- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Triple Negative Breast Cancer (TNBC) is the most aggressive breast cancer subtype suffering from limited targeted treatment options. Following recent reports correlating Fibroblast growth
factor-inducible 14 (Fn14) receptor overexpression in Estrogen Receptor (ER)-negative breast cancers with metastatic events, we show that Fn14 is specifically overexpressed in TNBC patients
and associated with poor survival. We demonstrate that constitutive Fn14 signalling rewires the transcriptomic and epigenomic landscape of TNBC, leading to enhanced tumour growth and
metastasis. We further illustrate that such mechanisms activate TNBC-specific super enhancers (SE) to drive the transcriptional activation of cancer dependency genes via chromatin looping.
In particular, we uncover the SE-driven upregulation of Nicotinamide phosphoribosyltransferase (NAMPT), which promotes NAD+ and ATP metabolic reprogramming critical for filopodia formation
and metastasis. Collectively, our study details the complex mechanistic link between TWEAK/Fn14 signalling and TNBC metastasis, which reveals several vulnerabilities which could be pursued
for the targeted treatment of TNBC patients.
Breast cancer (BC) is the leading malignancy in women (which comprises 11.7% of total cases), accounting for 6.9% of all cancer related deaths globally1,2,3,4. Additionally, the
heterogeneity of this disease makes it challenging for both diagnosis and treatment5,6. There are five distinct molecular subtypes of BC: Luminal A, Luminal B, HER2-enriched,
Basal-like/Triple negative and Normal-like7,8,9. Amongst them, TNBC is the most aggressive and heterogenous subtype that exhibits enhanced proliferative and metastatic capacity, poorer
prognosis and higher disease recurrence compared to the other subtypes. In addition, TNBCs are insensitive to endocrine and HER2-targeted therapy due to the absence of all three hormonal
receptors which limits treatment options to standard chemotherapeutic regimens, such as taxanes, anthracyclines and platinum-based agents, alongside recent combination treatment with
immuno-therapeutics10,11,12. However, despite various treatment options, fewer than 30% of patients achieve pathologic complete response10,13. Consequently, there is an urgent need to
identify effective molecular markers or driver factors specific to TNBC patients for targeted therapies.
To discover new therapeutic targets, it is crucial to elucidate the oncogenic signalling mechanisms and their requisite gene regulatory programmes which sustain TNBC malignancy. The Tumour
necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK)/Fibroblast growth factor-inducible 14 (Fn14) pathway is one such signalling cascade implicated in the pathogenesis of TNBC and
other solid tumours. The TWEAK cytokine, TNFSF12, is a member of the TNF superfamily that is widely expressed in several tissues and cell types including fibroblasts, immune, mesenchymal,
endothelial and tumour cells14,15,16,17,18. Through binding to its cognate receptor, Fn14 (or TNFRSF12A), TWEAK activates multiple cellular responses including modulation of cell death,
proliferation, migration, angiogenesis and production of proinflammatory mediators19,20. While TWEAK and Fn14 expression are typically low in most normal tissues, their aberrant expression
has been detected during tissue injury or chronic inflammation, and in various tumours and metastases such as gliomas, prostate and BCs21,22,23,24,25. Notably, TWEAK and Fn14 have been found
to be overexpressed in ER-negative BCs relative to the ER-positive subtype and activation of this pathway has been further correlated with increased breast tumour invasion and metastasis26.
Aberrant TWEAK/Fn14 signalling has been shown to activate several pro-oncogenic pathways such as NF-κB, JNK, ERK and TRAF signalling27,28,29,30. Many of the transcription factors activated
downstream of these pathways have been reported to mediate tumour cell invasion, metastasis and cancer stem cell phenotypes in multiple cancer types31,32,33,34,35. Additionally, some factors
such as those from the AP-1 or NF-κB family are known to regulate the epigenome in various disease or inflammatory settings36,37,38,39,40. However, the clinical significance and biological
roles of deregulated TWEAK/Fn14 signalling in regulating the epigenetic plasticity and transcriptome of TNBCs remain elusive. In this study, we performed integrative transcriptomic and
epigenomic analyses of TNBC cell lines and patient samples to characterise the gene regulatory roles of TWEAK/Fn14 pathway in remodelling the SE landscape of TNBC tumours. We identify a
distinct TWEAK-induced transcriptional signature in basal-like tumours that is enriched in pro-metastatic and metabolic genes. We further demonstrate that constitutive TWEAK/Fn14 signalling
activates a significant proportion of TNBC-specific SEs targeting several of these genes through chromatin interactions. Importantly, deleting one of the TNBC-specific SEs targeting a TNBC
dependency gene, NAMPT, abrogated the TWEAK/Fn14-driven tumorigenesis and metastasis of TNBC xenografts. Moreover, TWEAK/Fn14-induced NAMPT expression resulted in nicotinamide adenine
dinucleotide (NAD) metabolic rewiring that is critical for the filopodia formation and invasion of TNBC cells. Collectively, our findings reveal a previously unrecognised role for TWEAK/Fn14
pathway in reprogramming the SE landscape of TNBC tumours to drive the expression of metastasis and metabolic genes critical for TNBC progression. Our work thereby highlights the
therapeutic potential of targeting the TWEAK/Fn14 signalling cascade and future characterisation of the oncogenic functions of its downstream gene targets for plausible intervention.
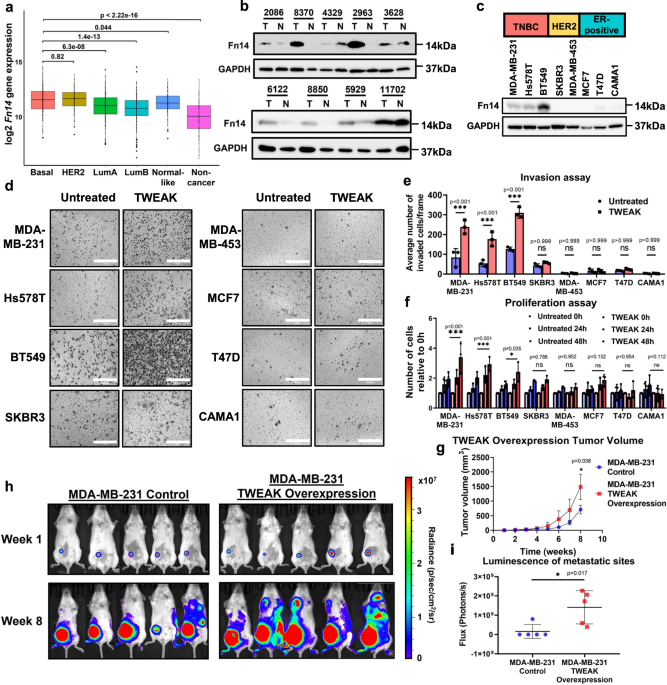
The expression of Fn14 in breast tumours has previously been shown to be positively correlated with HER2 expression as well as the lack of ER status26,41. To evaluate the prevalence of
aberrant TWEAK/Fn14 signalling in patients with TNBC, we interrogated the TCGA BRCA RNA-seq dataset which revealed the overexpression of Fn14 in basal-like tumours, the predominant molecular
subtype of TNBC, relative to ER-positive tumours (Fig. 1a). Further analysis of primary TNBC patient samples demonstrated higher Fn14 protein expression levels in TNBC tumours relative to
their adjacent normal tissues (6 out of 9 malignant samples) (Fig. 1b). The elevated expression of Fn14 was also consistent in TNBC cell lines compared to ER-positive cell lines.
Surprisingly, HER2 cell lines exhibited low Fn14 protein expression (Fig. 1c), suggesting a possible unreported mechanism for the post-transcriptional regulation of Fn14 in the HER2 subtype.
Further single cell RNA-seq (scRNA-seq) analysis of TWEAK and Fn14 expression in a TNBC primary tumour and its matched lymph node metastases revealed that in both primary and metastatic
settings, Fn14 was most highly expressed in epithelial cells. In contrast, TWEAK was most highly expressed in endothelial cells in the primary tumour and in monocytes and macrophages in the
lymph node metastases42 (Supplementary Fig. 1). Together, this suggests that in TNBCs, TWEAK/Fn14 activation occurs through a paracrine mechanism.
a Relative Fn14 gene expression levels in Basal-like (n = 229), HER2 (n = 158), Luminal A (n = 300), Luminal B (n = 304), Normal-like (n = 106) and non-cancer (n = 113) patient samples from
the TCGA BRCA RNA-seq dataset. Box plot depicts the first quartile, median and third quartile of values. Fn14 protein expression analysed by western blotting in (b) TNBC patient tumours (T)
and their matched normal samples (N), and in (c) TNBC, HER2 and ER-positive breast cancer cell lines. d Transwell invasion assay was performed in MDA-MB-231, Hs578T, BT549, SKBR3,
MDA-MB-453, MCF7, T47D and CAMA1 cells with and without TWEAK treatment. Representative images from n = 3 biological replicates are shown. Scale bars: 400 µm. e Plot depicts the average
number of invaded cells/frame (mean ± s.d) from n = 3 biological replicates, across four fields per replicate. f Proliferation assay plot depicts the average number of cells counted relative
to 0 h (mean ± s.d) in MDA-MB-231, Hs578T, BT549, SKBR3, MDA-MB-453, MCF7, T47D and CAMA1 cells, treated with and without TWEAK from n = 3 biological replicates. g Tumour growth plot
depicts the weekly average tumour volume (mean ± s.d) from mice injected with MDA-MB-231 cells overexpressing luciferase and overexpression control or TWEAK. Performed in n = 5 biological
replicates. h IVIS tracking of mice injected with MDA-MB-231 cells overexpressing luciferase and overexpression control or TWEAK. Representative bioluminescent images of the animals were
taken at 1 and 8 weeks after orthotopic xenograft. i Plot depicts total flux (mean ± s.d) at the metastatic sites of each animal after 8 weeks in n = 5 biological replicates. Western blot
samples were derived from the same experiment and on the same gels for Fn14 and GAPDH. Experiments involving cell lines were performed 3 times independently, each time on different days.
Two-sided Wilcoxon signed-rank test was used for differential gene expression. Two-sided two-way ANOVA was used for in vitro proliferation and invasion assay. Two-sided t test was used for
in vivo assays. *P








