- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Diabetic kidney disease (DKD) is the main cause of chronic kidney disease worldwide. While injury to the podocytes, visceral epithelial cells that comprise the glomerular filtration
barrier, drives albuminuria, proximal tubule (PT) dysfunction is the critical mediator of DKD progression. Here, we report that the podocyte-specific induction of human _KLF6_, a
zinc-finger binding transcription factor, attenuates podocyte loss, PT dysfunction, and eventual interstitial fibrosis in a male murine model of DKD. Utilizing combination of snRNA-seq,
snATAC-seq, and tandem mass spectrometry, we demonstrate that podocyte-specific KLF6 triggers the release of secretory ApoJ to activate calcium/calmodulin dependent protein kinase 1D
(CaMK1D) signaling in neighboring PT cells. CaMK1D is enriched in the first segment of the PT, proximal to the podocytes, and is critical to attenuating mitochondrial fission and restoring
mitochondrial function under diabetic conditions. Targeting podocyte-PT signaling by enhancing ApoJ-CaMK1D might be a key therapeutic strategy in attenuating the progression of DKD. SIMILAR
CONTENT BEING VIEWED BY OTHERS INTRINSIC TGF-Β SIGNALING ATTENUATES PROXIMAL TUBULE MITOCHONDRIAL INJURY AND INFLAMMATION IN CHRONIC KIDNEY DISEASE Article Open access 03 June 2023 PODOCYTE
OTUD5 ALLEVIATES DIABETIC KIDNEY DISEASE THROUGH DEUBIQUITINATING TAK1 AND REDUCING PODOCYTE INFLAMMATION AND INJURY Article Open access 27 June 2024 METTL10 ATTENUATES ADRIAMYCIN-INDUCED
PODOCYTE INJURY BY TARGETING CELL DEDIFFERENTIATION Article Open access 07 January 2025 INTRODUCTION Diabetic kidney disease (DKD) is the leading cause of chronic kidney disease (CKD) and
end-stage kidney disease (ESKD) worldwide1,2. Endothelial injury, glomerular hypertrophy, podocyte foot process effacement, eventual podocyte loss, and the accompanying albuminuria are key
features of early DKD3, with tubular injury and interstitial fibrosis occurring in later stages of DKD4. While a significant proportion of individuals with diabetes develop CKD, a majority
of these individuals do not progress to ESKD4,5. Recent studies demonstrate that while glomerular injury is the initial driver of early injury to the kidney, factors mediating proximal
tubule (PT) dysfunction is the key determinant of DKD progression6,7. Krüppel-like factors (KLFs) are a family of zinc-finger transcription factors that play a critical role in fundamental
cellular processes in multiple tissues, including the kidney, to maintain homeostasis as well as in development and in disease8. Among the 17 members of the KLF family, Krüppel-like factor 6
(KLF6) is an early-inducible responsive gene expressed with cell-specific diverse roles. Specifically in the podocyte, KLF6 is a key regulator of mitochondrial function and the conditional
loss of _Klf6_ in podocytes reduces mitochondrial complex IV assembly, which exacerbates glomerular injury in murine models of Focal Segmental Glomerulosclerosis (FSGS) and DKD9,10. Based on
these previous studies, we sought to investigate whether podocyte-specific induction of human _KLF6_ will attenuate podocyte injury in a murine model of DKD. Here, we demonstrate that this
induction of _KLF6_ specifically in podocytes attenuates podocyte injury and glomerulosclerosis, and improves PT injury and interstitial fibrosis under diabetic conditions in mice. While we
initially suspected this improvement in PT injury was a result of reduction in podocyte injury, unbiased single nuclei RNA-sequencing (snRNA-seq) and Assay for Transposase-Accessible
Chromatin with sequencing (ATAC-seq) demonstrate that the induction of podocyte _KLF6_ under basal conditions, primes transcriptional changes in the first segment of the PT, proximal to the
podocytes, through the induction of calcium/calmodulin dependent protein kinase ID (CaMK1D) signaling. We report that CaMK1D is highly enriched in the first segment of the PT and
preconditions the proximal tubule against mitochondrial fission under diabetic conditions. In addition, unbiased tandem mass spectrometry showed that the induction of _KLF6_ in podocytes
triggers the release of secretory Apolipoprotein J (ApoJ), which subsequently undergoes cellular uptake by low-density lipoprotein-related protein 2 (Lrp2)/megalin to activate CaMK1D
signaling in the PT to attenuate mitochondrial fission. To date, this is the first study to demonstrate a potential mechanism by which the podocyte preconditions the PT against injury
through ApoJ-CaMK1D signaling under diabetic conditions. RESULTS GENERATION AND CHARACTERIZATION OF _KLF6_ _PODTA_ MICE UNDER BASAL CONDITIONS Since the podocyte-specific loss of _Klf6_
increases susceptibility to DKD10, we sought to ascertain whether the podocyte-specific induction of _KLF6_ conversely attenuates the progression of DKD. Mice with podocyte-specific
expression of human _KLF6_ (_KLF6_) were initially generated using the “tet-on” system, where the binding of chimeric tetracycline transactivator protein (rtTA) to tet-operator and gene
activation only occurs in the presence of doxycycline (DOX). Specifically, _TRE-KLF6_ mice were bred with the _Podocin-rtTA_ (_PODTA_) mice to generate mice with podocyte-specific expression
of _KLF6_ (_KLF6__PODTA_) in the setting of DOX administration. To assess the specificity of _KLF6_ expression, relative _KLF6_ mRNA abundance was initially measured in isolated kidney
cortex, glomeruli, podocyte, and non-podocyte glomeruli fractions from _KLF6__PODTA_ and _NPHS2-rtTA_ mice (Supplementary Fig. 1a). While _KLF6_ expression remained completely undetected in
_NPHS2-rtTA_ mice across glomeruli, cortex, podocyte and non-podocyte glomeruli fractions, expression in the _KLF6__PODTA_ mice was primarily enriched in the podocyte and glomeruli
fractions, with a lower level of expression detectable in the kidney cortex and non-podocyte glomerular fractions (Supplementary Fig. 1a). Although mouse KLF6 is expressed in both glomerular
and non-glomerular cells, immunostaining for KLF6 confirmed the higher expression of podocyte-specific KLF6 in _KLF6__PODTA_ mice as compared to _NPHS2-rtTA_ mice (Supplementary Fig. 1b,
c). In addition, _mKlf6_ expression remained similar in both _KLF6__PODTA_ and _NPHS2-rtTA_ mice (Supplementary Fig. 1d). Furthermore, _KLF6__PODTA_ mice were viable and fertile with no
significant difference in albuminuria or kidney injury as compared to _NPHS2-rtTA_ mice (Supplementary Fig. 1e). PODOCYTE-SPECIFIC INDUCTION OF _KLF6_ ATTENUATES KIDNEY INJURY UNDER DIABETIC
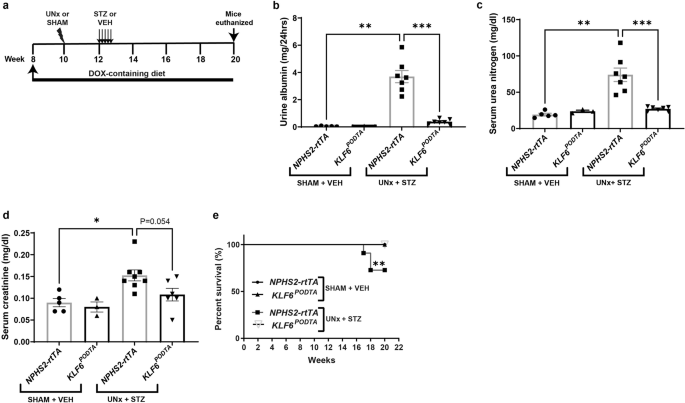
CONDITIONS To test whether the podocyte-specific induction of _KLF6_ attenuates DKD, _NPHS2-rtTA_ and _KLF6__PODTA_ mice underwent uninephrectomy (UNx) with subsequent low-dose
streptozotocin (STZ) treatment (Fig. 1a). SHAM + vehicle-treated (SHAM-VEH) _NPHS2-rtTA_ and _KLF6__PODTA_ mice were used as non-diabetic controls. The combination of UNx + STZ has been
shown to accelerate the development of glomerular lesions representative of DKD11, thereby making it a suitable model to test the potential renoprotective effects of podocyte _KLF6_
induction. All diabetic mice exhibited high blood glucose levels (>600 mg/dl) post-treatment, which was maintained throughout the experimental period (Supplementary Table 1). Blood
glucose levels of non-diabetic groups were within the normal range throughout the experimental period (Supplementary Table 1). Kidney weights and kidney to body weight ratios were
significantly increased for all diabetic groups compared to the non-diabetic groups, with no significant differences between _KLF6__PODTA_ and _NPHS2-rtTA_ mice under either condition
(Supplementary Table 1). While the diabetic _NPHS2-rtTA_ mice exhibited an increase in albuminuria, serum urea nitrogen, and serum creatinine as compared to the non-diabetic _NPHS2-rtTA_
mice and the diabetic _KLF6__PODTA_ mice, no significant differences were observed between the diabetic and non-diabetic _KLF6__PODTA_ mice (Fig. 1b–d). In addition, we observed an
improvement in survival in the diabetic _KLF6__PODTA_ mice as compared to the diabetic _NPHS2-rtTA_ mice (Fig. 1e). Subsequent staining with Periodic-acid Schiff (PAS) showed a significant
increase in glomerular volume, mesangial expansion, % sclerotic glomeruli in all diabetic mice as compared to their respective controls (Fig. 2a–c, Supplementary Table 2). However, the
diabetic _KLF6__PODTA_ mice exhibited less glomerular hypertrophy, mesangial expansion, % sclerotic glomeruli with proteinaceous casts as compared to the diabetic _NPHS2-rtTA_ mice (Fig.
2a–c). Immunostaining for Wilms tumor 1 (WT1), a podocyte-specific marker, showed that the diabetic _NPHS2-rtTA_ mice had fewer podocytes per glomerular area as compared to non-diabetic
mice. In comparison, the podocyte number was preserved in the diabetic _KLF6__PODTA_ mice as compared to the diabetic _NPHS2-rtTA_ mice (Fig. 2d). Furthermore, synaptopodin, critical for
podocyte actin cytoskeleton, expression was reduced in the diabetic _NPHS2-rtTA_ mice as compared to the diabetic _KLF6__PODTA_ mice and non-diabetic mice (Fig. 2e). We also investigated the
ultrastructural changes using transmission electron microscopy (TEM) and found that the diabetic _NPHS2-rtTA_ mice had an increase in foot process effacement and glomerular basement
membrane (GBM) thickness as compared to the non-diabetic _NPHS2-rtTA_ mice, which was attenuated in the diabetic _KLF6__PODTA_ mice (Fig. 2a, f, g). Hematoxylin & eosin (H&E) with
histopathological scoring by M.P.R., renal pathologist, in a blinded fashion showed an increase in interstitial inflammation in both diabetic groups as compared to their respective
non-diabetic controls, with an increasing trend (non-statistically significant) in the diabetic _NPHS2-rtTA_ mice as compared to the diabetic _KLF6__PODTA_ mice (Supplementary Fig. 2a,
Supplementary Table 2). While both diabetic groups also had some loss of lotus lectin staining, suggesting PT brush border loss, the diabetic _NPHS2-rtTA_ mice had a significant reduction as
compared to the diabetic _KLF6__PODTA_ mice (Supplementary Fig. 2a, b). In addition, picrosirius red, and α-SMA staining showed an increase in staining in both diabetic groups, which was
improved in the diabetic _KLF6__PODTA_ mice (Supplementary Fig. 2a, c). Interestingly, histopathological scoring noted significant tubular injury and interstitial fibrosis only in the
diabetic _NPHS2-rtTA_ mice (Supplementary Fig. 2a, Supplementary Table 2). Collectively, these data suggest that the podocyte-specific induction of _KLF6_ attenuated glomerular and
tubulointerstitial injury under diabetic conditions. PODOCYTE-SPECIFIC INDUCTION OF _KLF6_ RESTORES PODOCYTE DIFFERENTIATION MARKERS UNDER DIABETIC CONDITIONS To assess the potential
mechanism that mediates the renoprotective effects of _KLF6__PODTA_ under diabetic conditions at the single cell level, we initially performed snRNA-seq on the kidney cortex. Rationale for
choosing snRNA-seq versus scRNA-seq was based on recent studies12. After clearing all quality control checks, we initially generated single nuclear transcriptomes for 78,979 nuclei
(Supplementary Fig. 3a, b). Unsupervised clustering analysis subsequently identified 23 cell clusters (Supplementary Fig. 3c) and the top significant marker genes were compared to previously
reported cell type markers13 to assign cell type specific identity to these clusters. Clusters with similar cell type identity were combined to generate 18 unique clusters (Fig. 3a, b). The
nuclei count for each cluster, with respect to each group was reported (Supplementary Data 1). To identify differentially expressed genes (DEGs) in the setting of podocyte-specific _KLF6_
induction with and without diabetes, we initially performed differential expression analysis on all the clusters (Supplementary Data 2). Interestingly, in the podocyte cluster, there were
very few DEGs (0 upregulated and 2 downregulated) in the _KLF6__PODTA_ group vs _NPHS2-rtTA_ under non-diabetic conditions (Supplementary Data 2). However, under diabetic conditions, we
observed a significant number of DEGs (59 upregulated and 42 downregulated) in the _KLF6__PODTA_ as compared to the _NPHS2-rtTA_ mice (Fig. 3c, d). Subsequent pathway enrichment analysis on
these DEGs was conducted on the podocyte cluster from the diabetic _KLF6__PODTA_ mice using _Enrichr_14,15. Enrichment analysis using Reactome16, WikiPathways17, and KEGG pathways18 for the
upregulated DEGs demonstrated key pathways involving N-linked glycosylation, axon guidance, nephrin and semaphorin interactions, as well as vascular endothelial growth factor (VEGF)
signaling pathways (Fig. 3c, e). In addition, the reduced expression of podocyte structural and differentiation markers such as _Nephrin (Nphs1), Synaptopodin (Synpo), Podocalyxin (Podxl)_
and _Dachshund family transcription factor 1 (Dach1)_ in the diabetic _NPHS2-rtTA_ mice were all significantly restored in the diabetic _KLF6__PODTA_ mice (Fig. 3c, e). Conversely, the
downregulated DEGs were enriched for pathways involving focal adhesion, integrin-mediated cell adhesion, complement activation and inflammation signaling pathways (Fig. 3d, f). To determine
the cell specificity of the DEGs in the podocyte cluster, we plotted the expression of these genes on a dot plot showing their expression across all the cell types (Supplementary Fig. 4a,
b). We report that while the downregulated DEGs were not necessarily specific to one cell type, the upregulated genes were predominantly specific to the podocyte cluster (Supplementary Fig.
4a, b). This suggests that KLF6 might play a key role in maintenance of the mature podocyte differentiation markers in the setting of cell stress under diabetic conditions. To explore the
potential direct and indirect mechanism by which KLF6 might regulate gene expression, we initially conducted _in-silico_ analysis by matching the DEGs in the podocyte cluster with previously
reported KLF6 ChIP-seq expression arrays19 obtained from the _Encyclopedia of DNA Elements (ENCODE)_ project (Supplementary Fig. 5a). Class 0 DEGs were defined as having at least 1 KLF6
binding site within ±1 kb of the transcription start site (TSS), class 1 DEGs have at least 1 KLF6 binding site between ±1 and 10 kb of the TSS and class 2 DEGs as having no KLF6 binding
site within ±10 kb of the TSS. Of the 59 upregulated DEGs, there were 11 class 0, 5 class 1, and 43 class 2 DEGs (Supplementary Fig. 5b), while the downregulated DEGs were 9 class 0, 2 class
1, and 31 class 2 DEGs (Supplementary Fig. 5c). To examine the contribution of the class 0 DEGs to enriched pathways, we compared the statistical significance of pathway enrichment between
class 0 and class 2 DEGs (Supplementary Fig. 5d, e). Despite having fewer numbers of DEGs, upregulated class 0 DEGs were more highly enriched for N-linked glycosylation pathways, axon
guidance, and nephrin interaction pathways, suggesting a potentially more direct transcriptional regulation by KLF6 (Supplementary Fig. 5d). In comparison, class 0 downregulated DEGs were
enriched for pathways involving integrin and non-integrin dependent ECM interactions as compared to class 2 downregulated DEGs (Supplementary Fig. 5e). To further characterize the potential
mechanism by which podocyte-specific _KLF6_ might mediate its salutary effects, we performed snATAC-seq on kidney cortex samples from _NPHS2-rtTA_ and _KLF6__PODTA_ mice to ascertain the
degree of chromatin accessibility in the podocyte cluster. We successfully generated chromatin accessibility libraries for 9,894 nuclei that passed all quality control checks (Supplementary
Fig. 6a–c). Dimensionality reduction was performed using R-packages _Signac 1.8.0_20 and _Seurat 4.3.0_21, and clusters were identified and assigned using anchors transferred from the
snRNA-seq data. Label transfer was very successful in mapping the snATAC-seq data as indicated by the high prediction scores (Supplementary Fig. 6d). Nuclei from all 18 clusters in our
snRNA-seq data were identified in the snATAC-seq data (Fig. 4a). To compare conformity between the snRNA and snATAC data, we plotted gene activity metrics for the clusters using the top
significant marker genes from the snRNA-seq data (Fig. 4b). Gene activity score predicts the level of gene expression based on the accessibility of regulatory elements in the vicinity of the
gene22. Majority of the clusters showed exemplary correlations between gene expression of the selected marker gene and its corresponding gene activity score (Fig. 4b). We also observed that
all the clusters exhibited unique cell type-specific chromatin accessibility (Fig. 4c). To explore the potential chromatin accessibility changes induced by _KLF6_ induction in the podocyte
cluster, we performed differential accessibility analysis in the podocyte cluster between both groups (Fig. 4d, Supplementary Data 3). We also identified several chromatin organization genes
such as Nuclear Receptor Coactivator 1 (_Ncoa1_), Nuclear Receptor Coactivator 2 (_Ncoa2_), Bromodomain Containing 1 (_Brd1_), Lysine Demethylase 5 C (_Kdm5c_), Lysine Demethylase 2 A
(_Kdm2a_), PHD Finger Protein 20(_Phf20_), MSL Complex Subunit 2 (_Msl2_), GATA Zinc Finger Domain Containing 2B (_Gatad2b_) and, Polybromo 1 (Pbrm1) are differentially expressed in
_KLF6__PODTA_ podocytes (Supplementary Fig. 5f), suggesting that KLF6 might regulate chromatin reorganization. In addition, the _KLF6__PODTA_ podocytes possessed chromatin regions that were
more accessible compared to the _NPHS2-rtTA_ podocytes (Fig. 4d). Motif enrichment analysis of the differentially more accessible chromatin regions in the _KLF6__PODTA_ podocytes showed a
high enrichment for several classes of transcription factors involved in podocyte differentiation such as WT1, KLF15, Transcription Factor 21 (TCF21), including KLF6, among others (Fig. 4e,
Supplementary Data 4). Subsequent gene ontology (GO) enrichment analysis for the differential accessible chromatin regions showed an enrichment for biological processes involving the actin
cytoskeleton such as cell-cell communication, Ca2+ signaling, VEGF signaling, receptor for tyrosine kinase signaling, and syndecan interactions, with higher statistical significance in the
_KLF6__PODTA_ accessible chromatin regions (Fig. 4f). Collectively, these data suggest that podocyte-specific induction of _KLF6_ preserves podocyte health by increasing the accessibility to
podocyte pro-differentiation transcription factors. PODOCYTE-SPECIFIC INDUCTION OF _KLF6_ INDUCES CAMK1D SIGNALING IN THE 1ST SEGMENT OF THE PROXIMAL TUBULE Initial clustering of snRNA-seq
data from all 4 groups demonstrated a transcriptionally distinct cluster resembling some, but not all components of the known segments of the PT, which we labeled “preconditioned-PT” (Fig.
3a). Interestingly, this cluster was predominant in both the nondiabetic and diabetic _KLF6__PODTA_ groups, suggesting these transcriptional changes in this preconditioned-PT cluster are
driven primarily by the induction of _KLF6_ in the podocytes (Fig. 5a). In addition, the UMAP plots for all 4 groups showed a relative shift in cell population between the preconditioned-PT
and the other PT groups (PTS1-S2, PTS1-S3, PTS3). To characterize this preconditioned-PT cluster and explore its functional significance, we initially determined the top DEGs for this
cluster (Supplementary Data 2). Key DEGs in this cluster included calcium/calmodulin dependent protein kinase ID (_Camk1d_), Cms1 ribosomal small subunit homolog (_Cmss1_), aminoacylase 3
(_Acy3_), and glutathione peroxidase 3 (_Gpx3_) (Fig. 5b). We subsequently interrogated our preconditioned-PT cluster using previous reported markers for PT-S1-S3, repairing/Injured PT and
failed repair PT cell markers23,24,25,26,27 and found none of the repairing/injured or “failed repair” cell markers are expressed by the preconditioned-PT cell cluster, suggesting that this
cluster is distinct from previously reported post-injury PT clusters (Fig. 5c). Subsequent pathway enrichment analysis using WikiPathways17 for these upregulated DEGs demonstrated an
enrichment in metabolic pathways, such as electron transport chain, glycolysis & gluconeogenesis, amino acid metabolism, tricarboxylic acid cycle (TCA) cycle, and peroxisome
proliferator-activated receptors (PPAR) signaling, tryptophan metabolism, and fatty acid metabolism, which are collectively pathways known to be enriched in the PT segments under basal
conditions (Fig. 5d). Furthermore, among the upregulated DEGs in the preconditioned-PT cluster, _Camk1d_ and _Cmss1_ were significantly upregulated in the diabetic _KLF6__PODTA_ as compared
to the other groups (Fig. 5e). To explore the potential transcriptional paths between the tubular segments and the preconditioned-PT, we performed trajectory analysis on the snRNA sequencing
data using monocle (Supplementary Fig. 7a). While the other PT segments, namely PT(S1-S2), PT(S1-S3), PT(S3) and PT(S3)/LH(DL), showed similar ordering across pseudotime constrained mainly
on one side of branch point 1, the preconditioned-PT spanned across both sides of branch point 1, showing both its similarity as well as uniqueness from the other PT segments (Supplementary
Fig. 7b). Expression of the key DEGs of the preconditioned-PT (_Camk1d_), PT(S1-S2) (_Slc5a12_), PT(S1-S3) (_Erc2_), PT(S3) (_Keg1_), PT(S3)/LH(DL) (_Cyp7b1_) and PEC/Proliferating PT
(_Cd44_), localized similarly to their pseudotime ordering patterns (Supplementary Fig. 7d). We further interrogated the DEGs responsible for the branch-dependent expression across branch
point 1 and the pathways they are involved in using clusterprofiler (Supplementary Fig. 7c, e). We found fatty acid beta-oxidation, glycolysis and gluconeogenesis, oxidative phosphorylation,
and electron transport chain among the significant pathways (Supplementary Fig. 7e). These data suggest that a shift towards a highly metabolically active PT characterizes the transcriptome
of the preconditioned-PT cluster. To determine the spatial location of these unique PT cell populations, we costained for CaMK1D (high enrichment in the preconditioned-PT cluster) and lotus
lectin in _NPHS2-rtTA_ and _KLF6__PODTA_ mice, which showed granular and apical staining specifically in the 1st segment of the PT, proximal to the podocytes (Fig. 5f, Supplementary Fig.
7f). Furthermore, this PT-specific CaMK1D expression was increased in the _KLF6__PODTA_ mice as compared to _NPHS2-rtTA_ mice (Fig. 5f, g). We also validated that _Camk1d_ mRNA expression
was enriched in PT cells in isolated primary PT cells as compared to neighboring parietal epithelial cells (PECs), and podocytes (Fig. 5h). In addition, CaMK1D was enriched in the PT
segments with coexpression of CaMK1D in PHA-E+ve cells in healthy donor nephrectomies (Fig. 5i). Interestingly, this PT-specific CaMK1D expression was reduced in kidney biopsies with early
(<30% fibrosis), prior to significant loss of PT segments, as well as late (>30% fibrosis) DKD as compared to control specimens (Fig. 5i, j). Based on these data, the sole induction of
podocyte-specific _KLF6_ triggers CaMK1D signaling and pro-metabolic pathways in the 1st segment of PT, which might precondition the PT cells against diabetic injury. THE REQUISITE ROLE OF
CAMK1D IN THE PROXIMAL TUBULE In the setting of podocyte-specific _KLF6_ induction, we report an increase in CaMK1D expression and an enrichment in DEGs involving oxidative phosphorylation
and electron transport chain in the 1st segment of PT, proximal to the podocytes. To ascertain the role of CaMK1D in the kidney, we initially knocked down _CAMK1D_ in HK2 cells
(_CAMK1D-shRNA_). _CAMK1D_ knockdown was confirmed using qPCR, western blot and immunofluorescence staining (Fig. 6a–c). _CAMK1D-shRNA_ cells exhibited a reduction in cell viability (cell
count over time) as compared to _EV-shRNA_ cells (Fig. 6d). These findings were validated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Fig. 6e). In
addition, _CAMK1D-shRNA_ cells had reduced mitochondrial membrane potential as compared to _EV-shRNA_ cells (Fig. 6f). To assess for changes in mitochondrial structure, we initially stained
for translocase of the outer mitochondrial membrane (TOM20), which demonstrated an increase in mitochondrial fragmentation in _CAMK1D-shRNA_ cells as compared to _EV-shRNA_ cells, as
demonstrated by an increase in the % of cells with fragmented mitochondria as compared to tubular mitochondria (Fig. 6g, h). Since mitochondrial fission is inhibited with the phosphorylation
of Dynamin related protein 1 (pDRP1) at Ser63728, we observed that _CAMK1D-shRNA_ cells exhibited a reduction in pDRP1(ser637) expression as compared to _EV-shRNA_ cells (Fig. 6i). To
validate this detrimental effect of the loss of _CAMK1D_ on mitochondria specifically in primary PT cells, we pharmacologically inhibited CaMK1D with STO-609, which inhibits CAMKK, leading
to a loss of phosphorylation and subsequent inactivation of CaMK1D29,30. Primary PT cells treated with STO-609 at 20 µg/ml and 50 µg/ml resulted in reduced cell viability (Fig. 6j) as well
as a reduction in basal respiration, ATP production, maximal respiration, and spare respiratory capacity as compared to DMSO-treated cells (Fig. 6k, l). Furthermore, STO-609-treated cells
also exhibited a reduction in basal respiration, ATP production, maximal respiration, and spare respiratory capacity as compared to DMSO-treated cells under normal glucose (NG, 5 mM)
conditions, which was further exacerbated under high glucose (HG, 30 mM) conditions (Fig. 6m, n). TOM20 staining also showed an increase in mitochondrial fragmentation in STO-609-treated
cells as compared to DMSO-treated cells under HG conditions (Fig. 6o, p). In addition, STO-609 reduced pDRP1(ser637) expression in primary PT cells as compared to DMSO under HG conditions,
suggesting an increase in mitochondrial fission in the presence of pharmacological CaMK1D inhibition (Fig. 6q). Under diabetic conditions, we also observed that STO609 increased PT injury
(loss of brush border staining, tubular dilatation, and reduced lotus lectin expression) as compared to DMSO treatment in the _KLF6__PODTA_ mice (Supplementary Fig. 8a, b). STO609-treated
mice also had an increase in interstitial fibrosis (increase in picrosirius red and α-SMA expression) as compared to DMSO-treated mice, suggesting that CaMK1D inhibition exacerbated kidney
injury under diabetic conditions (Supplementary Fig. 8a–c). INCREASED RELEASE OF APOJ FROM THE KLF6+ PODOCYTES TRIGGERS PT CAMK1D SIGNALING, LEADING TO A REDUCTION IN PT INJURY To ascertain
the mechanism by which podocyte-specific _KLF6_ triggers CaMK1D signaling in PT cells, primary PT cells were initially treated with conditioned media from primary mouse podocytes isolated
from _KLF6__PODTA_ mice as compared to _NPHS2-rtTA_ mice under NG and HG conditions. While the _NPHS2-rtTA_ podocyte secretome reduced cell respiration in PT cells, this was restored with
exposure to the _KLF6__PODTA_ podocyte secretome under HG conditions (Fig. 7a, b). Tandem mass spectrometry was subsequently performed on the conditioned media, after removal of cell debris,
to determine the differentially secreted proteins between _KLF6__PODTA_ and _NPHS2-rtTA_ podocytes. A total of 838 proteins were identified, with 321 proteins significantly increased [fold
change (FC > 1.2)] and 132 proteins significantly decreased (FC < 0.77) in the _KLF6__PODTA_ compared with the _NPHS2-rtTA_ conditioned medium (Fig. 7c, Supplementary Data 5). Key
proteins such as ApoJ, calreticulin (CALR), clathrin light chain A (CLTA), clathrin light chain B (CLTB), collagen type IV alpha 1 (COL4A1) were differentially expressed in the conditioned
medium from _KLF6__PODTA_ as compared to _NPHS2-rtTA_ podocytes. In comparison, a total of 142 proteins were identified, with 30 proteins significantly increased (FC > 1.3) in the urine
proteome of _KLF6__PODTA_ as compared to _NPHS2-rtTA_ mice (Fig. 7d, Supplementary Data 5). Interestingly, ApoJ was uniquely enriched in both the podocyte secretome and the urine proteome of
_KLF6__PODTA_ as compared to the _NPHS2-rtTA_ mice, suggesting an increase in the release of ApoJ from the podocytes in presence of _KLF6_ induction. ApoJ, also known as clusterin, is a
glycoprotein that has been previously reported to have a salutary effect in the kidney31,32,33,34. Specifically, pre-treating podocytes with recombinant ApoJ has shown to be protective
against apoptosis by reducing oxidative-stress induced under diabetic conditions33. To determine the expression of _ApoJ_ in the glomeruli under diabetic conditions, we interrogated previous
reported microarrays using _Nephroseq_ to compare human biopsy specimens with DKD as compared to healthy donor nephrectomies35,36. _ApoJ_ expression was significantly increased in
microdissected glomeruli in DKD patients compared to healthy donors and it inversely corelates with estimated GFR (Supplementary Fig. 9a, b). To ascertain potential ligand-receptor
interactions between podocytes and PT cells, a previously validated database of known ligand-receptor interactions37 was interrogated to predict potential ligands from the podocyte secretome
and urine proteome as well as corresponding receptors from the DEGs in the preconditioned-PT cell cluster (Fig. 7e). ApoJ, which was identified in the podocyte secretome and the urine
proteome of _KLF6__PODTA_ mice, demonstrated a potential ligand-receptor interaction with Lrp2 in the preconditioned-PT cluster (Fig. 7e). Interestingly, depending on the degree of
glycosylation, the secretory form of ApoJ has been previously reported to serve as a molecular chaperone by binding to specific cell surface receptors to mediate its biological effects, such
as endocytosis38. Immunohistochemistry confirmed that ApoJ was enriched in the podocytes and the apical portion of the 1st PT segment in _KLF6__PODTA_ as compared to _NPHS2-rtTA_ mice (Fig.
7f). Additionally, we costained ApoJ with podocyte marker, synaptopodin, and found increased colocalization of ApoJ in podocytes (Supplementary Fig. 9c). While ApoJ is present at high
levels in the plasma and prevents complement deposition39, we did not observe an increase in complement deposition (C3 and C5b-9 expression) in _KLF6__PODTA_ as compared to _NPHS2-rtTA_ mice
(Supplementary Fig. 9d). To determine whether the PT-protective effects of the _KLF6__PODTA_ podocyte secretome are driven largely by ApoJ, the conditioned media from _KLF6__PODTA_ and
_NPHS2-rtTA_ podocytes were incubated with goat anti-ApoJ antibody (1:100) as compared to goat IgG antibody for 24 h prior to being administered to primary PT cells. While the _KLF6__PODTA_
podocyte secretome improved PT basal respiration, maximal respiration, and spare respiratory capacity, blocking of ApoJ attenuated these protective effects on the mitochondria (Fig. 7g, h).
To further validate these findings, we initially generated human podocytes with stable _APOJ_ overexpression (_Lenti-ORF_–_APOJ)_ as compared to control podocytes (_Lenti-ORF-control_)
(Supplementary Fig. 10a, b). PT cells treated with the conditioned media from _Lenti-ORF-APOJ_ podocytes had higher basal respiration, ATP production, maximal respiration, and spare
respiratory capacity as compared to _Lenti-ORF-control_ podocytes under HG conditions (Fig. 7i, j). Interestingly, the sole induction of _APOJ_ also increased canonical podocyte markers of
differentiated podocytes such as nephrin and synaptopodin, suggesting that ApoJ might also have salutary effects on podocyte health (Supplementary Fig. 10c). Since calmodulin is critical
component of CaMK1D activation and signaling, we tested the interaction between ApoJ and calmodulin by incubating the primary PT cell fractions that have been exposed to _KLF6__PODTA_ and
the _NPHS2-rtTA_ podocyte secretome under NG and HG conditions using calmodulin-sepharose beads. While the ApoJ-calmodulin interaction was reduced in the PT cells when exposed to
_NPHS2-rtTA_ podocyte secretome, this was restored with the _KLF6__PODTA_ podocyte secretome under HG conditions (Fig. 7k). In addition, immunofluorescence staining demonstrated ApoJ
colocalized with CaMK1D (Fig. 7l). Lrp2 has been reported previously as the endocytic receptor for ApoJ40. Since the molecular weight of Lrp2 limits co-immunoprecipitation studies, we
conducted immunofluorescence staining with quantification using _CellProfiler_ to demonstrate that ApoJ colocalizes with Lrp2 in the proximal tubules (Fig. 7m, Supplementary Fig. 11). We
also treated the PT cells with recombinant ApoJ which led to an increase in PT basal respiration, ATP respiration, and maximal respiration. Finally, concurrent treatment with cilastatin,
inhibitor of Lrp2 receptor activity41,42,43, attenuated the salutary effects of ApoJ in PT cells as demonstrated by a decrease in basal respiration, ATP-linked, and maximal respiration (Fig.
7n, o). Collectively, these data suggest that the kidney-protective effects of podocyte-specific KLF6 might, in part, be mediated through ApoJ-CaMK1D signaling in PT cells. DISCUSSION A
large body of literature has focused on factors that drive kidney injury in diabetes, but little is known about mechanisms that confer resistance to progression of DKD. These unexplored
mechanisms that delay progression of DKD might serve as potential targets for therapy in those individuals that progress rapidly. While podocyte injury contributing to glomerular dysfunction
is an early indicator of kidney injury in diabetes, PT injury correlates with the decline in kidney function3,44. To date, mechanisms mediating podocyte to PT injury remain elusive in DKD.
In this study, we demonstrate that the podocyte-specific induction of _KLF6_ attenuates kidney injury in a murine model of diabetes. In addition to the salutary effects in the podocyte, the
induction of podocyte-specific _KLF6_ preconditions the PT from injury under diabetic conditions. By utilizing a combination of snRNA-seq, snATAC-seq, and LC-MS/MS, we report that
podocyte-specific KLF6 triggers the release of ApoJ, which subsequently activates CaMK1D-mediated preservation of mitochondrial dynamics and function in the first segment of PT, proximal to
podocytes (Fig. 7p). To date, this is the first study to demonstrate a novel mechanism by which the podocyte directly regulates PT mitochondrial health to attenuate the progression of DKD.
Our previous studies demonstrated the detrimental effects of podocyte-specific knockdown of _Klf6_ in murine models of glomerular disease (i.e., Focal Segmental Glomerulosclerosis and
DKD)9,10. Here, conversely, the induction of KLF6 specifically in podocytes ameliorated albuminuria and improved kidney function as well as histological features consistent with DKD. We also
previously showed that induction of KLF6 in cultured podocytes attenuated detrimental effects of adriamycin in cultured podocytes9. However, similar to other tissues, this reno-protective
effect of KLF6 appears to be cell-context dependent in the kidney. For instance, we recently reported the detrimental role of PT-specific KLF6 in a murine model of post-DNA damage in PT
cells45. Furthermore, this contrasting cell-specific role of KLF6 is not restricted to the kidney, with opposing roles in the liver (hepatocytes versus hepatic stellate cells)46 as well as
in the heart (cardiac myocytes versus cardiac fibroblasts)47. One potential explanation in the kidney is that _Klf6_ is expressed at a higher level in podocytes as compared to PT cells under
basal conditions48, thereby suggesting a requisite physiological role in the podocytes. Nonetheless, studies investigating the mechanisms regulating cell-specific contrasting roles of KLF6
will be important to understand its biology in kidney health and disease. CaMKs (CaMK1, CaMK2, CaMK4, CaMKK) are multifunctional calcium/calmodulin-dependent protein kinases that have wide
specificity but regulate several critical cellular functions in multiple cell types49,50,51,52. While CaMKs are well studied in the brain, their biology in the kidney is not well understood.
A few studies have reported on the detrimental role of CaMK2 and CaMK4 activation in the kidney53,54,55,56, but CaMK1D remains unexplored in the kidney. In genome-wide association studies
(GWAS), single nucleotide polymorphisms in the _CAMK1D_ loci are associated with increased risk of Type 2 Diabetes Mellitus57,58,59,60,61,62. Validation of our snRNA-seq with immunostaining
demonstrates an enrichment of CaMK1D expression in the first segment of PT, proximal to podocytes. Interestingly, genetic and pharmacological inhibition of CaMK1D reduced pDRP1(S637)
expression, leading to an increase in mitochondrial fragmentation with a reduction in mitochondrial membrane potential and respiration. DRP1 is a critical regulator of mitochondrial dynamics
and posttranslational modifications are critical to its function63. Previous studies report that CaMKI might be involved in the regulation of DRP1 activity64, but the mechanism(s) mediating
this process remains to be investigated. Furthermore, future studies are needed to examine the interplay between CaMK1D and other isoforms of CaMKI signaling on pDRP1 activity and
mitochondrial health64,65,66. Nonetheless, this is the first study, to date, to demonstrate the protective role of CaMK1D in the kidney. Another key finding in this study is the role of ApoJ
in mediating the salutary effects of podocyte-specific KLF6 on CaMK1D activation in the PT. ApoJ undergoes posttranslational modification, largely glycosylation, in the endoplasmic
reticulum and golgi apparatus prior to being released extracellular space. Secretory ApoJ serves as a molecular chaperone to regulate various cellular processes such as lipid transport, cell
differentiation, membrane cycling, apoptosis, and cell-cell interactions67. Several studies have implicated ApoJ in a host of human diseases68,69,70,71,72, ranging from myocardial injury73
to post-ischemic brain injury74 to Alzheimer’s disease75. In the kidney, ApoJ expression has been previously shown to increase post-injury76,77,78,79. Specifically, both protein and mRNA
expression levels of ApoJ are increased in podocytes in diabetic mice and in kidney biopsies with early DKD33. Similarly, we validated this in expression arrays deposited in _Nephroseq_. In
addition, ApoJ levels in the urine have been associated with increased tubular damage in patients with diabetes80. We also observed an enrichment in ApoJ levels in both the urine and
podocyte secretome from _KLF6__PODTA_ as compared to _NPHS2-rtTA_ mice by LC-MS/MS. Interestingly, this increase in levels of secretory ApoJ might be in response to podocyte injury, since we
observed that blocking ApoJ in the _KLF6__PODTA_ podocyte secretome attenuated the salutary effects in the PT cells under HG conditions. Furthermore, _ApoJ_ knockout mice develop
glomerulopathy with aging76 and individuals with glomerular disease demonstrate an overall depletion in the pool of ApoJ with progressive disease32. In addition, salutary effects of ApoJ
have been shown in other models of kidney injury81,82. However, to date, the mechanism by which secretory ApoJ attenuates kidney injury remains poorly understood83. Here, we demonstrate that
induction of KLF6 triggers the release of secretory ApoJ, which undergoes cellular uptake by Lrp2, triggering the activation of CaMK1D by binding to calmodulin in PT cells. snRNA-seq
analysis demonstrated an enrichment in pathways associated with N-glycosylation in the DEGs with KLF6 binding sites. Glycosylated intracellular ApoJ forms may act as a redox sensor under
oxidative stress conditions and are essential for its chaperone activity as well as release of ApoJ into the extracellular space84,85. Therefore, the induction of KLF6 could potentially
increase glycosylation of ApoJ, thereby triggering the release of secretory ApoJ from the podocytes. However, additional studies are required to test the mechanism by which KLF6 leads to
glycosylation of ApoJ. Interestingly, ApoJ has been previously reported to serve as a ligand for Lrp2-facilitated endocytosis in the brain40,86,87 and has a putative calmodulin binding
domain containing three motifs (two 1–12, one 1–14)88. A recent study using Ingenuity Pathway Analysis also showed KLF6 motifs in the regulatory regions of _ApoJ_ as well as potential
interactions between calmodulin and Lrp289. Calcium and calmodulin also play a role in supporting the endocytosis of ApoJ as calmodulin has been previously reported to serve as a calcium
sensor for endocytosis in synapses90. Furthermore, the proximity of podocyte secretory ApoJ to Lrp2 and calmodulin-CaMK1D in the first segment of the PT, in combination with the direction of
glomerular filtrate, could enhance the feasibility of this interaction. ApoJ has also been reported to protect endothelial cells by suppressing mitochondrial fission under diabetic
conditions91. In addition, ApoJ has been reported to facilitate mitochondrial respiration in the healthy brain92 and overexpression of _ApoJ_ attenuated Drp1 activation, thereby inhibiting
mitochondrial fission93. Based on our findings, we postulate that ApoJ-mediated activation of CaMK1D-pDRP1 signaling protects the PT against injury by inhibiting mitochondrial fission under
diabetic conditions. Collectively, these studies uncover a previously unreported mechanism by which the podocyte preconditions the proximal tubule to attenuate mitochondrial injury and
subsequent deterioration of kidney function under diabetic conditions. In addition to this requisite role of podocyte-specific KLF6 in podocytes, we also provide evidence for a new
downstream signaling pathway involving secretory ApoJ-Lrp2-CaMK1D-pDrp1 between podocytes and the first segment of PT that might be critical for kidney health. Finally, maintaining secretory
ApoJ from podocytes or enhancing CaMK1D signaling in the PT might be a key therapeutic strategy in attenuating the progression of DKD. METHODS GENERATION AND VALIDATION OF _KLF6_ _PODTA_
MICE All animal studies were approved by the Stony Brook Animal Care and Use Committee and carried out in accordance with the National Institutes of Health standards. To generate
_KLF6__PODTA_ mice, _NPHS2-rtTA_ mice (FVB/N-Tg(NPHS2-rtTA2*M2)1Jbk/J, The Jackson Laboratory) were bred with the _TRE-KLF6_ mice to generate mice with both transgenes on the _FVB/N_
background. The _TRE-KLF6_ transgene contained the (TetO)7/CMV regulatory element driving the full-length human KLF6 coding sequence (ORF021674) followed by the polyadenylation signal45.
Transgene was purified from plasmid vector sequences and microinjected into the pronucleus of _FVB/N_ single-celled embryos to generate _TRE-KLF6_ mice. DNA extraction by Extracta DNA prep
(Quanta Biosciences) from tails at 2 weeks of age and PCR to confirm the genotype. Primers for genotyping are provided in Supplementary Table 3. To induce transgene expression, _KLF6__PODTA_
mice were fed TestDiet Modified Rodent Diet 5001 containing 0.15% doxycycline (DOX; El-Mel). _KLF6__PODTA_ mice and littermate controls remained on DOX continuously starting at 8 weeks of
age. To validate induction of podocyte-specific _KLF6_ in _KLF6__PODTA_ mice, we generated fluorescent labeled _TRE-GFP_ mice and bred them with _KLF6__PODTA_ mice or _NPHS2-rtTA_ mice. All
mice were treated with DOX. Glomeruli were subsequently isolated from these mice using iron oxide and the glomerular and tubular fraction was separated using a magnet after digestion with
collagenase for 45 min at 37 °C as previously described94. The purity of glomeruli was verified under microscopy. Both fractions were digested using collagenase, DNase and trypsin to make a
single cell suspension. The single cell suspension for the glomerular fractions was sorted using fluorescence-activated cell sorting (FACS) to isolate GFP(+ve) cells. GFP(-ve) glomerular and
tubular cells were also collected for further analysis. Podocyte-specific _KLF6_ expression was determined in GFP(+ve) glomerular cells as compared to GFP(-ve) glomerular and tubular cells.
ANIMAL EXPERIMENTS The animals in this study were housed in our animal facility with free access to chow (Lab diet 5053/Dox diet) and water and 12 h day/light cycle, in ambient temperature
and humidity conditions. Baseline urine was collected from the _NPHS2-rtTA_ and _KLF6__PODTA_ mice. Mice were anesthetized and UNx was performed as previously described95. In brief, the
blood vessels of the left kidney were ligated, and the kidney removed surgically, and the mice were monitored for a week after surgery. To induce diabetes, mice were administered STZ (50
mg/kg) in 50 mmol/L sodium citrate buffer (pH 4.5) by intraperitoneal injection over the course of 5 days10,96. On day 14 after the STZ injections, blood glucose was measured from the tail
vein using a OneTouch glucometer (LifeScan)94. Diabetes was defined as sustained fasting blood glucose level > 250 mg/dL. All mice were euthanized using intraperitoneal injection of
Ketamine/Xylazine (150/20 mg/kg) at 20 weeks of age. For the STO-609 experiments, UNx-STZ _KLF6__PODTA_ mice were treated with DMSO or STO-609 (30 µm/kg) daily intraperitoneal injection for
2 weeks. The mice were euthanized using intraperitoneal injection of Ketamine/Xylazine (150/20 mg/kg) at 16 weeks of age. MEASUREMENT OF ALBUMINURIA, SERUM UREA NITROGEN AND CREATININE The
animals were housed in a single mouse metabolic cage (Tecniplast) with free access to food and water, and urine was collected after 24 h. Albumin concentration was measured using ELISA assay
kit (Bethyl Laboratory). Twenty-hour urine albumin concentration was calculated by multiplying total volume of urine collected and albumin concentration. Serum urea nitrogen levels were
measured by a colorimetric detection method (Arbor Assay) according to the manufacturer’s protocol. Serum creatinine levels were measured using the isotope dilution liquid
chromatography-tandem mass spectrometer at the University of Alabama at the Birmingham O’Brien Core Center. REAL-TIME PCR Total RNA was extracted using TRIzol (Gibco) for cells and RNA easy
kit (Qiagen) for kidney tissue. First-strand cDNA was prepared from total RNA using the SuperScript III First-Strand Synthesis Kit (Life Technologies), and cDNA was amplified using SYBR
GreenER qPCR Supermix on ABI QuantStudio 3 (Applied Biosystems). Primers were designed using National Center for Biotechnology Information Primer-BLAST and validated for efficiency before
application. Primer sequences are listed in supplemental information (Supplementary Table 4). Light cycler analysis software was used to determine crossing points using the second derivative
method. Data was normalized to the housekeeping gene (_Actb_ or _ACTB_) and presented as a fold increase compared to the control group using the 2-ΔΔCT method. HISTOPATHOLOGY AND
MORPHOMETRIC STUDIES BY BRIGHT-FIELD LIGHT MICROSCOPY Mice were perfused with phosphate buffer saline (PBS) and the kidneys were fixed in 10% phosphate buffered formalin overnight and
switched to 70% ethanol prior to processing for histology. Kidney tissue was embedded in paraffin by histology core facility at Stony Brook University and 4 μm thick sections were stained
with periodic acid-Schiff (PAS) (Sigma-Aldrich), hematoxylin and eosin (H&E), and picrosirius red, according to previously reported protocols, and mounted in permanent mounting
medium45,94. Mesangial expansion, and glomerular volume were quantified as previously described10,94. In brief, images were scanned, and glomerular areas were traced using ImageJ. Mean
glomerular tuft volume (GV) was determined from mean glomerular cross-sectional area (GA) by light microscopy. GA was calculated based on average area of 20 glomeruli in each group and GV
was calculated based on the following equation: GV \(=\,\frac{\beta }{\kappa }\,\times \,{{GA}}^{3/2}\) [β = 1.38, the shape coefficient of spheres (the idealized shape of glomeruli), and
_κ_ = 1.1, the size distribution coefficient]. Mesangial expansion was defined as the PAS-positive and nuclei-free area in the mesangium. Quantification of mesangial expansion was based on
20 glomeruli cut at the vascular pole in each group. Histological scoring for sclerotic glomeruli, tubular injury, interstitial fibrosis, and inflammation was performed in a blinded fashion
using a semiquantitative scale from 0 to 3 (0 indicates none); 1 = mild (≤25%), 2 = moderate (>25%–50%) and 3 = severe (>50%) by the kidney histopathologist (M.P.R.).
IMMUNOFLUORESCENCE STAINING AND IMMUNOHISTOCHEMISTRY All kidney sections from mice were prepared for immunofluorescence staining in identical fashion as previously described94.
Immunofluorescence staining was performed using mouse anti-WT1 (Santa Cruz, sc-7385, 1:50 dilution), goat anti-synaptopodin (Santa Cruz, sc21537, 1:200 dilution), rabbit anti-KLF6 (Santa
Cruz, AP6588B, 1:150 dilution), mouse anti-α-SMA (Sigma-Aldrich, A1978, 1:10,000 dilution), rabbit anti-CaMK1D (Invitrogen, PA5-21957, 1:100 dilution), goat anti-ApoJ (Novus Biologicals,
NBP1-06027, 1:100 dilution), mouse anti-Lrp2 (Novus Biologicals, NB110-96417, 1:100 dilution) and rabbit anti-TOM20 (Abcam, ab78547, 1:100 dilution), Fluorescein-conjugated goat IgG to mouse
complement C3 (MP Biomedicals, 085500, 1:100 dilution) and mouse anti-C5b-9 (Santa Cruz, sc-66190, 1:100 dilution) antibodies. After washing, sections were incubated with a
fluorophore-linked secondary antibody (Alexa Fluor 647 Donkey anti-mouse, Fluor 488 Goat anti-rabbit, or Fluor 568 Donkey anti-rabbit from Life Technologies, 1:300 dilution). After
counterstaining with fluorescein-labeled lotus lectin (Vector Labs, 1:100 dilution) and/or Hoechst (Invitrogen, 1:1000 dilution), slides were mounted in ProLong gold antifade mounting media
(Invitrogen) and photographed under a Nikon Eclipse i90 microscope and DS-Qi1Mc camera. Immunohistochemistry was conducted for ApoJ as previously described9 using goat anti-ApoJ antibody
(R&D, AF2747, 1:100 dilution). Briefly, slides were dewaxed, followed by rehydration with decreasing concentrations of ethanol (100%, 90%, 70%). After antigen retrieval using sodium
citrate buffer at 120 °C, the endogenous peroxidases were blocked using hydrogen peroxide (3%) in methanol for 20 min, followed by blocking with 2% non-fat milk. The sections were incubated
overnight with primary antibodies at 4 °C followed by anti-goat horseradish peroxidase secondary (Sigma Aldrich, AP200P, 1:300 dilution). The slides were then incubated with diaminobenzidine
solution (Betazoid DAB Chromogen Kit, Biocare Medical) for 5 min, slides were then dehydrated in ethanol and xylene followed by mounting with permanent mounting media. De-identified human
biopsy specimens from University of Utah were categorized into early-stage (<30%) and late-stage (>30%) chronic tubulointerstitial fibrosis by a renal pathologist (M.P.R.). Control
kidney biopsy specimens were acquired from the unaffected pole of kidneys that were removed because of renal cell carcinoma. The study was approved by the Stony Brook University
Institutional Review Board (#798611). Podocyte number was determined by dividing the number of WT1(+ve) Hoechst(+ve) podocytes per glomerular cross-sectional area. Glomerular synaptopodin
expression was determined by measuring the percent area stained for synaptopodin in the glomerular cross-sectional area. Quantifications of lotus lectin and α-SMA in the cortex were
determined by measuring the percentage area stained per cross-sectional area of a high-power field. All quantification was conducted using 20X high-power digitized images in ImageJ.
Immunofluorescence staining and quantification of mitochondrial fragmentation were done using TOM20 staining in cultured cells as previously described9,97. Briefly, mitochondrial morphology
was categorized in each cell, by an investigator blinded to the experimental conditions, as tubular (>75% of mitochondria with tubular length > 5 mm), intermediate (25%–75% of
mitochondria with tubular length > 5 mm), or fragmented (<25% of mitochondria with tubular length > 5 mm). Quantification of colocalization of ApoJ with Lrp2 was done using
_CellProfiler_98. The colocalization pipeline with minor modifications was utilized to measure the colocalization between fluorescently labeled ApoJ and Lrp2 to measure the degree of overlap
between them. Briefly, the images were split into two channels (ApoJ and Lrp2), followed by illumination correction for both channels. The images were aligned to ensure accurate positioning
of the features in both images (Supplementary Fig. 11b), and primary objects were identified for each channel along with the outliers as shown in Supplementary Fig. 11c, d. The primary
objects from ApoJ channel that colocalize with the Lrp2 channel were measured (Supplementary Fig. 11e). A mask image indicating the colocalized areas was generated and the area occupied by
the colocalized objects and Lrp2 was used for quantifications. HISTOPATHOLOGY BY TRANSMISSION ELECTRON MICROSCOPY Mice were perfused with PBS and then immediately fixed in 2.5%
glutaraldehyde for transmission electron microscopy (TEM). Sections were mounted on a copper grid and photographed under a FEI BioTwinG2 transmission electron microscope. Briefly, negatives
were digitized, and images with a final magnitude of ~ 10,000X were obtained9. The quantification of podocyte effacement and glomerular basement membrane (GBM) thickness was performed as
previously described96,99. ImageJ was used to measure the length of the peripheral GBM and the number of slit pores overlying this GBM length was counted. The arithmetic mean of the foot
process width (WFP) was calculated using the following: WFP = \(\frac{\pi }{4}\) \(\times \,\frac{\sum {GBM\; Length}}{\sum {slits}}\) ; where ∑GBM length indicates the total GBM length
measured in one glomerulus, ∑ slits indicates the total number of slits counted, and = \(\frac{\pi }{4}\) is the correction factor for the random orientation by which the foot processes were
sectioned99. Quantification of GBM thickness was performed as described100,101. The thicknesses of multiple capillaries were measured in 3–4 glomeruli per mouse. A mean of 120 measurements
was taken per mouse (from podocyte to endothelial cell membrane) at random sites where GBM was displayed in the best cross section. CELL CULTURE 1° mouse podocytes were isolated from
GFP-labeled _KLF6__PODTA_ and _NPHS2-rtTA_ male mice using FACS and were maintained on DOX (1 μg/ml) throughout the experimental conditions. Serum free media was collected from these
podocytes to carry out proteomic analysis. Mouse 1° PT cells were isolated from the kidney cortex of male mice after cardiovascular perfusion with PBS as previously described102. In brief,
the cortex is digested using collagenase A at 37 οC for 1 h. The digested tissue is filtered, followed by multiple washing and resuspension in complete PT cell media (Dulbecco’s modified
Eagle’s medium:F12 with 10 ng/L epidermal growth factor, 5 pM T3, 3.5 mg/L ascorbic acid, 25 µg/L prostaglandin E1, 25 µg/L hydrocortisone, 1 × insulin transferrin selenium supplement, 100
units/ml penicillin, and 100 µg/ml streptomycin). Human kidney (HK2, ATCC, CRL-2190) cells with _CAMK1D_ knockdown were generated using the Genecopoeia lentiviral shRNA system with the
following construct, MSH079040-LVRU6GP-c, GGTGCTGTATATAAGAATCTT. In brief, lentiviral particles were produced by transfecting HEK 293 T cells with a combination of lentiviral expression
plasmid DNA, pCD/NL-BH ΔΔΔ packaging plasmid, and VSV-G–encoding pLTR-G plasmid. For infection, viral supernatants were supplemented with 8 μg/ml polybrene and incubated with cells for 24 h.
Cells expressing shRNA were selected with puromycin for 2–3 weeks before use in all studies. Real-time PCR, western blot, and immunofluorescence staining were performed to confirm _CAMK1D_
knockdown. Mouse 1° PT cells were treated with DMSO (vehicle) or CaMK1D inhibitor, STO-609 (5, 10, 20 and 50 μg/ml), for 24 h under normal glucose conditions. After dose optimization, the 1°
PT cells were treated with 20 µg/ml of STO-609 for 24 h under normal (5 mM) and high glucose (30 mM) conditions. For measuring the effect of conditioned media on the 1° PT cells, the cells
were treated with 5% conditioned media from 1° podocytes from _KLF6__PODTA_ and _NPHS2-rtTA_ mice under normal glucose (NG) and high glucose (HG) conditions for 24 h. For the blocking
experiments, the conditioned media (CM) was incubated with goat anti-clusterin antibody (R&D, AF2747) or goat IgG-antibody (1:100) for 24 h at 4 °C. The CM containing the blocking
antibody was used to treat the 1° PT cells under NG and HG conditions. The _LentiORF-APOJ_ clone was purchased from Genecopoeia, and stable _APOJ_ overexpression was achieved by lentiviral
delivery. Cells expressing _LentiORF-APOJ_ were selected with puromycin for 2–3 weeks prior to use in all studies. _LentiORF-control_ serves as the GFP control vector. qPCR and western was
performed to confirm _APOJ_ overexpression. Mouse 1° PT cells were treated with recombinant mouse ApoJ-His-tag (His-ApoJ) (20 µg/ml) (SinoBiological, 50485-M08H) and Cilastatin (0.1 mg/ml)
(MedChemExpress, HY-A0166A), for 24 h under NG conditions. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a Seahorse XFe96 Analyzer (Agilent)
in the presence or absence of mitochondrial function inhibitors as previously reported for all the different experimental conditions45. In brief, cells were seeded into 96 well plates at 2 ×
106 cells per well, and 24 h later, the growth media was removed, cells were washed with PBS. DMSO, STO-609 or conditioned media was added with or without NG or HG, and 24 h later the media
was removed and replaced with serum-free Seahorse DMEM, pH 7.4, supplemented with 1 mM glutamine and 2 mM pyruvate, NG or HG. After incubation in a CO2 free incubator for 45–60 min, OCR and
ECAR were measured at baseline or after acute injections of 1.5 μM oligomycin (Agilent), 3 μM carbonyl cyanide-p-(trifluoromethoxy) phenylhydrazone (FCCP) (Agilent), and 0.5 μM
rotenone/antimycin A (Agilent) using the Seahorse XF Cell Mito Stress Test Kit (Agilent). Mitochondrial membrane potential was measured using MitoProbe DiIC1(5) Assay Kit (Invitrogen). In
brief, cells were trypsinized, washed with PBS, and incubated with 1,1′,3,3,3′,3′-hexamethylindodicarbo-cyanine iodide (DilC1) alone or with DilC1 and carbonyl cyanide 3-
chlorophenylhydrazone (CCCP) and the difference in fluorescence intensity was measured between the groups using FACS. Cell numbers were determined using the Countess 3 cell counter at days
0, 2, 3 and 6. Cell proliferation was also measured using the 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) assay. GFP-labeled primary mouse podocytes were obtained
using FACS sorting. Other cell lines were validated in previous publications or by manufacturer’s website WESTERN BLOT ASSAY Protein lysates were collected from HK2 cells and 1° PT cells
with a buffer containing 4% SDS, and protease phosphatase inhibitor. Protein lysates were subjected to immunoblot analysis using rabbit anti-CaMK1D (Proteintech, 13613-1-AP, 1:1000
dilution), rabbit anti-DRP1 (Invitrogen, MA5-26255, 1:1000 dilution), rabbit anti-phospho-DRP1(Ser637) (Invitrogen, PA5-101038, 1:1000 dilution), goat anti-ApoJ (R&D, AF2747, 1:1000
dilution), rabbit anti-ApoJ (Cell Signaling technology, 42143, 1:1000 dilution), and mouse anti-β-actin (Sigma-Aldrich, A1978, 1:5000 dilution) antibodies. Uncropped and unprocessed scans
are included in the Source data file. IMMUNOPRECIPITATION Protein lysates were collected from 1° PT cells treated with 5% conditioned media with Pierce IP Lysis Buffer (Thermo Fisher
Scientific, 87788) and Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 78438). Pull-down was performed using calmodulin (CaM)-sepharose beads (Abcam, ab286869) as
previously described103. Briefly, protein lysates from 1° PT cells were incubated with CaM-sepharose beads at 4 °C for 3 h on a shaker, centrifuged, unbound protein was removed, and the
beads were incubated with elution buffer at room temperature for 30 min on a shaker. The beads were then centrifuged, and the bound protein was subjected to western blot analysis to
determine the amount of ApoJ pulled down with CaM beads. SINGLE-NUCLEI ISOLATION, SEQUENCING, DATA PROCESSING, AND ANALYSIS Nuclei were isolated from the mouse kidney cortex based on
previous protocol94,104. In brief, mice were perfused with PBS and kidney cortex was stored in RNAlater for snRNA-seq, whereas for snATAC-seq, kidney cortex without any RNAlater at -80 °C. A
2 mm3 section of tissue was rinsed briefly with PBS and minced before adding 1 mL of lysis buffer containing 20 mM Tris-HCl (pH 8), 320 mM sucrose, 5 mM CaCl2, 3 mM MgAc2, 0.1 mM EDTA, 0.1%
TritonX-100, 0.1% RNase Inhibitor and 0.1% DAPI. The tissue was initially dissociated by pipetting up and down 10X with a p-1000 tip and then being passed through a 25 G syringe 10X. The
tissue was incubated on ice for 10 min and then passed through a 30 µm CellTrics filter. Nuclei were pelleted by centrifugation (5 min, 500 g, 4 °C) and washed with PBS after removal of
supernatant. Nuclei were pelleted again and resuspended in 1 mL of PBS containing 0.04% BSA and RNAse inhibitor (0.2 u/µL) and then passed through a 40 µm FlowMI filter, followed by a 5 μm
pluriStrainer Mini filter before generating counts with hemocytometer. Nuclei were then diluted and prepared for snRNA-seq and snATAC-seq with the 10X Chromium System according to the
manufacturer’s instructions (10X Genomics). Sequencing was performed using a NovaSeqS4 platform. Raw sequencing data was demultiplexed and aligned to a mouse pre-mRNA reference genome using
Cell Ranger 3.1.0-v2 on _SeaWulf_, the HPC Cluster at Stony Brook University. For snRNA-seq, quality control, dimensionality reduction and clustering were performed using the R-package
_Seurat 4.3.0_21. Genes expressed in a minimum of 3 cells were retained. Cells expressing < 200 or > 7500 genes were excluded. Cells expressing > 5% mitochondrial genes were also
excluded. For the snATAC-seq, Cell Ranger atac-2.0.0, _Signac 1.8.0_20 and _Archr 1.0.2_22 were used for subsequent analysis. Quality control for each single cell were conducted based on
peak_region_fragments>100, peak_region_fragments <60000, pct_reads_in_peaks>5, blacklist_ratio<0.05, nuclesosome_signal<4, and TSS.enrichment>1. For chromatin
immunoprecipitation (ChIP)-enrichment analysis, KLF6 binding site data were obtained from ChIP-sequencing data deposited in the Gene Expression Omnibus database (accession no. GSE96355), and
locations of binding sites were determined using the Genomic Regions Enrichment of Annotations Tool (GREAT)105 “basal plus extension” approach, with a maximum extension of 10 kb from any
transcription start site (TSS). Heat maps were generated using Morpheus software (https://software.broadinstitute.org/morpheus) and genes were clustered using the one minus Pearson
correlation method. Pathway enrichment analyses of the significantly differentially expressed genes were undertaken using _Enrichr_14,15 libraries of KEGG 2019 (mouse) pathways18,
WikiPathways 2019 (mouse)17, and Reactome pathways16. Trajectory analysis was performed using the R package, Monocle-v2106,107. Ligand receptor interactions were shown in the circle plot
using R package, _edgebundler 0.1.4_108. Pathway enrichment analyses of the genes in the trajectory analysis and for differentially accessible regions (DARs) was carried out using
_clusterProfiler 4.6.2_109,110. PROTEOMICS Tandem mass spectrometry was performed on the supernatant from mouse primary podocytes isolated from _KLF6__PODTA_ and _NPHS2-rtTA_ mice (with and
without doxycycline) using a label-free proteomic approach. After the cells reached confluency, cells were washed five times with 1 × PBS and medium was replaced with phenol-red,
insulin-transferrin-selenium (ITS), and serum-free RPMI. After 48 additional hours, the CM harvested and centrifuged for 5 min at 500 g and then for 10 min at 1500 g. Samples were reduced
(dithiothreitol), alkylated (iodoacetamide), digested with trypsin, and cleaned using hydrophobic-lipophilic-balance (HLB) pack C-18. For the urine proteomics, the urine samples from
_NPHS2-rtTA_ and _KLF6__PODTA_ mice were spun at 2000 g for 10 min at 4 °C, the supernatant was mixed with a laemmli buffer and heated at 100 °C for 5 min. The protein amount was normalized
using creatinine. The samples were run in a 12% criterion TGX gel, the gel was fixed with 40% EtOH/10% acetic acid at room temperature for 15 min. Following a wash with water, the gel was
stained with coomassie overnight with gentle rocking. The gel bands were excised based on their molecular weight and cut into small pieces, before undergoing in-gel reduction
(dithiothreitol), alkylation (iodoacetamide) and destaining (ammonium bicarbonate/acetonitrile). The dried gel pieces were digested with trypsin and cleaned through a C-18 column. For both,
CM and urine proteomics, samples (4 μl) were injected onto a 20 cm-long ReproSil C-18 (3 μM particle) column and run on the Q Exactive HF Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo
Fisher Scientific) or TripleTOF 5600+ (Sciex). Analysis was carried out using Thermo Fisher Proteome Discoverer v2.4 and Scaffold. STATISTICAL ANALYSIS All statistical analysis was
performed using GraphPad Prism 9.0. Based on the distribution of the data, the appropriate parametric or nonparametric statistical tests were utilized. The exact test used for each
experiment is denoted in the figure legends and data presented as the mean ± SEM. STUDY APPROVAL All animal studies conducted were approved by the Stony Brook University Animal Institute
Committee (#564062). Consent was waived due to the de-identified human biopsy specimens and the study was approved by Stony Brook University Institutional Review Board (#798611). The
National Institutes of Health Guide for the Care and Use of Laboratory Animals was followed strictly. REPORTING SUMMARY Further information on research design is available in the Nature
Portfolio Reporting Summary linked to this article. DATA AVAILABILITY All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental Materials.
Source data are as a separate Source Data file. All raw data from snRNA-seq and ATAC-seq have been deposited in the Gene Expression Omnibus GSE171854 and GSE230681. Raw data from proteomics
have been deposited in Massive Database, MSV000092123 [https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=08d1896c2de340fca8cf078d9549aa01]. Previously reported datasets used in this study
includes accession no. GSE96355. Source data are provided with this paper. CODE AVAILABILITY R code and Seurat RDS data object are available through the following link:
https://github.com/MallipattuLab/KLF6_PODTA. REFERENCES * Stevens, P. E. & Levin, A. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global
outcomes 2012 clinical practice guideline. _Ann. Intern. Med._ 158, 825–831 (2013). Article PubMed Google Scholar * Jefferson, J. A., Shankland, S. J. & Pichler, R. H. Proteinuria in
diabetic kidney disease: a mechanistic viewpoint. _Kidney Int._ 74, 22–36 (2008). Article CAS PubMed Google Scholar * Mohandes, S. et al. Molecular pathways that drive diabetic kidney
disease. _J. Clin. Invest_. 133, e165654 (2023). * Alicic, R. Z., Rooney, M. T. & Tuttle, K. R. Diabetic kidney disease: challenges, progress, and possibilities. _Clin. J. Am. Soc.
Nephrol._ 12, 2032–2045 (2017). Article CAS PubMed PubMed Central Google Scholar * Centers for Disease Control and Prevention. _Chronic Kidney Disease in the United States._
https://www.cdc.gov/kidney-disease/media/pdfs/CKD-Factsheet-H.pdf (2023). * Faria, J., Gerritsen, K. G. F., Nguyen, T. Q., Mihaila, S. M. & Masereeuw, R. Diabetic proximal tubulopathy:
can we mimic the disease for in vitro screening of SGLT inhibitors? _Eur. J. Pharmacol._ 908, 174378 (2021). Article CAS PubMed Google Scholar * Xie, Y. et al. Reticulon-1A mediates
diabetic kidney disease progression through endoplasmic reticulum-mitochondrial contacts in tubular epithelial cells. _Kidney Int._ 102, 293–306 (2022). Article CAS PubMed PubMed Central
Google Scholar * Bialkowska, A. B., Yang, V. W. & Mallipattu, S. K. Krüppel-like factors in mammalian stem cells and development. _Development_ 144, 737–754 (2017). Article CAS
PubMed PubMed Central Google Scholar * Mallipattu, S. K. et al. Krüppel-like factor 6 regulates mitochondrial function in the kidney. _J. Clin. Invest._ 125, 1347–1361 (2015). Article
PubMed PubMed Central Google Scholar * Horne, S. J. et al. Podocyte-specific loss of Krüppel-like factor 6 increases mitochondrial injury in diabetic kidney disease. _Diabetes_ 67,
2420–2433 (2018). Article CAS PubMed PubMed Central Google Scholar * Uil, M. et al. Combining streptozotocin and unilateral nephrectomy is an effective method for inducing experimental
diabetic nephropathy in the ‘resistant’ C57Bl/6J mouse strain. _Sci. Rep._ 8, 5542 (2018). Article ADS PubMed PubMed Central Google Scholar * Wu, H., Kirita, Y., Donnelly, E. L. &
Humphreys, B. D. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. _J. Am. Soc. Nephrol._ 30, 23–32
(2019). Article CAS PubMed Google Scholar * Lake, B. B. et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. _Nat.
Commun._ 10, 2832 (2019). Article ADS PubMed PubMed Central Google Scholar * Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. _BMC
Bioinform._ 14, 128 (2013). Article Google Scholar * Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. _Nucleic Acids Res._ 44, W90–W97
(2016). Article CAS PubMed PubMed Central Google Scholar * Jassal, B. et al. The reactome pathway knowledgebase. _Nucleic Acids Res._ 48, D498–D503 (2020). CAS PubMed Google Scholar
* Slenter, D. N. et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. _Nucleic Acids Res._ 46, D661–D667 (2018). Article CAS PubMed Google
Scholar * Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. _Nucleic Acids Res._ 28, 27–30 (2000). Article CAS PubMed PubMed Central Google Scholar *
Partridge, E. C. et al. Occupancy maps of 208 chromatin-associated proteins in one human cell type. _Nature_ 583, 720–728 (2020). Article ADS CAS PubMed PubMed Central Google Scholar *
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. _Nat. Methods_ 18, 1333–1341 (2021). Article CAS PubMed PubMed
Central Google Scholar * Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and
species. _Nat. Biotechnol._ 36, 411–420 (2018). Article CAS PubMed PubMed Central Google Scholar * Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell
chromatin accessibility analysis. _Nat. Genet_. 53, 403–411 (2021). * Gerhardt, L. M. S. et al. Lineage tracing and single-nucleus multiomics reveal novel features of adaptive and
maladaptive repair after acute kidney injury. _J. Am. Soc. Nephrol._ 34, 554–571(2023). * Kirita, Y., Wu, H., Uchimura, K., Wilson, P. C. & Humphreys, B. D. Cell profiling of mouse acute
kidney injury reveals conserved cellular responses to injury. _Proc. Natl Acad. Sci. USA_ 117, 15874–15883 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Li, H.,
Dixon, E. E., Wu, H. & Humphreys, B. D. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. _Cell Metab._
34, 1977–1998.e1979 (2022). Article CAS PubMed PubMed Central Google Scholar * Gerhardt, L. M. S., Liu, J., Koppitch, K., Cippà, P. E. & McMahon, A. P. Single-nuclear
transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. _Proc. Natl Acad. Sci. USA_ 118, e2026684118 (2021). Article CAS PubMed
PubMed Central Google Scholar * Balzer, M. S. et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. _Nat. Commun._
13, 4018 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Knott, A. B., Perkins, G., Schwarzenbacher, R. & Bossy-Wetzel, E. Mitochondrial fragmentation in
neurodegeneration. _Nat. Rev. Neurosci._ 9, 505–518 (2008). Article CAS PubMed PubMed Central Google Scholar * Tokumitsu, H. et al. STO-609, a specific inhibitor of the
Ca2+/calmodulin-dependent protein kinase kinase. _J. Biol. Chem._ 277, 15813–15818 (2002). Article CAS PubMed Google Scholar * Kukimoto-Niino, M. et al. Crystal structure of the
Ca2+/Calmodulin-dependent protein kinase kinase in complex with the inhibitor STO-609. _J. Biol. Chem._ 286, 22570–22579 (2011). Article CAS PubMed PubMed Central Google Scholar *
Nguan, C. Y., Guan, Q., Gleave, M. E. & Du, C. Promotion of cell proliferation by clusterin in the renal tissue repair phase after ischemia-reperfusion injury. _Am. J. Physiol. Renal
Physiol._ 306, F724–F733 (2014). Article CAS PubMed Google Scholar * Ghiggeri, G. M. et al. Depletion of clusterin in renal diseases causing nephrotic syndrome. _Kidney Int._ 62,
2184–2194 (2002). Article CAS PubMed Google Scholar * He, J. et al. Glomerular clusterin expression is increased in diabetic nephropathy and protects against oxidative stress-induced
apoptosis in podocytes. _Sci. Rep._ 10, 14888 (2020). Article CAS PubMed PubMed Central Google Scholar * Saunders, J. R. et al. Clusterin depletion enhances immune glomerular injury in
the isolated perfused kidney. _Kidney Int._ 45, 817–827 (1994). Article CAS PubMed Google Scholar * Woroniecka, K. I. et al. Transcriptome analysis of human diabetic kidney disease.
_Diabetes_ 60, 2354–2369 (2011). Article CAS PubMed PubMed Central Google Scholar * Ju, W. et al. Defining cell-type specificity at the transcriptional level in human disease. _Genome
Res._ 23, 1862–1873 (2013). Article CAS PubMed PubMed Central Google Scholar * Ramilowski, J. A. et al. A draft network of ligand–receptor-mediated multicellular signalling in human.
_Nat. Commun._ 6, 7866 (2015). Article ADS CAS PubMed Google Scholar * Seo, J. A. et al. Apolipoprotein J is a hepatokine regulating muscle glucose metabolism and insulin sensitivity.
_Nat. Commun._ 11, 2024 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Yamada, K. et al. Clusterin is up-regulated in glomerular mesangial cells in complement-mediated
injury. _Kidney Int._ 59, 137–146 (2001). Article CAS PubMed Google Scholar * Kounnas, M. Z. et al. Identification of glycoprotein 330 as an endocytic receptor for apolipoprotein
J/clusterin. _J. Biol. Chem._ 270, 13070–13075 (1995). Article CAS PubMed Google Scholar * Pérez, M. et al. Inhibition of brush border dipeptidase with cilastatin reduces toxic
accumulation of cyclosporin A in kidney proximal tubule epithelial cells. _Nephrol. Dial Transplant._ 19, 2445–2455 (2004). Article PubMed Google Scholar * Hori, Y. et al. Megalin
blockade with cilastatin suppresses drug-induced nephrotoxicity. _J. Am. Soc. Nephrol._ 28, 1783–1791 (2017). Article CAS PubMed PubMed Central Google Scholar * Goto, S., Hosojima, M.,
Kabasawa, H. & Saito, A. The endocytosis receptor megalin: from bench to bedside. _Int. J. Biochem. Cell Biol._ 157, 106393 (2023). Article CAS PubMed Google Scholar * Vallon, V. The
proximal tubule in the pathophysiology of the diabetic kidney. _Am. J. Physiol. Regul. Integr. Comp. Physiol._ 300, R1009–R1022 (2011). Article CAS PubMed Google Scholar * Piret, S. E.
et al. Krüppel-like factor 6–mediated loss of BCAA catabolism contributes to kidney injury in mice and humans. _Proc. Natl. Acad. Sci. USA_ 118, 1 (2021). Article Google Scholar *
Ghiassi-Nejad, Z. et al. Reduced hepatic stellate cell expression of krüppel-like factor 6 tumor suppressor isoforms amplifies fibrosis during acute and chronic rodent liver injury.
_Hepatol._ 57, 786–796 (2013). Article CAS Google Scholar * Sawaki, D. et al. Modulation of cardiac fibrosis by Krüppel-like factor 6 through transcriptional control of thrombospondin 4
in cardiomyocytes. _Cardiovasc. Res._ 107, 420–430 (2015). Article CAS PubMed PubMed Central Google Scholar * Park, J. et al. Single-cell transcriptomics of the mouse kidney reveals
potential cellular targets of kidney disease. _Science_ 360, 758–763 (2018). Article CAS PubMed PubMed Central Google Scholar * Skelding, K. A. & Rostas, J. A. The role of molecular
regulation and targeting in regulating calcium/calmodulin stimulated protein kinases. _Adv. Exp. Med. Biol._ 740, 703–730 (2012). Article CAS PubMed Google Scholar * Skelding, K. A.,
Rostas, J. A. & Verrills, N. M. Controlling the cell cycle: the role of calcium/calmodulin-stimulated protein kinases I and II. _Cell Cycle_ 10, 631–639 (2011). Article CAS PubMed
Google Scholar * Schulman, H. The multifunctional Ca2+/calmodulin-dependent protein kinases. _Curr. Opin. Cell Biol._ 5, 247–253 (1993). Article CAS PubMed Google Scholar * Skelding, K.
A. & Rostas, J. A. P. Regulation of multifunctional calcium/calmodulin stimulated protein kinases by molecular targeting. _Adv. Exp. Med. Biol._ 1131, 649–679 (2020). Article CAS
PubMed Google Scholar * Zhou, J. et al. NMDA receptor-mediated CaMKII/ERK activation contributes to renal fibrosis. _BMC Nephrol._ 21, 392 (2020). Article CAS PubMed PubMed Central
Google Scholar * Xiaocui, F. et al. The CaMKII inhibitory peptide AIP alleviates renal fibrosis through the TGF-β/Smad and RAF/ERK pathways. _J. Pharmacol. Exp. Ther._ 386, 310–322 (2023).
Article Google Scholar * Junho, C. V. C., Caio-Silva, W., Trentin-Sonoda, M. & Carneiro-Ramos, M. S. An overview of the role of calcium/calmodulin-dependent protein kinase in
cardiorenal syndrome. _Front Physiol._ 11, 735 (2020). Article PubMed PubMed Central Google Scholar * Mikhailov, A. V., Liu, Y., Cheng, H. J., Lin, J. J. & Cheng, C. P.
Calmodulin-dependent protein kinase II activation promotes kidney mesangial expansion in streptozotocin-induced diabetic mice. _Heliyon_ 8, e11653 (2022). Article CAS PubMed PubMed
Central Google Scholar * Haney, S. et al. RNAi screening in primary human hepatocytes of genes implicated in genome-wide association studies for roles in Type 2 diabetes identifies roles
for CAMK1D and CDKAL1, among others, in hepatic glucose regulation. _PLoS ONE_ 8, e64946 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Kooner, J. S. et al. Genome-wide
association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. _Nat. Genet._ 43, 984–989 (2011). Article CAS PubMed PubMed Central
Google Scholar * Zeggini, E. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. _Nat. Genet._
40, 638–645 (2008). Article CAS PubMed PubMed Central Google Scholar * Fromont, C. et al. Discovery of highly selective inhibitors of calmodulin-dependent kinases that restore insulin
sensitivity in the diet-induced obesity in vivo mouse model. _J. Med. Chem._ 63, 6784–6801 (2020). Article CAS PubMed PubMed Central Google Scholar * Xue, A. et al. Genome-wide
association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. _Nat. Commun._ 9, 2941 (2018). Article ADS PubMed PubMed Central Google Scholar *
Fogarty, M. P., Cannon, M. E., Vadlamudi, S., Gaulton, K. J. & Mohlke, K. L. Identification of a regulatory variant that Binds FOXA1 and FOXA2 at the CDC123/CAMK1D Type 2 diabetes GWAS
locus. _PLoS Genet._ 10, e1004633 (2014). Article PubMed PubMed Central Google Scholar * Chang, C. R. & Blackstone, C. Dynamic regulation of mitochondrial fission through
modification of the dynamin-related protein Drp1. _Ann. N. Y. Acad. Sci._ 1201, 34–39 (2010). Article ADS CAS PubMed PubMed Central Google Scholar * Han, X. J. et al. CaM kinase I
alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. _J. Cell Biol._ 182, 573–585 (2008). Article CAS PubMed PubMed Central Google Scholar * Xu, S. et al. CaMKII
induces permeability transition through Drp1 phosphorylation during chronic β-AR stimulation. _Nat. Commun._ 7, 13189 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Bo,
T. et al. Calmodulin-dependent protein kinase II (CaMKII) mediates radiation-induced mitochondrial fission by regulating the phosphorylation of dynamin-related protein 1 (Drp1) at serine
616. _Biochem. Biophys. Res. Commun._ 495, 1601–1607 (2018). Article CAS PubMed Google Scholar * Jones, S. E. & Jomary, C. Clusterin. _Int. J. Biochem. Cell Biol._ 34, 427–431
(2002). Article CAS PubMed Google Scholar * Pereira, R. M. et al. Protective molecular mechanisms of clusterin against apoptosis in cardiomyocytes. _Heart Fail. Rev._ 23, 123–129 (2018).
Article CAS PubMed Google Scholar * Ma, Y. et al. Apolipoprotein-J blocks increased cell injury elicited by ox-LDL via inhibiting ROS-CaMKII pathway. _Lipids Health Dis._ 18, 117
(2019). Article PubMed PubMed Central Google Scholar * Vargas, A., Yamamoto, K. L., Craft, C. M. & Lee, E.-J. Clusterin enhances cell survival by suppressing neuronal nitric-oxide
synthase expression in the rhodopsin S334ter-line3 retinitis pigmentosa model. _Brain Res._ 1768, 147575 (2021). Article CAS PubMed Google Scholar * Kim, H.-J. et al. Protective role of
clusterin/apolipoprotein J against neointimal hyperplasia via antiproliferative effect on vascular smooth muscle cells and cytoprotective effect on endothelial cells. _Arterioscler. Thromb.
Vasc. Biol._ 29, 1558–1564 (2009). Article CAS PubMed Google Scholar * Park, J. S. et al. Clusterin overexpression protects against Western diet-induced obesity and NAFLD. _Sci. Rep._
10, 17484 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * McLaughlin, L. et al. Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. _J.
Clin. Invest._ 106, 1105–1113 (2000). Article CAS PubMed PubMed Central Google Scholar * Han, B. H. et al. Clusterin contributes to caspase-3-independent brain injury following neonatal
hypoxia-ischemia. _Nat. Med._ 7, 338–343 (2001). Article CAS PubMed Google Scholar * May, P. C. et al. Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer’s
disease and in response to experimental lesions in rat. _Neuron_ 5, 831–839 (1990). Article CAS PubMed Google Scholar * Rosenberg, M. E. & Paller, M. S. Differential gene expression
in the recovery from ischemic renal injury. _Kidney Int._ 39, 1156–1161 (1991). Article CAS PubMed Google Scholar * Schlegel, P. N. et al. Clusterin production in the obstructed rabbit
kidney: correlations with loss of renal function. _J. Am. Soc. Nephrol._ 3, 1163–1171 (1992). Article CAS PubMed Google Scholar * Witzgall, R., Brown, D., Schwarz, C. & Bonventre, J.
V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a
large pool of mitotically active and dedifferentiated cells. _J. Clin. Invest._ 93, 2175–2188 (1994). Article CAS PubMed PubMed Central Google Scholar * Dvergsten, J., Manivel, J. C.,
Correa-Rotter, R. & Rosenberg, M. E. Expression of clusterin in human renal diseases. _Kidney Int._ 45, 828–835 (1994). Article CAS PubMed Google Scholar * Kim, S. S. et al. Urine
clusterin/apolipoprotein J is linked to tubular damage and renal outcomes in patients with type 2 diabetes mellitus. _Clin. Endocrinol._ 87, 156–164 (2017). Article CAS Google Scholar *
Girton, R. A., Sundin, D. P. & Rosenberg, M. E. Clusterin protects renal tubular epithelial cells from gentamicin-mediated cytotoxicity. _Am. J. Physiol. Renal Physiol._ 282, F703–F709
(2002). Article CAS PubMed Google Scholar * Weng, X. et al. Clusterin regulates macrophage expansion, polarization and phagocytic activity in response to inflammation in the kidneys.
_Immunol. Cell Biol._ 99, 274–287 (2021). Article CAS PubMed Google Scholar * Jung, G.-S. et al. Clusterin attenuates the development of renal fibrosis. _J. Am. Soc. Nephrol._ 23, 73–85
(2012). Article CAS PubMed Google Scholar * Rohne, P., Prochnow, H., Wolf, S., Renner, B. & Koch-Brandt, C. The chaperone activity of clusterin is dependent on glycosylation and
redox environment. _Cell. Physiol. Biochem._ 34, 1626–1639 (2014). Article CAS PubMed Google Scholar * Foster, E. M., Dangla-Valls, A., Lovestone, S., Ribe, E. M. & Buckley, N. J.
Clusterin in Alzheimer’s disease: mechanisms, genetics, and lessons from other pathologies. _Front. Neurosci_. 13, 164 (2019). * Byun, K. et al. Clusterin/ApoJ enhances central leptin
signaling through Lrp2-mediated endocytosis. _EMBO Rep._ 15, 801–808 (2014). Article CAS PubMed PubMed Central Google Scholar * Tarrant, J. 4.15 - emerging translatable safety
biomarkers. _Comprehensive Medicinal Chemistry III_ (eds. Chackalamannil, S., Rotella, D. P., Ward, S. E.) 1-4369 (PublisherElsevier Inc., 2017). * O’Day, D. H. & Huber, R. J. Calmodulin
binding proteins and neuroinflammation in multiple neurodegenerative diseases. _BMC Neurosci._ 23, 10 (2022). Article PubMed PubMed Central Google Scholar * Kovács, P., Pushparaj, P.
N., Takács, R., Mobasheri, A. & Matta, C. The clusterin connectome: emerging players in chondrocyte biology and putative exploratory biomarkers of osteoarthritis. _Front. Immunol_. 14,
1103097 (2023). * Wu, X. S. et al. Ca2+ and calmodulin initiate all forms of endocytosis during depolarization at a nerve terminal. _Nat. Neurosci._ 12, 1003–1010 (2009). Article CAS
PubMed PubMed Central Google Scholar * Ren, L. et al. Clusterin ameliorates endothelial dysfunction in diabetes by suppressing mitochondrial fragmentation. _Free Radic. Biol. Med._ 145,
357–373 (2019). Article CAS PubMed Google Scholar * Rodríguez-Rivera, C., Garcia, M. M., Molina-Álvarez, M., González-Martín, C. & Goicoechea, C. Clusterin: Always protecting.
Synthesis, function and potential issues. _Biomed. Pharmacother._ 134, 111174 (2021). Article PubMed Google Scholar * Tang, S. et al. Clusterin alleviates Cr(VI)-induced mitochondrial
apoptosis in L02 hepatocytes via inhibition of Ca2+-ROS-Drp1-mitochondrial fission axis. _Ecotoxicol. Environ. Saf._ 205, 111326 (2020). Article CAS PubMed Google Scholar * Gujarati, N.
A. et al. Loss of functional SCO2 attenuates oxidative stress in diabetic kidney disease. _Diabetes_ 71, 142–156 (2021). Article PubMed PubMed Central Google Scholar * Zhong, F. et al.
Reduced krüppel-like factor 2 aggravates glomerular endothelial cell injury and kidney disease in mice with unilateral nephrectomy. _Am J Pathol_ 186, 2021–2031 (2016). Article CAS PubMed
PubMed Central Google Scholar * Mallipattu, S. K. et al. Expression of HIV transgene aggravates kidney injury in diabetic mice. _Kidney Int._ 83, 626–634 (2013). Article CAS PubMed
PubMed Central Google Scholar * Huang, H. et al. piRNA-associated germline nuage formation and spermatogenesis require mitoPLD profusogenic mitochondrial-surface lipid signaling. _Dev.
Cell_ 20, 376–387 (2011). Article CAS PubMed PubMed Central Google Scholar * Lamprecht, M. R., Sabatini, D. M. & Carpenter, A. E. CellProfiler™: Free, versatile software for
automated biological image analysis. _BioTechniques_ 42, 71–75 (2007). Article CAS PubMed Google Scholar * Koop, K. et al. Expression of podocyte-associated molecules in acquired human
kidney diseases. _J. Am. Soc. Nephrol._ 14, 2063–2071 (2003). Article CAS PubMed Google Scholar * Mallipattu, S. K. et al. Krüppel-like factor 15 (KLF15) is a key regulator of podocyte
differentiation. _J. Biol. Chem._ 287, 19122–19135 (2012). Article CAS PubMed PubMed Central Google Scholar * Reiniger, N. et al. Deletion of the receptor for advanced glycation end
products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. _Diabetes_ 59, 2043–2054 (2010). Article CAS PubMed PubMed Central Google Scholar * Piret,
S. E. et al. Loss of proximal tubular transcription factor krüppel-like factor 15 exacerbates kidney injury through loss of fatty acid oxidation. _Kidney Int._ 100, 1250–1267 (2021). Article
CAS PubMed PubMed Central Google Scholar * Kaleka, K. S., Petersen, A. N., Florence, M. A. & Gerges, N. Z. Pull-down of calmodulin-binding proteins. _J. Vis. Exp_. 23, 3502 (2012).
* Pace, J. A. et al. Podocyte-specific KLF4 is required to maintain parietal epithelial cell quiescence in the kidney. _Sci. Adv._ 7, eabg6600 (2021). Article ADS CAS PubMed PubMed
Central Google Scholar * Bejerano, G. et al. GREAT improves functional interpretation of cis-regulatory regions. _Nat. Biotechnol._ 28, 495–501 (2010). Article PubMed PubMed Central
Google Scholar * Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. _Nat. Biotechnol._ 32, 381–386 (2014).
Article CAS PubMed PubMed Central Google Scholar * Qiu, X. et al. Single-cell mRNA quantification and differential analysis with Census. _Nat. Methods_ 14, 309–315 (2017). Article CAS
PubMed PubMed Central Google Scholar * David, S. _edgebundle: Algorithms for Bundling Edges in Networks and Visualizing Flow and Metro Maps._ https://github.com/schochastics/edgebundle
(2022). * Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. _OMICS J. Integr. Biol._ 16, 284–287 (2012). Article
CAS Google Scholar * Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. _Innovation_ 2, 100141 (2021). CAS PubMed PubMed Central Google
Scholar Download references ACKNOWLEDGEMENTS This work was supported by funds from the National Institute of Diabetes and Digestive and Kidney Diseases Grant (DK112984, DK121846), Veterans
Affairs (1I01BX003698, 1I01BX005300) and Dialysis Clinic Inc. to S.K.M. The authors thank Northport Veterans Affairs Medical Center-Stony Brook University Single Cell Genomic Core for single
nuclei library generation. AUTHOR INFORMATION Author notes * These authors contributed equally: Nehaben A. Gujarati, Bismark O. Frimpong. AUTHORS AND AFFILIATIONS * Division of Nephrology
and Hypertension, Department of Medicine, Stony Brook University, Stony Brook, NY, USA Nehaben A. Gujarati, Bismark O. Frimpong, Malaika Zaidi, Robert Bronstein, Yiqing Guo & Sandeep K.
Mallipattu * Department of Pathology, University of Utah, Salt Lake City, UT, USA Monica P. Revelo * Department of Pharmacology, Stony Brook University, Stony Brook, NY, USA John D. Haley *
Division of Endocrinology, Department of Medicine, Stony Brook University, Stony Brook, NY, USA Igor Kravets * Renal Section, Northport VA Medical Center, Northport, NY, USA Sandeep K.
Mallipattu Authors * Nehaben A. Gujarati View author publications You can also search for this author inPubMed Google Scholar * Bismark O. Frimpong View author publications You can also
search for this author inPubMed Google Scholar * Malaika Zaidi View author publications You can also search for this author inPubMed Google Scholar * Robert Bronstein View author
publications You can also search for this author inPubMed Google Scholar * Monica P. Revelo View author publications You can also search for this author inPubMed Google Scholar * John D.
Haley View author publications You can also search for this author inPubMed Google Scholar * Igor Kravets View author publications You can also search for this author inPubMed Google Scholar
* Yiqing Guo View author publications You can also search for this author inPubMed Google Scholar * Sandeep K. Mallipattu View author publications You can also search for this author
inPubMed Google Scholar CONTRIBUTIONS N.A.G., B.O.F., and S.K.M. designed experiments. N.A.G., B.O.F., and S.K.M. wrote the draft of the manuscript. N.A.G., B.O.F., M.Z., R.B., M.P.R., J.H.,
I.K., and Y.G. performed experiments and analysis. CORRESPONDING AUTHOR Correspondence to Sandeep K. Mallipattu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interest. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is
available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4
SUPPLEMENTARY DATA 5 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link
to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Gujarati, N.A., Frimpong, B.O., Zaidi, M. _et al._ Podocyte-specific KLF6 primes proximal tubule CaMK1D signaling to attenuate diabetic
kidney disease. _Nat Commun_ 15, 8038 (2024). https://doi.org/10.1038/s41467-024-52306-5 Download citation * Received: 05 September 2023 * Accepted: 02 September 2024 * Published: 13
September 2024 * DOI: https://doi.org/10.1038/s41467-024-52306-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative