- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The escape of mitochondrial double-stranded dsRNA (mt-dsRNA) into the cytosol has been recently linked to a number of inflammatory diseases. Here, we report that the release of
mt-dsRNA into the cytosol is a general feature of senescent cells and a critical driver of their inflammatory secretome, known as senescence-associated secretory phenotype (SASP). Inhibition
of the mitochondrial RNA polymerase, the dsRNA sensors RIGI and MDA5, or the master inflammatory signaling protein MAVS, all result in reduced expression of the SASP, while broadly
preserving other hallmarks of senescence. Moreover, senescent cells are hypersensitized to mt-dsRNA-driven inflammation due to their reduced levels of PNPT1 and ADAR1, two proteins critical
for mitigating the accumulation of mt-dsRNA and the inflammatory potency of dsRNA, respectively. We find that mitofusin MFN1, but not MFN2, is important for the activation of the
mt-dsRNA/MAVS/SASP axis and, accordingly, genetic or pharmacologic MFN1 inhibition attenuates the SASP. Finally, we report that senescent cells within fibrotic and aged tissues present dsRNA
foci, and inhibition of mitochondrial RNA polymerase reduces systemic inflammation associated to senescence. In conclusion, we uncover the mt-dsRNA/MAVS/MFN1 axis as a key driver of the
SASP and we identify novel therapeutic strategies for senescence-associated diseases. SIMILAR CONTENT BEING VIEWED BY OTHERS APOPTOTIC STRESS CAUSES MTDNA RELEASE DURING SENESCENCE AND
DRIVES THE SASP Article Open access 11 October 2023 TXNRD1 DRIVES THE INNATE IMMUNE RESPONSE IN SENESCENT CELLS WITH IMPLICATIONS FOR AGE-ASSOCIATED INFLAMMATION Article 24 January 2024 P53
ENHANCES DNA REPAIR AND SUPPRESSES CYTOPLASMIC CHROMATIN FRAGMENTS AND INFLAMMATION IN SENESCENT CELLS Article Open access 05 March 2025 INTRODUCTION Cellular senescence is an evolutionary
conserved and stereotypic response characterized by an inability to proliferate and by the secretion of pro-inflammatory factors, the latter collectively known as the senescence-associated
secretory phenotype (SASP)1. Cellular senescence is elicited by multiple stresses, including aberrant oncogene expression, DNA damage, reactive oxygen species, and telomeric erosion1. In
young, healthy, organisms, senescent cells are short-lived due to their immune clearance, mainly by macrophages and natural killer (NK) cells2. However, in aged organisms or in
immunosuppressive environments, immune clearance fails and senescent cells accumulate, as it happens in a broad variety of diseases and in aged tissues1,2,3. Senescent cells directly
contribute to the aggravation and progression of multiple diseases and this is mainly due to their continued release of the SASP4. Importantly, there is growing evidence, both in animal
models and also in patients, that senescence-targeting strategies are efficient against multiple diseases1,3,4. Emerging evidence has pointed to the mitochondria of senescent cells as one of
the principal modulators of the SASP5. Specifically, cells with severe disruption of mitochondrial functions, such as inhibition of the electron transport chain, or ablation of
mitochondrial protein chaperones, undergo an atypical senescence that lacks the full SASP secretome6. In an even more striking demonstration of this concept, cells devoided of mitochondria
due to forced mitophagy fail to activate the SASP upon exposure to senescence-inducing triggers7. The nature of the SASP-inducing signals emanating from the mitochondria is only partially
understood. The mitochondria of senescent cells produce high levels of ROS that activate innate immune signaling hubs8. More recently, it has been reported that senescent cells release
mitochondrial double-stranded DNA (mt-dsDNA) into the cytosol through transient BAX/BAK pores, and this cytosolic mt-dsDNA contributes to the SASP via activation of the cGAS/STING pathway9.
Moreover, during aging, senescent cells release mitochondrial DNA fragments detectable in the serum, which also contribute to systemic inflammation10. The above findings on senescent cells
are part of a larger body of evidence linking the release of mt-dsDNA to the cytosol as a critical inflammatory event of relevance in multiple contexts and diseases11,12,13. Mitochondria
have the peculiarity of generating double-stranded RNA (dsRNA) molecules as a result of the normal process of bidirectional and overlapping transcription of its circular genome. There are
dedicated mechanisms to separate the two strands and ensure the stability of the H strand, which is almost entirely coding, and the degradation of the L strand, which is largely
non-coding14. The majority of the L strand undergoes rapid decay by the RNA degradosome complex containing the ribonuclease PNPase (encoded by _PNPT1_)14. Importantly, perturbations in
mitochondrial RNA processing may cause abnormal accumulation of mt-dsRNA, its release through BAX/BAK pores, and the activation of innate immune signaling pathways15. This is the case of
cells from patients with Aicardi-Goutières or Leigh syndromes carrying loss-of-function mutations in the _PNPT1_ gene and, as a consequence, presenting aberrant constitutive activation of
interferon type I signaling15. The release of mt-dsRNA has also been reported in osteoarthritis16, renal injury17, and Huntington's disease18. Also, the inflammatory response elicited
by genotoxic agents has been reported to be mostly due to the damage inflicted on the mt-dsDNA, which in turn results in the cytosolic release of mitochondrial RNA through BAX/BAK pores19.
Another gene linked to the Aicardi-Goutières syndrome is _ADAR_, which encodes a dsRNA-binding protein with adenosine deaminase activity, and whose main function is to reduce the
inflammatory activity of cytosolic dsRNA20. The presence of mt-dsRNAs in the cytosol is a potent inflammatory trigger through the activation of sensors RIGI and MDA5, and the oligomerization
of MAVS on the mitochondrial surface, which serves as a platform to activate the interferon and NFκB signaling pathways21. Here, we show that mt-dsRNA-driven inflammation is a general
feature of cellular senescence and, therefore, relevant to multiple aging-associated diseases. We also identify a number of drug-actionable targets whose inhibition strongly reduces the
pro-inflammatory secretome of senescent cells. RESULTS SENESCENT CELLS RELEASE MITOCHONDRIAL DSRNA INTO THE CYTOSOL Given the large body of evidence linking mt-dsRNA with
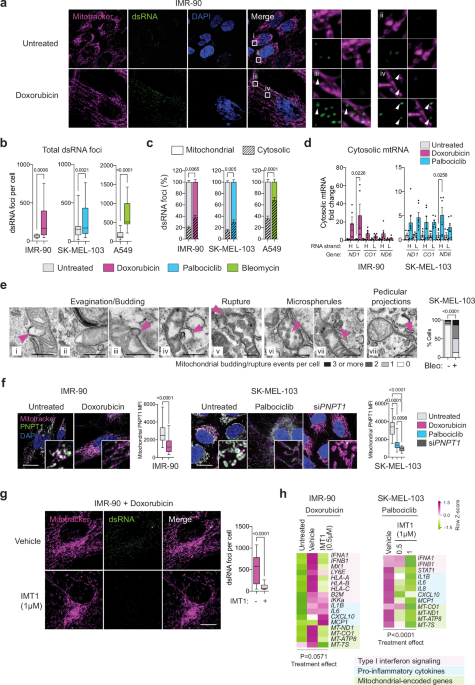
inflammation23,24,25,26,27,28,29, we hypothesized that mt-dsRNA could also be released into the cytosol from senescent mitochondria. To test this, we detected dsRNA by immunofluorescence
using monoclonal antibody (J2)22. To confirm the specificity of J2 against dsRNA, we treated proliferating cells with the general transcriptional inhibitor actinomycin D (ActD) and this
completely eliminated J2 foci, while siRNA-mediated inhibition of _PNPT1_ (encoding a key enzyme that prevents the accumulation of mt-dsRNA15) resulted in a robust increase in J2 foci
(Supplementary Fig. 1a). Then, we induced senescence in three different human cell types, IMR-90 lung fibroblasts using the DNA damaging agent doxorubicin, SK-MEL-103 melanoma cells treating
them with the CDK4/6 inhibitor palbociclib, and A549 lung adenocarcinoma cells exposing them to the genotoxic agent bleomycin. These cellular models were chosen for their efficiency in
inducing senescence, and also because they cover a variety of cell types and senescence triggers. Senescence was confirmed by senescence-associated β-galactosidase (SABG) staining and by
measuring the expression of senescence-associated genes _CDKN1A_ and _IL6_ by RT–qPCR (Supplementary Fig. 1b, c). As previously described5, we observed that senescent cells have an increased
mitochondrial mass, increased mitochondrial DNA copy number, and decreased mitochondrial membrane potential (Supplementary Fig. 1d–f). Interestingly, staining with J2 revealed a significant
accumulation of dsRNA foci in senescent cells compared to their proliferative counterparts (Fig. 1a, b; Supplementary Fig. 1g–h). A tendency to accumulate mt-dsRNA is detectable as early as
day 3 after the senescence-inducing treatment and a significant increase was observed at day 7, coincident with the full establishment of senescence (Supplementary Fig. 1i). By co-staining
cells with a mitochondria-specific probe, we quantified the proportion of J2 foci localized within or outside of mitochondria. Interestingly senescent cells presented a significant increase
in extra-mitochondrial dsRNA compared to proliferative cells (Fig. 1c). To assess the possible mitochondrial origin of these cytosolic dsRNA foci, we isolated RNA from mitochondria-free
cytosolic fractions of proliferative and senescent cells. Of note, our cytosolic fractions lacked detectable mitochondrial protein AIF, suggesting the absence of mitochondrial contamination
and also that the fractionation method did not cause major mitochondrial membrane rupture (Supplementary Fig. 1j). By strand-specific quantitative PCR, we observed that the cytosolic
fractions from senescent cells contained higher levels of each of the two mitochondrial RNA strands, H and L, compared to proliferative cells (Fig. 1d). Given the importance of the above
observations, we confirmed them in additional models of cellular senescence, in particular, irradiation-induced senescence and replicative senescence in human and mouse fibroblasts
(Supplementary Fig. 2a–e). The release of mt-dsDNA in acutely damaged cells occurs through large mitochondrial evaginations that rupture and extrude the mitochondrial matrix content into the
cytosol23,24. These evaginations could conceivably allow the release of mt-dsRNA and, for this reason, we wondered if we could detect mitochondrial evaginations in senescent cells. Electron
microscopy analysis revealed that the mitochondria of senescent cells present budding structures, some including the matrix and even others showing evidence of rupture (Fig. 1e). In
addition to identifying evagination and budding structures, we observed pedicular projections on the membrane of mitochondria in senescent cells, as well as the formation of “microspherules”
within the matrix and their potential release into the cytosol (Fig. 1e). These abnormalities were observed in the majority of senescent cells (87% of the cells with at least one
mitochondrial budding event) which was in contrast with the low frequency in the parental non-senescent cells (13% of cells with at least one event). Interestingly, we observed a significant
reduction in the protein levels of PNPT1 detected by immunofluorescence in senescent cells compared to non-senescent cells (Fig. 1f). Reduced levels of PNPT1 could be one of the factors
that contribute to the accumulation of mt-dsRNA in senescent cells. Collectively, we conclude that the mitochondria of senescent cells accumulate abnormally high levels of dsRNA that are
released to the cytosol. PHARMACOLOGIC INHIBITION OF MITOCHONDRIAL RNA POLYMERASE REDUCES THE SASP Considering the link between mt-dsRNA release and the constitutive inflammation that
characterizes the Aicardi-Goutières and the Leigh syndromes20,25, we wondered if the release of mt-dsRNA could also underlie the inflammatory phenotype of senescent cells. We took advantage
of a highly specific inhibitor of mitochondrial RNA polymerase, named IMT126. Senescent cells were further exposed to IMT1 for 48 h prior to analysis. As expected, inhibition of
mitochondrial RNA polymerase activity resulted in a profound reduction in the levels of dsRNAs (detected by J2) (Fig. 1g) and, mitochondrial-encoded mRNAs (_MT-ND1_, _MT-CO1_, and _MT-ATP8_)
(Fig. 1h). Importantly, treatment of senescent cells with IMT1 also reduced the mRNA levels of SASP factors in SK-MEL-103 cells and type-I interferons in IMR-90 cells (Fig. 1h). At
increasing concentrations of IMT1, mitochondrial RNA levels were progressively reduced together with a proportional reduction in _IFNA1_ mRNA levels (Supplementary Fig. 3a, b). In contrast
to mitochondrial RNA, the mRNA levels of nuclear-encoded mitochondrial genes were not affected by IMT1 (Fig. 1h; Supplementary Fig. 3a–c). Also, the levels of _CDKN1A_ (encoding the
cell-cycle inhibitor and senescence marker p21) were not affected by the presence of IMT1 (Supplementary Fig. 3c). This is in agreement with the well-established concept that inhibition of
the SASP does not impair other features of senescent cells, including the inhibition of the cell cycle27,28. Of note, mitochondrial DNA levels were not affected by IMT1 during the 48 h
treatment period (Supplementary Fig. 3d, e). To explore the contribution of nuclear-derived dsRNAs to the pool of dsRNAs present in senescent cells, we have focused on repeated retroelements
due to their known ability to generate stable dsRNAs21. Previous reports have indicated increased expression of retroelements LINE1 and ERVs in senescent cells29,30. We have confirmed these
observations in our senescent cells by RT–qPCR (ERVV1, ERV31, LINE1, or ALU elements). However, the majority of the retroelements tested showed no decrease, or even elevation, in their
expression following IMT1 treatment (Supplementary Fig. 3c). These observations indicate that mitochondria are the main source of the elevated dsRNA foci present in senescent cells and that
mt-dsRNA is an important contributor to the SASP. SENESCENT CELLS ARE SENSITIZED TO DSRNA-DEPENDENT INNATE IMMUNE SIGNALING Nuclear as well as cytosolic dsRNAs are subject to modifications
that reduce their pro-inflammatory activity. In particular, the adenosine deaminase ADAR1 is a key RNA modifying enzyme that reduces the capacity of dsRNAs to activate innate immune
signaling31. Interestingly, compared to proliferative cells, senescent cells presented a marked reduction in the levels of the two ADAR1 isoforms (both encoded by the same gene _ADAR_)31,
namely, p150 (which shuttles between nucleus and cytosol) and p110 (mostly nuclear) (Fig. 2a–c; Supplementary Fig. 4a–c). To ascertain whether ADAR1 is functional in senescent cells, despite
its lower levels, we treated IMR-90 and SK-MEL-103 cells with si_ADAR_ (or scrambled control si_SCR_) and then we induced senescence. Notably, the presence of si_ADAR_ resulted in elevated
levels of MAVS, as detected by immunofluorescence (Supplementary Fig. 4d), and in a dramatically enhanced induction of _IFNA1_ and _IFNB1_ and pro-inflammatory cytokines (Supplementary Fig.
4e). Conversely, we overexpressed each of the two ADAR1 isoforms (p110 o p150) in senescent cells and this led to a reduction in the SASP of senescent IMR-90 cells, while only p150, but not
p110 decreased the SASP in senescent SK-MEL-103 cells (Fig. 2d; Supplementary Fig. 4f, g). Together, these observations indicate that senescent cells, by downregulating ADAR1 protein levels,
are hypersensitized to the inflammatory activity of cytosolic dsRNAs. We then focused on the main cytoplasmic dsRNA sensors. In particular, RIGI (encoded by gene _DDX58_) and MDA5 (encoded
by gene _IFIH1_) were upregulated at the mRNA level soon after treatment with senescence-inducing agents (Fig. 2e). This was in contrast to sensors LGP2 (_DHX58_) and PKR (_EIF2AK2_) that
remained unchanged in senescent cells (Fig. 2e). The signaling integrator MAVS (_MAVS_) was significantly upregulated in senescent SK-MEL-103, but not in senescent IMR-90 cells (Fig. 2e).
Notably, IMR-90 cells with combined siRNA-mediated depletion of RIGI and MDA5 underwent doxorubicin-induced senescence with reduced expression of cytokines (Fig. 2f; Supplementary Fig. 4h),
whereas depletion of PKR did not reduce the expression of the cytokines tested (Supplementary Fig. 4i). In agreement with previous reports29,32,33,34, combined siRNA-mediated depletion of
dsDNA sensor cGAS and STING, followed by induction of senescence, resulted in reduced expression of cytokines (Fig. 2f; Supplementary Fig. 4h). To reinforce the concept that RIGI and MDA5
are involved in the SASP, we measured the presence of a set of cytokines directly in the extracellular medium and confirmed that many of the cytokines overproduced upon senescence were
normalized upon combined depletion of RIGI and MDA5 (Fig. 2g). In the case of mt-dsDNA, its release from senescent cells involves BAX and BAK9. Interestingly, compared to the depletion of
RIGI/MDA5 or cGAS/STING, combined depletion of BAX/BAK resulted in a more profound reduction in the expression of SASP-related cytokines (Fig. 2f). The involvement of BAX and BAK in the
release of mt-dsRNA in senescent cells was further supported by using mouse embryo fibroblasts doubly-deficient in _Bax_ and _Bak_ (_Bax_/_Bak_-DKO). Interestingly, while mtRNA was clearly
elevated in the cytosolic fraction of senescent wild-type (WT) MEFs, this was not observed in the cytosolic fraction of senescent _Bax_/_Bak_-DKO MEFs (Fig. 2h). Accordingly, and consistent
with a previous report9, IFN-induced genes and SASP factors were reduced in senescent _Bax_/_Bak_-DKO MEFs treated with doxorubicin compared to senescent WT MEFs (Supplementary Fig. 4j). In
summary, senescent cells downregulate the dsRNA editing machinery and upregulate the dsRNA sensing machinery, together rendering senescent cells highly responsive to cytosolic dsRNA. Our
data, together with a previous report9, are consistent with a model in which the mitochondria of senescent cells release mt-dsDNAs and mt-dsRNAs to the cytosol in a BAX/BAK-dependent manner,
activating the cGAS/STING and RIGI/MDA5 sensors, respectively, triggering inflammatory signaling and the SASP. MAVS IS ACTIVATED IN SENESCENT CELLS AND IT IS A KEY INDUCER OF THE SASP The
mitochondrial antiviral signaling (MAVS) protein is a signaling hub that integrates multiple inflammatory inputs, most notably those elicited by dsRNA and its sensors RIGI and MDA521.
Western blot analysis revealed that MAVS protein levels increased in senescent cells (Fig. 3a). MAVS activation results in large prion-like aggregates in the outer surface of mitochondria
detectable by immunofluorescence35. Fluorescence microscopy analysis of MAVS revealed large discrete dots along the mitochondria of senescent IMR-90, SK-MEL-103 and A549 cells (Fig. 3b–d;
Supplementary Fig. 5a). This signal was completely lost in senescent cells generated in the presence of a siRNA against _MAVS_ mRNA (Fig. 3c, d; Supplementary Fig. 5a). We directly confirmed
increased MAVS oligomerization in senescent cells both by dissucinimidyl suberate (DSS) cross-linking followed by acrylamide electrophoresis (Fig. 3e) and by semi-denaturing detergent
agarose electrophoresis (SDD-AGE) (Fig. 3f). To evaluate the impact of MAVS on the transcriptome of senescent cells, we performed RNA-seq in senescent cells (IMR-90 and SK-MEL-103) generated
in the presence of si_MAVS_ and compared them to senescent cells with scrambled siRNA. Interestingly, among the top downregulated pathways were inflammatory signatures (Broad Hallmarks)
(Fig. 3g, h). These observations were confirmed by RT–qPCR; indeed, senescent IMR-90 cells generated in the presence of si_MAVS_ were completely devoided of the expression of the tested
cytokines and other components of the SASP, such as _PAI1_ (also known as _SERPINE1_) and _B2M_ (Fig. 3i and Supplementary Fig. 5b). Of note, consistent with the idea that SASP inhibition
does not affect other features of senescence, depletion of _MAVS_ did not affect the levels of _CDKN1A_ (Fig. 3i). We conclude that MAVS aggregation on the outer surface of mitochondria is a
distinctive feature of senescent cells and an important mediator of the SASP. ANTAGONISTIC ROLE OF MITOFUSINS ON THE SASP To better understand the role of mitochondria in senescence, we
focused on the fact that senescent cells present highly fused mitochondria5 (Fig. 4a), and inhibition of mitochondrial fusion is anti-inflammatory36,37. Based on this, we decided to study
the effect of mitofusin inhibition on the SASP. First, we confirmed previous reports indicating that senescent cells present elevated protein levels of mitofusin 1 (MFN1) and mitofusin 2
(MFN2)38 (Supplementary Fig. 6a, b) together with a reduction in the levels of fission-inducing phospho-DRP1(S616)39 (Supplementary Fig. 6c, d). We then downregulated MFN1 and MFN2, alone or
combined, by transfecting siRNAs in IMR-90 cells, and this was followed by exposure of cells to doxorubicin to induce senescence. Senescence was allowed to develop for 7 days
post-doxorubicin and then we confirmed the downregulation of _MFN1_ and _MFN2_ (Supplementary Fig. 6e, f). Downregulation of mitofusins, alone or combined, caused fragmentation of
mitochondria, although fragmentation was much more extensive in the case of si_MFN2_ compared to si_MFN1_ in the three cell lines tested (IMR-90, SK-MEL-103 and A549) (Fig. 4b; Supplementary
Fig. 6g). Importantly, mitochondria fragmentation did not affect the development of senescence features such as SABG staining (Supplementary Fig. 6h). Gene set enrichment analysis (GSEA) of
the transcriptome of senescent IMR-90 and SK-MEL-103 cells with combined inhibition of MFN1 and MFN2 (si_MFN1_+_2_) revealed cytokine and interferon signaling among the most downregulated
signatures (Fig. 4c). These results were validated by RT–qPCR analysis. In particular, we focused on cytokines characteristic of the SASP and IFN-signaling targets related to the MHC-I40,41,
both of which were significantly downregulated compared to senescent cells treated with scrambled siRNA (Fig. 4d; Supplementary Fig. 6i, j). Recent reports have uncovered a complex
interplay between MFN1 and MFN2 in relation to MAVS and inflammation. In particular, although the combined inhibition of the two mitofusins is anti-inflammatory36,37, this is not the case
for their single inhibition: single downregulation of MFN1 is anti-inflammatory42, whereas single downregulation of MFN2 is pro-inflammatory43,44. The mechanistic basis for these
differential effects on inflammation remains to be elucidated; but the fact that the combined inhibition has the same phenotype as single MFN1 inhibition suggests that MFN1 acts downstream
of MFN2. We observed the same pattern in relation to the SASP of senescent IMR-90 cells: si_MFN1_ significantly decreased the SASP and IFN responsive genes; si_MFN2_ further elevated the
expression of these genes; and combined inhibition of both mitofusins recapitulated the same effect as the inhibition of MFN1 alone (Fig. 4d). It is interesting to note that, while both
si_MFN1_ and si_MFN2_ induce mitochondrial fragmentation, only si_MFN1_ suppresses the SASP, thus disconnecting fragmentation from inflammation. In an effort to further characterize the role
of mitofusins in senescence-driven inflammation, we observed that si_MFN1_ strongly reduced the amount of dsRNA (J2) foci and MAVS aggregates, while si_MFN2_ elevated the amount of both of
them (Fig. 4e,f; Supplementary Fig. 7a–c). In fact, si_MFN1_ reduces the levels of pIRF3 and RIGI, both of which are increased in senescent cells (Supplementary Fig. 7d). Moreover, the
individual mRNA levels of _IL6_, _IL1A_, _IFNA1_, and protein levels of HLA-A/B/C determined by cytometry were positively correlated with the levels of dsRNA (J2) foci and mitochondrial MAVS
aggregates (Supplementary Fig. 7e, f). These observations indicate that mitofusins modulate the levels of mt-dsRNA foci in senescent cells in an antagonistic manner, with the inhibition of
MFN1 being dominant and anti-inflammatory. PHARMACOLOGIC INHIBITION OF MITOFUSINS REDUCES THE SASP Building upon the finding that combined depletion of mitofusins leads to a reduction in the
SASP, we aimed to achieve similar results using pharmacological tools. We have recently reported small molecules rationally designed to destabilize or stabilize the anti-tethering,
non-fusogenic, conformation of mitofusins45. In particular, compound MASM7 (abbreviated here as MFNa) activates both MFN1 and MFN2, whereas compound MFI8 (abbreviated as MFNi) inhibits both
mitofusins45. Senescence was induced for 7 days in IMR-90 and SK-MEL-103 cells and then they were treated with MFNa or MFNi for 48 h. As expected, MFNi induced fragmentation, both in
senescent and proliferative IMR-90 and SK-MEL-103 cells (Fig. 4g; Supplementary Fig. 8a, b). Senescent cells present high basal levels of fusion and, therefore, treatment with MFNa only
modestly increased fusion (Supplementary Fig. 8b). At the drug concentrations used, we did not observe cell death after a period of 48 h (Supplementary Fig. 8c). To capture the
transcriptional programs most sensitive to pharmacological activation or inhibition of mitofusins, we compared the transcriptome of senescent IMR-90 cells treated for 48 h with MFNa or MFNi.
Although only a limited number of pathways presented enrichment in MFNa-treated compared to MFNi-treated senescent cells, the elevation of IFN targets emerged as a significant upregulated
Broad Hallmark (Fig. 4h). Moreover, analysis by RT–qPCR confirmed the inhibition of SASP factors upon treatment with MFNi in senescent SK-MEL-103 and A549 cancer cells (Supplementary Fig.
8d, e). The extracellular medium of senescent cells treated with MFNi also presented decreased concentration at the protein level of those SASP factors (Supplementary Fig. 8f). These
observations support the idea that it is possible to suppress the SASP of senescent cells by pharmacological inhibition of MFN1. MT-DSRNA ACCUMULATES IN SENESCENT CELLS IN VIVO Finally, we
aimed to extend our results on the mt-dsRNA/MAVS/SASP axis to cellular senescence in in vivo contexts. We began by examining xenografts of human SK-MEL-103 cells grown in nude mice and
treated with palbociclib, which efficiently induces senescence in vivo46. Foci of dsRNA (J2) were clearly detected in histological sections of palbociclib-treated tumors, but not in
untreated tumors (Fig. 5a; Supplementary Fig. 9a). We then focused on fibrotic lesions because of their strong association with senescent cells47. We directed our attention towards the heart
and used a mouse model of myocardial ischemia by permanently blocking the left anterior descending (LAD) coronary artery, which closely mimics the pathophysiology of myocardial infarction.
As expected48, one week after heart ischemia, there was a significant increase in collagen deposition, iron levels, p21, and SABG-positive cells in the infarcted tissue (Supplementary Fig.
9b). Interestingly, we observed a significant increase in dsRNA levels detected by the J2 antibody in infarcted hearts (Fig. 5b), and a positive correlation between the individual levels of
SABG and dsRNA (J2) per infarcted heart analyzed (Fig. 5c). Multiple studies have demonstrated that senescent cells accumulate during aging49. We therefore analyzed SABG activity and dsRNA
in tissues from young and aged mice. We observed an accumulation of dsRNA (J2) in the brain and liver sections of old mice compared to young mice, and the total levels observed per mouse
sample correlated with the total levels of SABG (Fig. 5d; Supplementary Fig. 9c, d). We also detected increased expression of mtRNAs from both H and L strands in the brain of old mice
(Supplementary Fig. 9e), supporting an accumulation of mt-dsRNA with aging. Importantly, when we analyzed histological sections of the liver and kidney, we observed a good colocalization
between areas with dsRNA (J2) accumulation and SABG positivity (Fig. 5e; Supplementary Fig. 9f). These results are consistent with the elevated expression of type I interferon response
genes, p16 (_Cdkn2a_) and dsRNA sensors in old mice across different tissues (Fig. 5f). To gain insight into the potential implications in humans, we examined the expression of dsRNA sensors
and mitochondrial genes in a publicly available dataset of healthy volunteers at different ages50. Interestingly, the levels of the pro-inflammatory genes _RIGI (DDX58)_, _MDA5 (IFIH1)_ and
_MAVS_ clearly increased with aging, while the anti-inflammatory genes _ADAR_ and _ADARB1_ decreased with age (Fig. 5g). We also observed an upregulation of mitochondrial-encoded genes
(_MT-ND1_ and _MT-ATP8_), while nuclear-encoded mitochondrial genes (_SDHA, ATP5A1_) did not increase (Fig. 5g). Sensors for dsDNA _CGAS_, _STING_ or _TLR9_ also increased with age. These
expression patterns are reminiscent of senescent cells. As additional supportive data, mitochondrial mRNA transcripts circulating in a cell-free form in the serum are among the most
significantly elevated during aging in humans51 (Fig. 5h). Finally, to establish a direct association between dsRNA foci and senescence in vivo, we treated old mice with the senolytic drug
navitoclax for 14 days. This regimen of navitoclax efficiently reduces the burden of senescent cells in the kidney (Supplementary Fig. 9g). We observed that senescent cell removal in the
kidney also caused a reduction in dsRNA (J2) foci (Supplementary Fig. 9h). To reinforce the in vivo link between mt-dsRNA and inflammation, we induced systemic senescence in mice by
treatment with doxorubicin. As expected, doxorubicin significantly increased the serum levels of many cytokines. Interestingly, however, concomitant treatment with doxorubicin and IMT1
attenuated the systemic inflammatory response in vivo (Fig. 5i). These observations provide in vivo evidence of the link between mt-dsRNA and senescence-associated inflammation. DISCUSSION
Here, we provide evidence that mt-dsRNA is released during cellular senescence by different cell types, non-cancerous and cancerous, and in response to different types of triggers, including
genotoxic agents such as doxorubicin, inhibitors of cyclin-dependent kinases, such as palbociclib, and replicative senescence. We observed an increase in dsRNA foci in vivo in tumors
treated with chemotherapeutic agents, in fibrotic lesions associated with cellular senescence, such as post-infarction heart, and also in aged tissues, such as liver, brain, and kidney. We
have found suggestive colocalization of dsRNA foci and the senescence marker senescence-associated β-galactosidase (SABG) in the kidney and liver of old mice. Moreover, treatment of old mice
with the senolytic agent navitoclax reduced the detection of dsRNA, thereby linking in vivo the presence of dsRNA and senescence. Importantly, we show that a highly selective inhibitor of
the mitochondrial RNA polymerase, IMT1, almost completely abrogates the detection of dsRNA foci in senescent cells, and strongly reduces the senescence-associated secretory phenotype (SASP),
both in vitro and in vivo. IMT1 does not affect the expression levels of retroelements that could conceivably contribute to the formation of dsRNAs52,53. Indeed, a small fraction of dsRNA
foci (~10%) persist in senescent cells after IMT1 treatment and they could correspond to non-mitochondrial dsRNA sources, such as transcribed retroelements. Together, all the above evidence
directly involve mt-dsRNA as the primary source of dsRNA in senescent cells and as a key inducer of the SASP (Fig. 6). The inflammatory genetic syndromes Aicardi-Goutères and Leigh are
associated with mutations in _PNPT1_ and _ADAR_20,25. Interestingly, these two genes are functionally connected to mt-dsRNA. PNPT1 is critical for preventing the accumulation of mt-dsRNA15
and ADAR1 edits dsRNA, including mt-dsRNA, to reduce its inflammatory activity15,31. Interestingly, we have observed that senescent cells are characterized by very low protein levels of
PNTP1 and ADAR1, which makes them prone to accumulate and release inflammatory mt-dsRNA. In the case of cells lacking PNPT1 or undergoing caspase-independent cell death, mt-dsRNA escapes
from mitochondria through BAX/BAK pores15,54. BAX/BAK pores are also responsible, at least in part, for the escape of mt-dsDNA from the mitochondria of senescent cells9. We have found that
the depletion of BAX/BAK in senescent cells strongly reduces the SASP and this effect is more pronounced than the depletion of the dsDNA sensors cGAS/STING or the dsRNA sensors RIGI/MDA5.
This suggests a scenario in which the mitochondria of senescent cells release two critical inflammatory signals through BAX/BAK pores, namely, mt-dsDNA and mt-dsRNA. Moreover, we have found
that senescent cells upregulate the two main dsRNA sensors involved in the activation of inflammation RIGI and MDA5. In summary, our findings suggest that the downregulation of PNTP1 and
ADAR1, together with the upregulation of RIGI and MDA5, render senescent cells extremely prone to the inflammatory activity of mt-dsRNA (Fig. 6). Mitofusins interact with MAVS and regulate
inflammation in opposite directions42,43. In our work, we build on these concepts to expand their relevance to cellular senescence. Specifically, we found that MFN1 elimination reduces the
levels of dsRNA, inhibits the formation of active MAVS aggregates, and impairs the SASP, while MFN2 elimination has the opposite effect. We also show that the effect of MFN1 depletion on the
mt-dsRNA/MAVS axis is dominant over MFN2 depletion, as the double depletion of MFN1+2 recapitulates the same effects as single MFN1 elimination. Importantly, recently developed small
molecules that directly inhibit both mitofusins45 are able to reduce SASP expression (Fig. 6). This approach could have potential implications for developing novel interventions to mitigate
the deleterious effects of senescence in various disease contexts. In conclusion, we identify the release of mitochondrial dsRNA as an important mediator of the SASP in senescent cells.
Additionally, we demonstrate that the pharmacological elimination of mt-dsRNA or inhibition of mitofusins is efficient in attenuating the SASP. Understanding these molecular pathways opens
up new possibilities for targeted interventions to counteract cellular senescence and improve treatment outcomes in aging-related pathologies. METHODS ANIMAL EXPERIMENTATION MOUSE PROCEDURES
Male and female C57BL/6 mice between 2 and 4 months were used. For aging studies, male and female C57BL/6 mice between 2 and 3 months were used as young, and between 20 and 28 months were
used as old. Animals had _ad-libitum_ access to food (SAFE® A40) and water throughout the study period and maintained in a 12 h–12 h light-dark cycle. All animals were housed under specific
pathogen-free conditions in the mouse facility of the Institute for Research in Biomedicine (IRB) in accordance with the protocols approved by the Animal Care and Use Ethical Committee of
animal experimentation of the Barcelona Science Park (CEEA-PCB) and the Catalan Government. Euthanasia of animals was performed by CO2. ISCHEMIC HEART FIBROSIS Three-four months old male
C57BL/6J mice were used. Intraoperative analgesia was induced by treating mice with buprenorphine (0.1 mg/kg) 60 min prior to anesthesia. Anesthesia was induced with 5% isoflurane and it was
maintained at 2% with mechanical ventilation following endotracheal intubation (2% isoflurane/97% oxygen, 130–140 stroke rate, stroke volume initially 5 ml/kg-increased to 7.5 ml/kg
post-thoracotomy). Animals were placed on a heating table during the whole surgery and eyes were covered with ophthalmic gel (LACRYVISC 3 mg/g, Alcon) to protect the corneas. Neck hair and
hair in the upper thorax were removed using a depilatory cream. An incision was made, followed by a left-sided thoracotomy at the fourth intercostal space. The pericardium was removed and
the heart left anterior descending artery was located between the pulmonary artery and the left auricle and was ligated using an 8-0 Prolene suture (PremiCron®- B Braun Surgical, S.A). The
thoracic incision was closed in layers, using 6-0 Prolene sutures (PremiCron®- B Braun Surgical, S.A) to adapt the ribs and 4-0 Prolene sutures (PremiCron®- B Braun Surgical, S.A) to close
the skin. A heating lamp was used to warm the animals until they fully recovered from the anesthesia. Analgesic treatment continued during the 3 following days after surgery by
intraperitoneal injections of buprenorphine every 12 h. Sham-operated mice were used as control. SK-MEL-103 XENOGRAFT TUMORS 2 × 105 SK-MEL-103 cells were harvested, resuspended in a mixture
of PBS and matrigel (1:1), and subcutaneously injected in the flank of athymic nude female mice of 8–12 weeks. Once tumors were visible, approximately at day 7, the mice were randomly
assigned to the palbociclib-treated group, which received 100 mg/kg of palbociclib by oral gavage in 50 mM sodium lactate every day, or the control group, which received vehicle. Mice were
treated for 8 days. Tumor growth was monitored by caliper measurements and tumor volume was calculated using the formula volume = (length * width2)/2. The tumors were then extracted and
frozen in OCT for SABG or dsRNA staining as described below. ANALYSIS OF CYTOKINES IN MOUSE SERUM Two-months old immunocompetent C57BL/6J male and female mice, with a subcutaneous tumor by
injection of B16-F10 cells one week before the treatment, were used. One week later, animals were randomly assigned to the doxorubicin-treated group, which received 50 mg/kg of doxorubicin
intraperitoneally dissolved in water (day 0 and 3), or the control group, which received vehicle. Starting at day 0, mice were treated with IMT1 (100 mg/kg) or vehicle (10% DMSO, 40% PEG300,
5% Tween 80, 45% saline) by oral gavage until day 7. On day 7, mouse serum was extracted, centrifuged at 2000 × _g_ for 10 min at 4 °C, and then stored at −80 °C. Levels of cytokines,
chemokines, and other factors were quantified using Mouse Cytokine/Chemokine 44-Plex Discovery Assay Array (MD44) (Eve Technologies Corporation, Canada). SENOLYTIC TREATMENT IN VIVO Old
C57BL/6J male and female mice between 20 and 28 months were treated with navitoclax once daily for 2 weeks by oral gavage (50 mg/kg/day). Control mice were treated with vehicle (10% DMSO,
30% PEG400, 60% PHOSAL®PG). CELL CULTURE TREATMENTS CELL LINES SK-MEL-103 (human melanoma), IMR-90 (human fetal lung fibroblast), A549 (human non-small cell lung cancer), and B16-F10 (mouse
melanoma) were obtained from ATCC. Human foreskin fibroblasts were kindly provided by K. Raj (Altos Laboratories). Wild-type (WT) mouse embryonic fibroblasts (MEFs) were isolated from
13.5-day C57BL/6J mouse embryos. MEFs _Bax/Bak_-DKO were obtained from ATCC. Cells were cultured in standard DMEM supplemented with 10% heat-inactivated Fetal Bovine serum (FBS) (Gibco) and
1% antibiotics (penicillin/streptomycin 100 U/mL; (Gibco)). Cells were maintained in a humidified incubator at 37 °C and 5% CO2. IMR-90 cells were cultured under 2% O2. Cells were routinely
tested for mycoplasma contamination using standard PCR, and only negative cells were used. SENESCENCE INDUCTION IN VITRO Cells were plated subconfluent and cultured in the presence of 1 μM
palbociclib (PD033299, Pfizer Inc.), 200 nM doxorubicin (D1515, Sigma), or 10mU/ml bleomycin (B8416, Sigma) for 2 days, after which cells were washed, and replenished with complete culture
medium. Palbociclib was added to the medium with every replenishment. To induce senescence by irradiation, cells were plated subconfluent and exposed to a dose of 20 Gy irradiation. Seven
days after the treatment (unless specified), senescent cells were collected and used for experiments. SMALL MOLECULES TREATMENTS Experiments for in vitro assays were performed after
senescence induction unless indicated elsewhere, with the addition of IMT1 (0.1–1 μM, 48 h), MFI8 (20 μM, 48 h) or MASM7 (10 μM, 48 h). PCR, western blot, immunostaining, and SABG assay were
performed immediately after treatments. SIRNAS TRANSFECTION siRNA pools (siPOOLs) with a complexity of 30 optimally-designed siRNAs were acquired from siTOOLs Biotech GmbH. siRNA gene
knock-down was performed on untreated cells. One day after transfection, senescence was induced for 7 days as described above. Lipofectamine reagent (RNAiMAX; Cat. 13778075) was diluted in
Opti-MEM (Cat. 31985062) and used according to the manufacturer’s instructions. CONDITIONED MEDIUM ANALYSIS Cells were cultured in DMEM medium under standard conditions. Forty-eight hours
before medium collection, cells were incubated in the presence of MFI8 or MASM7 as described above. For cells transfected with siRNA, the medium was changed 48 h before collection without
any treatment. Conditioned medium (CM) was collected, centrifuged at 3000 × _g_, and then stored at −80 °C. Levels of cytokines, chemokines, and other factors were quantified using the Human
Cytokine 15-Plex Discovery Assay Array (HDF15) and Human Interferon 9-Plex Discovery Assay Array (HDIFN9) (Eve Technologies Corporation, Canada). CELL VIABILITY ASSAY To measure cell
survival after treatment with mitofusin activator (MFNa; 20 μM, 48 h) and inhibitor (MFNi; 20 μM, 48), we used CellTiter-Glo® Luminescent Cell Viability Assay (Promega). SEPARATION OF
CYTOPLASMIC AND MITOCHONDRIAL FRACTIONS Cytoplasmic and mitochondrial fractions were prepared using the Mitochondria Isolation Kit (89874, Thermo Fisher Scientific). The purity of fractions
was tested by western blot according to standard protocols. RNA was extracted using RNeasy Mini Kit (Qiagen) with DNase on-column treatment. WESTERN BLOTTING Cell pellets were lysed in 50 mM
Tris-HCl pH8; 1 mM EDTA; 150 mM NaCl; 1% NP40; 0.5% Triton X-100; 1.0% SDS, with freshly added protease inhibitors (87785, Thermo Fisher) and phosphatase inhibitors (4906837001, Roche). A
total protein of 15 or 30 μg was run on NuPAGE 4–12% gradient Bis-Tris gels and transferred to 0.2 μm nitrocellulose membranes (10600001, GE Healthcare). Blots were blocked with a LICOR
blocking buffer and incubated with the corresponding antibodies. β-ACTIN was used as a loading control. Revert™ 700 Total Protein Stain (962-11011, LICORbio) was used to normalize to total
protein. Antibodies used are in Supplementary Table 1. MAVS OLIGOMERIZATION ASSAY SDD-AGE For semi-denaturing detergent agarose electrophoresis (SDD-AGE), cells were harvested on day 7 after
senescence induction, then washed twice with PBS, resuspended in 1× sample buffer (0.5 × TBE, 10% glycerol, 2%SDS, and 0.0025% bromophenol blue), incubated for 5 min and loaded onto a 1.5%
agarose gel. After electrophoresis in the running buffer (1 × TBE and 0.1% SDS) for 45 min with a constant voltage of 100 V at 4 °C, the proteins were transferred overnight at 20 V for
immunoblotting. DSS CROSS-LINKING For dissucinimidyl suberate (DSS) cross-linking followed by acrylamide electrophoresis, cells were harvested at day 7 after senescence induction, then
washed twice with PBS before being lysed in CHAPS buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 1% CHAPS) with phosphatase inhibitor cocktail (Thermo Fisher #78440) and incubated for 30 min at 4
°C. Samples were then centrifuged for 10 min at 6000 × _g_. The pellet was washed once with HEPES 50 mM with a phosphatase inhibitor cocktail and then centrifuged for 10 min at maximum
speed. The pellet was crosslinked with 500 µl buffer (HEPES 50 mM, 150 mM NaCl), phosphatase inhibitor cocktail, disuccinimidyl suberate (DSS) (at 2 mM final concentration). Samples were
vortexed and incubated for 45 min in a 37 °C, 600 rpm heat block-shaker. Tris-HCl (pH 7.5) (at 20 mM final concentration) was added for 15 min to quench the reaction. Samples were
centrifuged for 15 min at 4 °C, maximum speed. The resulting pellet was resuspended in 1× Laemmli sample buffer (with 5% β-mercaptoethanol) and subsequently boiled for 5 min at 95 °C before
being run on a SDS-PAGE Bis-Tris 4–12% NuPage gel (Thermo Fisher Scientific). The primary antibody anti-MAVS used was Santa Cruz sc-166583. FLOW CYTOMETRY For analysis of cultured cell
lines, cells were digested into single cells by trypsinization (0.25% trypsin-EDTA, Invitrogen). To assess mitochondrial mass, cells were previously treated with MitoTracker™ Red CMXRos
(7512, Thermo Fisher) at 200 nM for 30 min at 37 °C. For HLA/B/C detection, cells were trypsinized, washed with PBS, and then incubated with anti-mouse CD16/CD32 at 1:400 (BD Fc BlockTM, BD
Biosciences #553142) for 10 min at 4 °C in FACS Buffer. Then, cells were washed and incubated with antibodies in Supplementary Table 2 for 30 min at 4 °C. Cell viability was assessed using
DAPI (0.1 μmol/L, Molecular Probes, #D1306) following the manufacturer’s instructions. Dead cells were excluded from the analysis. Cell suspensions were run on a FACSAriaTM Fusion Cell
Sorter (BD Biosciences). Autofluorescence signal from the unstained samples was obtained and subtracted from each sample in all experiments. Data were analyzed using FlowJo v10 software.
RT–QPCR: RNA EXTRACTION, CDNA SYNTHESIS AND PCR Total RNA was isolated from cell pellets or tissue using the Trizol (Invitrogen) or the RNeasy® Micro Kit (Quiagen) and cDNA was synthesized
using the iScript™ Advanced cDNA Synthesis Kit (BIO-RAD). qPCR was performed on the QuantStudio6K (Thermo Scientific) using the GoTaq, qPCR Master Mix (Promega), and specific primers.
Thermocycling conditions were as follows: initial step of 10 min at 95 °C, then 40 cycles of 15 s denaturation at 95 °C, 1 min annealing at 60 °C and 15 s extension at 72 °C. Reactions were
run in triplicate, and normalization of all the qRT-PCR data was done by the 2-△△Ct method (Yuan et al. 2006) using the housekeeping genes _B_-_ACTIN_ and _GAPDH_ as endogenous controls. The
primers used are in Supplementary Tables 3 and 4. RNA-SEQ LIBRARY PREPARATION, SEQUENCING, AND BIOINFORMATIC ANALYSIS Total RNA was extracted using the RNeasy® Mini Kit (Qiagen), following
the manufacturer’s instructions. LIBRARY PREPARATION AND SEQUENCING RNA extractions were quantified with a Nanodrop One (Thermo Fisher), and RNA integrity was assessed using the RNA
ScreenTape assay of the Tapestation 4200 platform (Agilent). The integrity of a restricted subset of samples was reconfirmed with the Bioanalyzer 2100 RNA Nano assay (Agilent). Libraries for
RNA-seq were prepared at IRB Barcelona Functional Genomics Core Facility. Briefly, mRNA was isolated from 760 ng of total RNA and used to generate dual-indexed cDNA libraries with the
Illumina Stranded mRNA ligation kit (Illumina) and UD Indexes Set A (Illumina). Ten cycles of PCR amplification were applied to all libraries. Sequencing-ready libraries were quantified
using the Qubit dsDNA HS assay (Invitrogen) and quality controlled with the Tapestation HS D5000 assay (Agilent). Three equimolar pools were prepared with the sixty libraries and submitted
for sequencing at the _Centro Nacional de Análisis Genómico_ (CNAG). A final quality control by qPCR was performed by the sequencing provider before paired-end 150 nt sequencing on a
NovaSeq6000 S4 (Illumina). More than 940 Gbp of reads were produced, with a minimum of 19 million paired-end reads per sample. RNA-SEQ DATA PREPROCESSING AND ALIGNMENT Stranded paired-end
reads were processed using Trimgalore (v0.6.7)55 using default parameters. Trimmed reads were aligned to the human reference genome version hg38 using STAR56 with default parameters. STAR
indexes were built using the ENSEMBL annotation version GRCh38.101. SAM files were converted to BAM and sorted using sambamba (v0.6.70)57. Gene counts were obtained with the featureCounts
function from the Rsubread package58 with the gtf file corresponding to ENSEMBL version GRCh38.101 and parameters set to isPairedEnd = TRUE and strandSpecific = 2. Technical replicates were
collapsed by adding the corresponding columns in the count matrix. RNA-SEQ DIFFERENTIAL EXPRESSION AND FUNCTIONAL ENRICHMENT All analyses were performed in the R programming language version
4.1.359 unless otherwise stated. Differential analyses were performed using the DESeq2 package60 with the experimental batch as a covariate. Functional enrichment was performed using the
roastgsa package61. Briefly, a rotation-based approach for enrichment62 implemented in the R package limma63 is used to represent the null distribution. The maxmean enrichment statistic
proposed in ref. 64, under restandardization, was considered for competitive testing. Gene set analysis was performed employing the regularized VST transformed matrix from DESeq2, in which
low variance genes (genes with standard deviation lower than 0.135) were filtered out. The Broad Hallmarks gene set collection65 was downloaded from refs. 65,66. GENE SET ENRICHMENT ANALYSIS
FOR MFNA AND MFI TREATMENTS GSEA Pre-ranked was used to perform a gene set enrichment analysis of annotations from the MsigDB Hallmarks with standard GSEA analysis settings. The input for
enrichment consisted of the LFC values. We used the GO biological process dataset as the reference gene annotation database. We used the list ranked by statistic, setting ‘gene set’ as the
permutation method, and ran it with 1000 permutations for Kolmogorov-Smirnoff correction for multiple testing, including only gene sets with more than 15 members and less than 500 members.
GSEA Enrichment data were obtained and ranked according to their FDR _q_-value. We considered significantly enriched those gene sets with FDR _q_-value <0.25. DETECTION OF MITOCHONDRIAL
HEAVY (H) AND LIGHT (L) STRANDS RNA IN CYTOSOLIC EXTRACTS FROM CELLS AND IN MOUSE TISSUES For RNA isolated from cytosolic fractions or extracted from brain samples, SuperScript IV reverse
transcriptase (18090050, InvitrogenTM) was used to synthesize the cDNA using primers in Supplementary Table 5. RT–qPCR was performed using strand-specific primers containing CMV promoter
sequences to target different genes as previously described67. Primers used are listed in Supplementary Table 5-6. MITOCHONDRIAL DNA CONTENT DNA was extracted from cell pellets using the
DNeasy Blood and Tissue Kit (Qiagen). From the DNA extract, 10 ng per reaction were used to amplify DNA using specific primers for mitochondrial DNA (mtDNA) or nuclear DNA (nDNA)
(Supplementary Table 7). Mitochondrial DNA content was calculated as follows: * a. ΔCT = (nDNA CT – mtDNA CT) * b. Relative mtDNA content = 2 × 2ΔCT Two primer sets were used for each sample
and the average was calculated. HUMAN GENE EXPRESSION ANALYSIS To analyze mRNA expression in human specimens, we used the voyAGEr database50. Data for mitochondrial RNAs present in a
cell-free form in the serum of aged individuals was taken from available data from previously published studies51 SENESCENCE-ASSOCIATED Β-GALACTOSIDASE (SABG) ACTIVITY For
senescence-associated ß-galactosidase staining, cells and OCT-frozen tissue sections were fixed in SABG fixation solution (PBS with 0.1 M EGTA, 1 M MgCl2, and 50% glutaraldehyde) for 15 min
at room temperature and washed 2 times with PBS. Then, a staining solution (PBS with 1 M MgCl2, 0.5 M K4[Fe(CN)6], and 0.5 M K3[Fe(CN)6]) at pH 6 with 1 mg/ml X-Gal diluted in DMF (all from
Sigma) was added and incubated overnight at 37 °C in a CO2-free incubator. Samples were then washed in PBS and visualized using a Nikon Eclipse TS2 brightfield microscope. IMMUNOFLUORESCENCE
AND SUPER-RESOLUTION MICROSCOPY IMAGE PROCESSING Cells were fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.1% triton, and blocked in 10% FBS + 0.1% triton for 30 min. For
MAVS staining, blocking and incubation were done in 5% milk. Primary antibodies were used as indicated in Supplementary Table 1 overnight at 4 °C. Cells and tissues were washed three times
with PBS and incubated with secondary antibody (1:500) for 1 h. They were then washed with PBS before mounting with Vectashield mounting medium with DAPI (Vector Laboratories). Confocal
images were obtained using the super-resolution Airyscan detector of the ZEISS Elyra 7 microscope at ×64 magnification and 1.8NA. Mitochondrial staining was achieved by incubating cells with
MitoTracker Red CMXRos (Thermo Fisher) at 200 nM for 30 min prior to fixation. The secondary antibodies used were Alexa Fluor 488, Alexa Fluor 568, and Alexa Fluor 647. Image processing and
quantifications were performed using ImageJ software. All quantifications were done on stacks of images. IMMUNOFLUORESCENCE OF DSRNA WITH J2 ANTIBODY Cells were grown in a Nunc Lab-Tek II
Chamber Slide System 24–48 h before treatment. For transcription inhibitor treatment, cells were treated with dimethylsulfoxide (DMSO), actinomycin D (1 μM, 2 h) or IMT1 at the indicated
times and concentrations. Cells were incubated with MitoTracker™ Red CMXRos (7512, Thermo Fisher) at 200 nM for 30 min at 37 °C before fixing in 3.7% PFA-DMEM for 15 min at 37 °C. Tissue
cryosections (OCT) were fixed in 4% paraformaldehyde for 10 min. Cells and tissues were washed three times with PBS and permeabilized with 0.25% Triton X-100 in PBS, washed with 0.05%
Tween20 in PBS, and incubated with 3% BSA in PBS for 30 min at room temperature. Primary antibody anti-dsRNA (J2) was used at 1:200 in 3% BSA in PBS for 1 h at room temperature. They were
washed three times with 0.05% Tween20-PBS and incubated with secondary goat anti-mouse IgG (H+L) conjugated with Alexa Fluor 488 at (1:300) concentration. Then, washed three times with 0.05%
Tween20-PBS, PBS, and mounted with a Vectashield mounting medium with DAPI (Vector Laboratories). Z-stack images were collected with a super-resolution Airyscan detector of ZEISS Elyra 7
microscope at ×64 magnification and 1.8NA. Images were analyzed using ImageJ. COLOCALIZATION OF DSRNA AND MAVS WITH MITOCHONDRIA Cells were stained as described in the “Immunofluorescence of
dsRNA with j2 antibody” or “Immunofluorescence and super-resolution microscopy image processing” sections for dsRNA and MAVS staining, respectively. Cells were incubated with MitoTracker™
Red CMXRos (7512, Thermo Fisher) at 200 nM for 30 min at 37 °C before fixing in 3.7% PFA-DMEM for 15 min at 37 °C. Z-stack confocal images of microscopic slides were obtained using the
super-resolution AiryScan detector of the ZEISS Elyra 7 microscope at ×64 magnification and 1.8NA. Image processing and quantifications were performed using ImageJ software. Colocalization
of j2 spots or MAVS staining with mitochondria was based on fluorescence intensity from Mitotracker. LIVE-CELL MICROSCOPY Cells were loaded with 200 nM Mitotracker Green (Life Technologies)
or JC-10 (AAT Bioquest) in DMEM for 30 min at 37 °C. Cells were then washed and image acquisition was performed using a Spinning Disk microscope (Andor Revolution xD, Andor) equipped with a
stage-top incubator and CO2 control system. A total of 10 µm in 21 z-stacks were acquired at 512 × 512 pixel of format resolution with a 1.4 NA/60× oil immersion objective. Fiji-/ImageJ
software was used to obtain a z projection of the z-stacks and quantified accordingly. MITOCHONDRIAL MORPHOLOGY ANALYSIS Cells were classified according to the following categories: (1)
fragmented, if the vast majority of their mitochondria look like “dots”; (2) intermediate, if they have a mixture of both fragmented and elongated mitochondria but with no apparent branches;
(3) elongated, if mitochondria are clearly long and have some branches; (4) hyperfused, if mitochondria form highly tubular networks very interconnected or if they form clusters. MEMBRANE
POTENTIAL MEASUREMENTS Cells were loaded with 10 μM JC-10 (Deltaclon) in a pre-warmed cell culture medium for 30 min at 37 °C. Cells were washed in a fresh medium and images acquired
according to “Live-cell microscopy” section. As a positive control, membrane potential was dissipated in cells by 10 μM FCCP treatment. HISTOLOGICAL ANALYSIS For formalin-fixed
paraffin-embedded (FFPE) samples, tissues were fixed overnight at 4 °C with neutral buffered formalin (Sigma-Aldrich, #HT501128). Paraffin-embedded tissue sections (2–3 μm) were air-dried
and dried overnight at 60 °C. SIRIUS RED STAINING Picrosirius Red Fast Green staining, tissue samples were incubated with the mordant Thiosemicarbazide 99% (TSC) (Sigma, T33405) for 10 min,
washed in distilled water, incubated with 0.1% Fast green (Sigma, FCF F7552) for 20 min and rinsed with 1% acetic acid (Sigma, 320099) for 1 min. In all cases, samples were dehydrated and
mounted with Mounting Medium, Toluene-Free (CS705, Dako, Agilent) using a Dako CoverStainer. IRON DETECTION BY ENHANCED PERL´S PRUSSIAN BLUE (EPPB) STAINING An Iron Stain Kit was used to
identify iron pigments (AR15811-2, Artisan, Dako, Agilent), with some modifications. To enhance iron detection, we adapted the EPPB staining protocol by adding a blocking step with 5% normal
goat serum (16210064, Life Technology) with 2.5% BSA (10735078001, Sigma) for 60 min followed with 30 min of peroxidase-blocking solution (S2023, Dako-Agilent) and a 30 min incubation with
Liquid DAB+ Substrate Chromogen System (K3468, Dako-Agilent) on sections that had been previously stained with the Iron Stain kit. IMMUNOHISTOCHEMISTRY Immunohistochemistry was performed
manually, in a Ventana Discovery XT platform and in a Leica Bond RX platform. Primary antibodies were incubated on sections in a dilution and for the time as follows: p21 clone HUGO 291H/B5
(CNIO) RTU 60 min. Antigen retrieval for p21 was performed with Cell Conditioning 1 (CC1) buffer (Roche, 950-124). Secondary antibodies used were the OmniMap anti-Rat HRP (Roche, 760-4457)
or OmniMapTM anti-Rb HRP (Roche, 760-4311). Blocking was done with Casein (Roche, 760-219). Antigen–antibody complexes were revealed with ChromoMap DAB Kit (Roche, 760-159). Washings were
performed using the Wash Solution AR (AR10211-2, Dako, Agilent). Quenching of endogenous peroxidase was performed by 10 min of incubation with Peroxidase-Blocking Solution at RT (S2023,
Dako, Agilent). Nonspecific unions were blocked using 5% of goat normal serum (16210064, Life technology) mixed with 2.5% BSA diluted in a wash buffer for 60 min at RT. Blocking of
nonspecific endogenous mouse Ig staining was also performed using Mouse on mouse (M.O.M) Immunodetection Kit – (BMK-2202, Vector Laboratories). Secondary antibodies used were the
BrightVision Poly-HRP-Anti Rabbit IgG Biotin-free, ready to use (DPVR-110 HRP, Immunologic), the HRP-Anti-Rat IgG (MP-7444, Vector) for 45 min or the Goat Anti-Mouse Immunoglobulins/HRP
(Dako-Agilent, P0447) at 1:100 for 30 min. Antigen–antibody complexes were revealed with 3-3′- diaminobenzidine (K346811, Dako) or the DAB (Polymer) (Leica, RE7230-CE) with the same time
exposure. Sections were counterstained with hematoxylin (CS700. Dako, Agilent) and mounted with Mounting Medium, Toluene-Free (CS705, Dako, Agilent) using a Dako CoverStainer. Specificity of
staining was confirmed staining with the following isotype controls: rabbit IgG, polyclonal (Abcam, ab27478), the mouse IgG1, Kappa (NCG01) (Abcam, ab81032), mouse IgG2a kappa (eBM2a)
(eBioscienceTM, 14-4724-82) and the rat IgG (R&D Systems, 6- 001-F). IMAGE ACQUISITION AND QUANTIFICATION OF HISTOLOGICAL SECTIONS Brightfield images were acquired with a NanoZoomer-2.0
HT C9600 digital scanner (Hamamatsu) equipped with a 20× objective. All images were visualized with a gamma correction set at 1.8 in the image control panel of the NDP.view 2 U12388-01
software (Hamamatsu, Photonics, France). Image quantification was performed in the QuPath software. ELECTRON MICROSCOPY ANALYSIS Cells were fixed at 4 °C with a mixture of 2% PFA and 2.5%
glutaraldehyde in 0.1 M phosphate buffer (PB), then scrapped, pelleted, washed in PB, then postfixed with 1% osmium tetroxide and 0.8% potassium ferrocyanide. After that, samples were
dehydrated with acetone and finally embedded in Spurr resin. Ultrathin sections (60 nm) were obtained using an Ultracut E (Reichert) and picked up on copper grids and observed with the JEM
1011 (JEOL, Japan) electron microscope, operating at 80 kV. Micrographs were taken with a camera (Orius 1200A; Gatan, USA) using the DigitalMicrograph software package (Gatan, USA). Electron
micrographs were processed using Adobe Photoshop CS6 (13.0.1) (Adobe Systems). All mitochondria in randomly selected cells showing nuclear section per EM field (each field is the square
hole of a 200mesh grid) were examined at a magnification of 60.000× and 120.000×. The proportion of cells containing MDV-related structures and the mean number of mitochondria per cell
showing these structures was estimated in a total of 30 control and 30 bleomycin-treated cells from five EM fields. STATISTICS ANALYSIS For mouse experiments, no specific blinding method was
used but mice in each simple group were selected randomly. The sample size (n) of each experimental group is described in each corresponding figure legend. Unless otherwise specified,
results from each group were averaged and expressed as means + standard error of the mean (represented as error bars). _T_-test was used for comparison between 2 groups and one-way ANOVA
analysis was used for comparisons among 3 or more groups. Two-way ANOVA analysis was used to examine the influence of two different independent variables. The Statistics program Prism
(GraphPad Prism 9) was used for analysis. Statistical significance was set at a _P_–value < 0.05 REPORTING SUMMARY Further information on research design is available in the Nature
Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The RNA-sequencing data generated in this study have been deposited in the GEO database under accession code GSE236521.
Previously published datasets that were used for analysis in the current study are GSE112289, RNA-seq of cell-free RNA in the serum of young and old humans; voyAGEr database50, mRNA
expression in human specimens. All of the data needed to reproduce the results presented here can be found in the manuscript, figures, and supplementary materials. Source data are provided
with this paper. REFERENCES * Abid, S. et al. _Cellular Senescence in Disease_ (eds Muñoz-Espín, D. & Serrano, M.) xv–xix (Academic Press, 2022). * Kale, A. et al. Role of immune cells
in the removal of deleterious senescent cells. _Immun. Ageing_ 17, 1–9 (2020). Article Google Scholar * Chaib, S., Tchkonia, T. & Kirkland, J. L. Cellular senescence and senolytics:
the path to the clinic. _Nat. Med._ 28, 1556–1568 (2022). Article CAS PubMed PubMed Central Google Scholar * Birch, J. & Gil, J. Senescence and the SASP: Many therapeutic avenues.
_Genes Dev._ 34, 1565–1576 (2020). Article CAS PubMed PubMed Central Google Scholar * Martini, H. & Passos, J. F. Cellular senescence: all roads lead to mitochondria. _FEBS J._ 290,
1186–1202 (2023). Article CAS PubMed Google Scholar * Wiley, C. D. et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. _Cell Metab._ 23, 303–314
(2016). Article CAS PubMed Google Scholar * Correia‐Melo, C. et al. Mitochondria are required for pro‐ageing features of the senescent phenotype. _EMBO J._ 35, 724–742 (2016). Article
PubMed PubMed Central Google Scholar * Vizioli, M. G. et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. _Genes
Dev._ 34, 428–445 (2020). Article CAS PubMed PubMed Central Google Scholar * Victorelli, S. et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. _Nature_
622, 627–636 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * Iske, J. et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs
following transplantation. _Nat. Commun._ 11, 4289 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T. & Galluzzi,
L. Mitochondrial control of inflammation. _Nat. Rev. Immunol._ 23, 159–173 (2023). Article CAS PubMed Google Scholar * Newman, L. E. & Shadel, G. S. Mitochondrial DNA release in
innate immune signaling. _Annu. Rev. Biochem._ 92, 299–332 (2023). Article CAS PubMed PubMed Central Google Scholar * Xian, H. & Karin, M. Oxidized mitochondrial DNA: a protective
signal gone awry. _Trends Immunol._ 44, 188–200 (2023). Article CAS PubMed Google Scholar * Xavier, V. J. & Martinou, J. C. Rna granules in the mitochondria and their organization
under mitochondrial stresses. _Int. J. Mol. Sci._ 22, 9502 (2021). Article CAS PubMed PubMed Central Google Scholar * Dhir, A. et al. Mitochondrial double-stranded RNA triggers
antiviral signalling in humans. _Nature_ 560, 238–242 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Kim, S. et al. Mitochondrial double-stranded RNAs govern the stress
response in chondrocytes to promote osteoarthritis development. _Cell Rep._ 40, 111178 (2022). Article CAS PubMed Google Scholar * Zhu, Y. et al. Polynucleotide phosphorylase protects
against renal tubular injury via blocking mt-dsRNA-PKR-eIF2α axis. _Nat. Commun._ 14, 1223 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * Lee, H. et al. Cell
type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. _Neuron_ 107, 891–908.e8 (2020). Article CAS PubMed
PubMed Central Google Scholar * Tigano, M., Vargas, D. C., Tremblay-Belzile, S., Fu, Y. & Sfeir, A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance.
_Nature_ 591, 477–481 (2021). Article ADS CAS PubMed Google Scholar * Rice, G. I. et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon
signature. _Nat. Genet._ 44, 1243–1248 (2012). Article CAS PubMed PubMed Central Google Scholar * Chen, Y. G. & Hur, S. Cellular origins of dsRNA, their recognition and
consequences. _Nat. Rev. Mol. Cell Biol._ 23, 286–301 (2022). Article CAS PubMed Google Scholar * Schonborn, J. et al. Monoclonal antibodies to double-stranded RNA as probes of RNA
structure in crude nucleic acid extracts. _Nucleic Acids Res._ 19, 2993–3000 (1991). Article CAS PubMed PubMed Central Google Scholar * Riley, J. S. et al. Mitochondrial inner membrane
permeabilisation enables mt DNA release during apoptosis. _EMBO J._ 37, e99238 (2018). Article PubMed PubMed Central Google Scholar * McArthur, K. et al. BAK/BAX macropores facilitate
mitochondrial herniation and mtDNA efflux during apoptosis. _Science_ 359, eaao6047 (2018). Article PubMed Google Scholar * Matilainen, S. et al. Defective mitochondrial RNA processing
due to PNPT1 variants causes Leigh syndrome. _Hum. Mol. Genet._ 26, 3352–3361 (2017). Article CAS PubMed Google Scholar * Bonekamp, N. A. et al. Small-molecule inhibitors of human
mitochondrial DNA transcription. _Nature_ 588, 712–716 (2020). Article ADS CAS PubMed Google Scholar * Laberge, R. M. et al. MTOR regulates the pro-tumorigenic senescence-associated
secretory phenotype by promoting IL1A translation. _Nat. Cell Biol._ 17, 1049–1061 (2015). Article CAS PubMed PubMed Central Google Scholar * Herranz, N. et al. mTOR regulates MAPKAPK2
translation to control the senescence-associated secretory phenotype. _Nat. Cell Biol._ 17, 1205–1217 (2015). Article CAS PubMed PubMed Central Google Scholar * De Cecco, M. et al. L1
drives IFN in senescent cells and promotes age-associated inflammation. _Nature_ 566, 73–78 (2019). Article ADS PubMed PubMed Central Google Scholar * Liu, X. et al. Resurrection of
endogenous retroviruses during aging reinforces senescence. _Cell_ 186, 287–304.e26 (2023). Article CAS PubMed Google Scholar * Song, B., Shiromoto, Y., Minakuchi, M. & Nishikura, K.
The role of RNA editing enzyme ADAR1 in human disease. _Wiley Interdiscip. Rev. RNA_ 13, e1665 (2022). Article CAS PubMed Google Scholar * Glück, S. et al. Innate immune sensing of
cytosolic chromatin fragments through cGAS promotes senescence. _Nat. Cell Biol._ 19, 1061–1070 (2017). Article PubMed PubMed Central Google Scholar * Dou, Z. et al. Cytoplasmic
chromatin triggers inflammation in senescence and cancer. _Nature_ 550, 402–406 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Takahashi, A. et al. Downregulation of
cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. _Nat. Commun._ 9, 1–12 (2018). Article Google Scholar * Ye, J. & Maniatis, T. A prion-like
trigger of antiviral signaling. _Cell_ 146, 348–350 (2011). Article CAS PubMed PubMed Central Google Scholar * Castanier, C., Garcin, D., Vazquez, A. & Arnoult, D. Mitochondrial
dynamics regulate the RIG-I-like receptor antiviral pathway. _EMBO Rep._ 11, 133–138 (2010). Article CAS PubMed Google Scholar * Kim, S. J. et al. Hepatitis C virus triggers
mitochondrial fission and attenuates apoptosis to promote viral persistence. _Proc. Natl Acad. Sci. USA_ 111, 6413–6418 (2014). Article ADS CAS PubMed PubMed Central Google Scholar *
Martínez, J. et al. Mitofusins modulate the increase in mitochondrial length, bioenergetics and secretory phenotype in therapy-induced senescent melanoma cells. _Biochem. J._ 476, 2463–2486
(2019). Article PubMed Google Scholar * Mai, S., Klinkenberg, M., Auburger, G., Bereiter-Hahn, J. & Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial
elongation in senescent cells and enhances resistance to oxidative stress through PINK1. _J. Cell Sci._ 123, 917–926 (2010). Article CAS PubMed Google Scholar * Marin, I. et al. Cellular
senescence is immunogenic and promotes antitumor immunity. _Cancer Discov._ 13, 410–431 (2023). Article CAS PubMed Google Scholar * Chen, H. A. et al. Senescence rewires
microenvironment sensing to facilitate antitumor immunity. _Cancer Discov._ 13, 433–453 (2023). Article CAS Google Scholar * Onoguchi, K. et al. Virus-infection or 5′ppp-RNA activates
antiviral signal through redistribution of IPS-1 mediated by MFN1. _PLoS Pathog._ 6, e1001012 (2010). Article PubMed PubMed Central Google Scholar * Yasukawa, K. et al. Mitofusin 2
inhibits mitochondrial antiviral signaling. _Sci. Signal._ 2, ra47 (2009). Article PubMed Google Scholar * Khodzhaeva, V. et al. Mitofusin 2 deficiency causes pro-inflammatory effects in
human primary macrophages. _Front. Immunol._ 12, 723683 (2021). Article CAS PubMed PubMed Central Google Scholar * Zacharioudakis, E. et al. Modulating mitofusins to control
mitochondrial function and signaling. _Nat. Commun._ 13, 3775 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Muñoz‐Espín, D. et al. A versatile drug delivery system
targeting senescent cells. _EMBO Mol. Med._ 10, 1–18 (2018). Article Google Scholar * Schafer, M. J., Haak, A. J., Tschumperlin, D. J. & LeBrasseur, N. K. Targeting senescent cells in
fibrosis: pathology, paradox, and practical considerations. _Curr. Rheumatol. Rep._ 20, 3 (2018). Article PubMed Google Scholar * Maus, M. et al. Iron accumulation drives fibrosis,
senescence and the senescence-associated secretory phenotype. _Nat Metab_ 5, 2111–2130 (2023). * Borghesan, M., Hoogaars, W. M. H., Varela-Eirin, M., Talma, N. & Demaria, M. A
senescence-centric view of aging: implications for longevity and disease. _Trends Cell Biol._ 30, 777–791 (2020). Article CAS PubMed Google Scholar * Schneider, A. L., Martins-Silva, R.,
Kaizeler, A., Saraiva-Agostinho, N. & Barbosa-Morais, N. L. voyAGEr, a free web interface for the analysis of age-related gene expression alterations in human tissues. _eLife_ 12, 1–19
(2024). Article CAS Google Scholar * Dluzen, D. F. et al. Extracellular RNA profiles with human age. _Aging Cell_ 17, e12785 (2018). Article PubMed PubMed Central Google Scholar *
Kim, Y. et al. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. _Mol. Cell_ 71, 1051–1063.e6 (2018). Article CAS PubMed Google Scholar *
Sun, T. et al. A small subset of cytosolic dsRNAs must be edited by ADAR1 to evade MDA5-mediated autoimmunity. Preprint at _bioRxiv_ https://doi.org/10.1101/2022.08.29.505 (2022). *
Killarney, S. T. et al. Executioner caspases restrict mitochondrial RNA-driven type I IFN induction during chemotherapy-induced apoptosis. _Nat. Commun_. 14, 1399 (2023). * Krueger, F.
TrimGalore: a wrapper around Cutadapt and FastQC to consistently apply adapter and quality trimming to FastQ files, with extra functionality for RRBS data. _GitHub_
https://github.com/FelixKrueger/TrimGalore (2017). * Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS PubMed Google Scholar *
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. _Bioinformatics_ 31, 2032–2034 (2015). Article CAS PubMed PubMed
Central Google Scholar * Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads.
_Nucleic Acids Res._ 47, e47 (2019). Article CAS PubMed PubMed Central Google Scholar * Team, R. C. _R: A Language and Environment for Statistical Computing_ (R Foundation for
Statistical Computing. Vienna, Austria, 2020). * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. _Genome Biol._ 15,
550 (2014). Article PubMed PubMed Central Google Scholar * Caballe Mestres, A., Berenguer Llergo, A. & Stephan-Otto Attolini, C. A comparison of rotation-based scores for gene set
analysis. _Bioinformatics_ 24, 408 (2023). CAS PubMed PubMed Central Google Scholar * Wu, D. et al. ROAST: Rotation gene set tests for complex microarray experiments. _Bioinformatics_
26, 2176–2182 (2010). Article CAS PubMed PubMed Central Google Scholar * Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies.
_Nucleic Acids Res._ 43, e47 (2015). Article PubMed PubMed Central Google Scholar * Efron, B. & Tibshirani, R. On testing the significance of sets of genes. _Ann. Appl. Stat._ 1,
107–129 (2007). Article MathSciNet Google Scholar * Liberzon, A. et al. The molecular signatures database hallmark gene set collection. _Cell Syst._ 1, 417–425 (2015). Article CAS
PubMed PubMed Central Google Scholar * Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. _Proc. Natl Acad.
Sci. USA_ 102, 15545–15550 (2005). Article ADS CAS PubMed PubMed Central Google Scholar * Kim, S., Yoon, J., Lee, K. & Kim, Y. Analysis of mitochondrial double-stranded RNAs in
human cells. _STAR Protoc._ 4, 102007 (2023). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We are grateful to the IRB Functional Genomics Unit
and CRG/CNAG for library preparation and genomic sequencing, to the IRB microscopy unit, and to the CCiUB flow cytometry facility for the technical support. We thank Ms. Tanya Liesel de
Silva from the Gavathiotis lab for her technical assistance. V.L.P. was the recipient of a predoctoral contract from the Spanish Ministry of Education (FPU-18/05917). M.M. received funding
from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement (No 794744), and from the Spanish Ministry of Science and Innovation
(MCIN) (RYC2020-030652-I /AEI /10.13039/501100011033 and PID2022-142205OB-I00). E.Z., F.D.M.M. and E.G. were funded by the National Institutes of Health grants P01AG031782, P30AG038072,
R01CA223243, and a Hevolution Foundation partnership grant. M.K. was funded by the Barcelona Institute of Science and Technology (BIST) and Asociación Española Contra el Cáncer (AECC;
POSTD18020SERR), and supported by the European Molecular Biology Organization (EMBO). Work in the laboratory of M.S. was funded by the IRB and “laCaixa” Foundation, and by grants from the
Spanish Ministry of Science co-funded by the European Regional Development Fund (ERDF) (SAF2017-82613-R), European Research Council (ERC-2014-AdG/669622), and Secretaria d’Universitats i
Recerca del Departament d’Empresa i Coneixement of Catalonia (Grup de Recerca consolidat 2017 SGR 282). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Institute for Research in Biomedicine
(IRB Barcelona), Barcelona Institute of Science and Technology (BIST), Barcelona, Spain Vanessa López-Polo, Mate Maus, Camille Stephan-Otto Attolini, Marta Kovatcheva & Manuel Serrano *
Vall d’Hebron Institute of Oncology (VHIO), Barcelona, Spain Mate Maus * Department of Biochemistry, Department of Medicine, Department of Oncology, Montefiore Einstein Comprehensive Cancer
Center, Institute for Aging Research, Wilf Family Cardiovascular Research Institute, Albert Einstein College of Medicine, Bronx, NY, USA Emmanouil Zacharioudakis, Francisco D. M. Marques
& Evripidis Gavathiotis * Department of Anatomy and Cell Biology and Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), University of
Cantabria-IDIVAL, Santander, Spain Miguel Lafarga * IFOM ETS – The AIRC Institute of Molecular Oncology, Milan, Italy Marta Kovatcheva * Altos Labs, Cambridge Institute of Science, Granta
Park, UK Manuel Serrano Authors * Vanessa López-Polo View author publications You can also search for this author inPubMed Google Scholar * Mate Maus View author publications You can also
search for this author inPubMed Google Scholar * Emmanouil Zacharioudakis View author publications You can also search for this author inPubMed Google Scholar * Miguel Lafarga View author
publications You can also search for this author inPubMed Google Scholar * Camille Stephan-Otto Attolini View author publications You can also search for this author inPubMed Google Scholar
* Francisco D. M. Marques View author publications You can also search for this author inPubMed Google Scholar * Marta Kovatcheva View author publications You can also search for this author
inPubMed Google Scholar * Evripidis Gavathiotis View author publications You can also search for this author inPubMed Google Scholar * Manuel Serrano View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS V.L.P. conceptualized, designed, performed, and analyzed all experiments, contributed to bioinformatic data analysis, and
co-wrote the manuscript. M.M. processed electron microscopy samples and helped with mouse experiments. E.Z., F.D.M.M. and E.G. contributed to experiments related to the activation or
inhibition of MAVS and mitofusins, development and validation of the small molecule MFN1/2 activator and inhibitor. M.L. analyzed and interpreted electron microscopy samples. C.S-O.A.
performed RNA-sequencing bioinformatics analysis. M.M., M.K., E.Z. and E.G. contributed to the general interpretation of the data. M.S. conceptualized, designed, and supervised the study,
secured funding, analyzed the data, and co-wrote the manuscript. All authors discussed the results and commented on the manuscript. CORRESPONDING AUTHOR Correspondence to Manuel Serrano.
ETHICS DECLARATIONS COMPETING INTERESTS M.S. is shareholder of Senolytic Therapeutics, Inc., Life Biosciences, Inc., Rejuveron Senescence Therapeutics, AG, and Altos Labs, Inc. M.S. has been
consultant, until the end of 2022, of Rejuveron Senescence Therapeutics, AG, and Altos Labs, Inc. M.K. has ongoing or completed research contracts with Galapagos NV, Rejuveron Senescence
Therapeutics, and mesoestetic®. E.Z. and E.G. are inventors in patents submitted by Albert Einstein College of Medicine that protect compounds, compositions, and methods for controlling
mitofusins for the treatment of diseases and disorders. E.G. has received compensation for consulting and has equity ownership from BaxGen Therapeutics, Comorin Therapeutics, BeanPod
Biosciences, Life Biosciences, and Stelexis Biosciences. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The
remaining authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Muhammet Gulen and the other, anonymous, reviewer(s) for their
contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN
ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you
modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in
this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE López-Polo, V., Maus, M., Zacharioudakis, E. _et
al._ Release of mitochondrial dsRNA into the cytosol is a key driver of the inflammatory phenotype of senescent cells. _Nat Commun_ 15, 7378 (2024).
https://doi.org/10.1038/s41467-024-51363-0 Download citation * Received: 20 October 2023 * Accepted: 06 August 2024 * Published: 27 August 2024 * DOI:
https://doi.org/10.1038/s41467-024-51363-0 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative