2018). Article PubMed PubMed Central Google Scholar * Didelot, X., Sarah Walker,
A., Peto, T. E., Crook, D. W. & Wilson, D. J. Within-host evolution of bacterial pathogens. _Nat. Rev. Microbiol._ 14, 150–162 (2016). Article CAS PubMed PubMed Central Google
Scholar * Zhao, S. et al. Adaptive evolution within gut microbiomes of healthy People. _Cell Host Microbe_ 25, 656–67 (2019). Article CAS PubMed PubMed Central Google Scholar * Goh, Y.
J., Goin, C., O’Flaherty, S., Altermann, E. & Hutkins, R. Specialized adaptation of a lactic acid bacterium to the milk environment: the comparative genomics of Streptococcus
thermophilus LMD-9. _Micro. Cell Fact._ 10, S22 (2011). Article Google Scholar * Hottes, A. K. et al. Bacterial adaptation through loss of function. _PLoS Genet._ 9, e1003617 (2013).
Article CAS PubMed PubMed Central Google Scholar * Ochman, H. & Moran, N. A. Genes lost and genes found: evolution of bacterial pathogenesis and symbiosis. _Science_ 292, 1096–1099
(2001). Article ADS CAS PubMed Google Scholar * Bogaard, A. et al. Reconsidering domestication from a process archaeology perspective. _World Archaeol._ 53, 56–77 (2021). Article
Google Scholar * Zeder, M. A. The domestication of animals. _J. Anthropol. Res._ 68, 161–190 (2012). Article Google Scholar * Grizon, A. et al. Genomic characterization of wild
lactobacillus delbrueckii strains reveals low diversity but strong typicity. _Microorganisms_ 12, 512 (2024). Article PubMed PubMed Central Google Scholar * Somerville, V. et al.
Extensive diversity and rapid turnover of phage defense repertoires in cheese-associated bacterial communities. _Microbiome_ 10, 137 (2022). Article CAS PubMed PubMed Central Google
Scholar * Somerville, V. et al. Long-read based de novo assembly of low-complexity metagenome samples results in finished genomes and reveals insights into strain diversity and an active
phage system. _BMC Microbiol._ 19, 143 (2019). Article PubMed PubMed Central Google Scholar * Oechslin, F. et al. Fermentation practices select for thermostable endolysins in phages.
_Mol. Biol. Evol._ 41, msae055 (2024). * El Kafsi, H. et al. Lactobacillus delbrueckii ssp. lactis and ssp. bulgaricus: a chronicle of evolution in action. _BMC Genom._ 15, 407 (2014).
Article Google Scholar * Molinet, J. et al. Wild Patagonian yeast improve the evolutionary potential of novel interspecific hybrid strains for lager brewing. _PLoS Genet._ 20, e1011154
(2024). Article CAS PubMed PubMed Central Google Scholar * Duan, S. F. et al. The origin and adaptive evolution of domesticated populations of yeast from Far East Asia. _Nat. Commun._
9, 2690 (2018). Article ADS PubMed PubMed Central Google Scholar * Walter, J. Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical
research. _Appl. Environ. Microbiol._ 74, 4985–4996 (2008). Article ADS CAS PubMed PubMed Central Google Scholar * Duar, R. M. et al. Lifestyles in transition: evolution and natural
history of the genus Lactobacillus. _FEMS Microbiol. Rev._ 41, S27–S48 (2017). Article ADS PubMed Google Scholar * De Filippis, F., Pasolli, E. & Ercolini, D. The food-gut axis:
lactic acid bacteria and their link to food, the gut microbiome and human health. _FEMS Microbiol. Rev._ 44, 454–489 (2020). Article PubMed PubMed Central Google Scholar * Fleischmann,
W. _Das Molkereiwesen: Ein Buch für Praxis und Wissenschaft. Zugleich als vierter Theil zu Otto-Birnbaun’s Lehrbuch der landwirthschaftlichen Gewerbe._ (F. Vieweg & Sohn, 1876). * Moser,
A., Berthoud, H., Eugster, E., Meile, L. & Irmler, S. Detection and enumeration of Lactobacillus helveticus in dairy products. _Int. Dairy J._ 68, 52–59 (2017). Article CAS Google
Scholar * Shani, N., Isolini, D., Marzohl, D. & Berthoud, H. Evaluation of a new culture medium for the enumeration and isolation of Streptococcus salivarius subsp. thermophilus from
cheese. _Food Microbiol._ 95, 103672 (2021). * Vingataramin, L. & Frost, E. H. A single protocol for extraction of gDNA from bacteria and yeast. _Biotechniques_ 58, 120–125 (2015).
Article CAS PubMed Google Scholar * Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. _EMBnet. J._ 17, 10 (2011). Article Google Scholar * Li, H.
& Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. _Bioinformatics_ 26, 589–595 (2010). Article PubMed PubMed Central Google Scholar * Bankevich, A.
et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. _J. Comput. Biol._ 19, 455–477 (2012). Article MathSciNet CAS PubMed PubMed Central Google
Scholar * Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long error-prone reads using repeat graphs. 37, 540–546 (2019). * Vaser, R., Sović, I., Nagarajan, N. &
Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. _Genome Res._ 27, 737–746 (2017). Article CAS PubMed PubMed Central Google Scholar * Garrison, E. &
Marth G. Haplotype-based variant detection from short-read sequencing. Preprint at _arXiv_ (2012). * Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. _Nucleic Acids Res._ 44,
6614–6624 (2016). Article CAS PubMed PubMed Central Google Scholar * Rodríguez Del Río, Á. et al. Functional and evolutionary significance of unknown genes from uncultivated taxa.
_Nature_ 626, 377–384 (2023). * Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear
species boundaries. _Nat. Commun._ 9, 5114 (2018). Article ADS PubMed PubMed Central Google Scholar * Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for
comparative genomics. _Genome Biol._ 20, 238 (2019). Article PubMed PubMed Central Google Scholar * Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM.
Preprint at _arXiv_ (2013). * Danecek, P. et al. The variant call format and VCFtools. _Bioinformatics_ 27, 2156–2158 (2011). Article CAS PubMed PubMed Central Google Scholar * Baets,
G. D. et al. SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. _Nucleic Acids Res._ 40, D935–D939 (2012). Article PubMed Google Scholar *
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. _Mol. Biol. Evol._ 30, 772–780 (2013). Article CAS PubMed
PubMed Central Google Scholar * Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic
inference. _Bioinformatics_ 35, 4453–4455 (2019). Article CAS PubMed PubMed Central Google Scholar * Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T. ggtree: an r package for
visualization and annotation of phylogenetic trees with their covariates and other associated data. _Methods Ecol. Evol._ 8, 28–36 (2017). Article Google Scholar * Ellegaard, K. M. &
Engel, P. Genomic diversity landscape of the honey bee gut microbiota. _Nat. Commun._ 10, 446 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Katoh, K., Misawa, K.,
Kuma, K. I. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. _Nucleic Acids Res._ 30, 3059–3066 (2002). Article CAS PubMed
PubMed Central Google Scholar * Didelot, X. & Wilson, D. J. ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. _PLOS Comput. Biol._ 11, e1004041 (2015).
Article ADS PubMed PubMed Central Google Scholar * Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. _Bioinformatics_ 35,
526–528 (2018). Article Google Scholar * Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. _Bioinformatics_ 28,
3211–3217 (2012). Article CAS PubMed Google Scholar * Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. _Bioinformatics_ 31,
166–169 (2014). Article PubMed PubMed Central Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.
_Genome Biol._ 15, 550 (2014). Article PubMed PubMed Central Google Scholar * Vaas, L. A. I., Sikorski, J., Michael, V., Göker, M. & Klenk, H. P. Visualization and curve-parameter
estimation strategies for efficient exploration of phenotype microarray kinetics. _PLoS ONE_ 7, e34846 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Borshchevskaya, L.
N., Gordeeva, T. L., Kalinina, A. N., & Sineokii, S. P. Spectrophotometric determination of lactic acid. _J. Anal. Chem._ 71, 755–758 (2016). * Bates, D., Mächler, M., Bolker, B. &
Walker, S. Fitting linear mixed-effects models using lme4. _J. Stat. Softw._ 67, 1–48 (2015). Article Google Scholar * Wilkinson, L. ggplot2: Elegant graphics for data analysis by WICKHAM,
H. _Biometrics_ 67, 678–679 (2011). Article Google Scholar Download references ACKNOWLEDGEMENTS We thank Meral Turgay and Elvira Wagner for the help with the mini-satellite PCR, Xavier
Didelot for the useful advice for calculating the molecular clock, Herwig Bachmann and Bas Teusink for the many discussions on these topics, Julien Marquis and Johann Weber from the Genomic
Technologies Facility in Lausanne, Alban Ramette from the IFIK for the sequencing. The authors would like to thank SNF Doc.Mobility P1LAP3_199476 (V.S.), SNF Postdoc.Mobility P500PB_214419
(V.S.). This work was supported as a part of NCCR Microbiomes, a National Centre of Competence in Research, funded by the Swiss National Science Foundation (grant number 180575). F.M. thanks
Uni. of Lausanne, NCCR Microbiome, SNF and Antoine Guisan. AUTHOR INFORMATION Author notes * These authors contributed equally: Florent Mazel, Philipp Engel. AUTHORS AND AFFILIATIONS *
Department of Fundamental Microbiology, University of Lausanne, Lausanne, Switzerland Vincent Somerville, Florent Mazel & Philipp Engel * Agroscope, Liebefeld, Switzerland Vincent
Somerville, Nadine Thierer, Remo S. Schmidt, Alexandra Roetschi, Lauriane Braillard, Monika Haueter, Hélène Berthoud, Noam Shani & Ueli von Ah * Université Laval, Quebec, Canada Vincent
Somerville * McGill, Montréal, Canada Vincent Somerville Authors * Vincent Somerville View author publications You can also search for this author inPubMed Google Scholar * Nadine Thierer
View author publications You can also search for this author inPubMed Google Scholar * Remo S. Schmidt View author publications You can also search for this author inPubMed Google Scholar *
Alexandra Roetschi View author publications You can also search for this author inPubMed Google Scholar * Lauriane Braillard View author publications You can also search for this author
inPubMed Google Scholar * Monika Haueter View author publications You can also search for this author inPubMed Google Scholar * Hélène Berthoud View author publications You can also search
for this author inPubMed Google Scholar * Noam Shani View author publications You can also search for this author inPubMed Google Scholar * Ueli von Ah View author publications You can also
search for this author inPubMed Google Scholar * Florent Mazel View author publications You can also search for this author inPubMed Google Scholar * Philipp Engel View author publications
You can also search for this author inPubMed Google Scholar CONTRIBUTIONS V.S.: Conceptualisation, formal analysis, Funding Acquisition, Visualisation, Writing – Review & Editing,
U.v.A., F.M., and P.E.: Funding Acquisition, Writing, Conceptualisation – Review & Editing. N.T., A.R., L.B., M.H., H.B., N.S., and R.S.S.: Methodology. CORRESPONDING AUTHOR
Correspondence to Vincent Somerville. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks
the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY
FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Somerville, V., Thierer, N., Schmidt, R.S. _et al._ Genomic and phenotypic
imprints of microbial domestication on cheese starter cultures. _Nat Commun_ 15, 8642 (2024). https://doi.org/10.1038/s41467-024-52687-7 Download citation * Received: 29 March 2024 *
Accepted: 16 September 2024 * Published: 05 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52687-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative