- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Anti-CD38 monoclonal antibodies like Daratumumab (Dara) are effective in multiple myeloma (MM); however, drug resistance ultimately occurs and the mechanisms behind this are poorly
understood. Here, we identify, via two in vitro genome-wide CRISPR screens probing Daratumumab resistance, KDM6A as an important regulator of sensitivity to Daratumumab-mediated
antibody-dependent cellular cytotoxicity (ADCC). Loss of _KDM6A_ leads to increased levels of H3K27me3 on the promoter of _CD38_, resulting in a marked downregulation in CD38 expression,
which may cause resistance to Daratumumab-mediated ADCC. Re-introducing CD38 does not reverse Daratumumab-mediated ADCC fully, which suggests that additional KDM6A targets, including CD48
which is also downregulated upon _KDM6A_ loss, contribute to Daratumumab-mediated ADCC. Inhibition of H3K27me3 with an EZH2 inhibitor resulted in CD38 and CD48 upregulation and restored
sensitivity to Daratumumab. These findings suggest _KDM6A_ loss as a mechanism of Daratumumab resistance and lay down the proof of principle for the therapeutic application of EZH2
inhibitors, one of which is already FDA-approved, in improving MM responsiveness to Daratumumab. SIMILAR CONTENT BEING VIEWED BY OTHERS LOSS-OF-FUNCTION MUTATIONS IN THE HISTONE
METHYLTRANSFERASE EZH2 PROMOTE CHEMOTHERAPY RESISTANCE IN AML Article Open access 12 March 2021 LOW NCOR2 LEVELS IN MULTIPLE MYELOMA PATIENTS DRIVE MULTIDRUG RESISTANCE VIA MYC UPREGULATION
Article Open access 04 December 2021 DYRK1A INHIBITION RESULTS IN MYC AND ERK ACTIVATION RENDERING _KMT2A-_R ACUTE LYMPHOBLASTIC LEUKEMIA CELLS SENSITIVE TO BCL2 INHIBITION Article Open
access 27 March 2025 INTRODUCTION Multiple myeloma (MM) is an incurable hematologic neoplasm characterized by the infiltration of aberrant plasma cells into the bone marrow1,2. In the past
decade, patient outcomes have improved, in part, due to immunotherapy3, including monoclonal antibodies (mAb)4,5,6, bi-specific T-cell engagers (BiTE)7,8,9, antibody-drug conjugates
(ADC)10,11 and chimeric antigen receptor T cells (CAR-T)12,13. Specifically, mAbs, which selectively target surface antigens that are highly expressed on MM cells, have achieved remarkable
responses, with three mAbs now approved by the U.S. Food and Drug Administration (FDA)14,15,16. Daratumumab (Dara) is the first-in-class humanized mAb targeting CD38. It has a high frequency
and depth of response in newly diagnosed and relapsed/refractory MM when used alone or in combination with other agents17,18,19. Dara induces MM cell death through different mechanisms,
including antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), complement-dependent cytotoxicity (CDC), direct cytotoxicity and
immunomodulatory effects. Among these effects, ADCC is the most important mechanism of MM cell killing. The sensitivity of Dara-mediated ADCC is strongly associated with the activity of
effector cells20 like natural killer (NK) cells and with the expression of CD38 on MM cells21,22. However, the development of Dara resistance and subsequent relapse is common. CD38 is a
transmembrane glycoprotein that is highly expressed in MM cells and lowly expressed in normal hematological cells including NK cells, B cells, and T cells23, and its expression on MM cells
is correlated with response to Dara treatment21. Clinically, CD38 expression on patient MM cells significantly decreases during Dara treatment21, and low levels of CD38 expression are also
observed during MM progression21. CD38 expression can be modulated by immunomodulatory drugs (IMiD)24, _all-trans_ retinoic acid (ATRA)25, histone deacetylase inhibitors (HDAC)26, IL-6 from
bone marrow stromal cells, and the JAK2 inhibitor ruxolitinib22. However, using these agents in the clinic has not achieved the desired effect, suggesting that the molecular mechanisms
underlying CD38 regulation are still not completely understood. In addition, NK cells play a crucial role in mAbs-mediated ADCC, so enhancing the activation of NK cells is an alternative
strategy27. Here, we perform two genome-wide CRISPR screens, one for genes that regulate Dara-mediated cytotoxicity and one for genes that regulate CD38 expression. We find that _KDM6A_ was
a hit in both screens and thus investigate its mechanisms, finding that the loss of _KDM6A_ increases the level of H3K27me3, resulting in downregulation of both CD38 and CD48 expression,
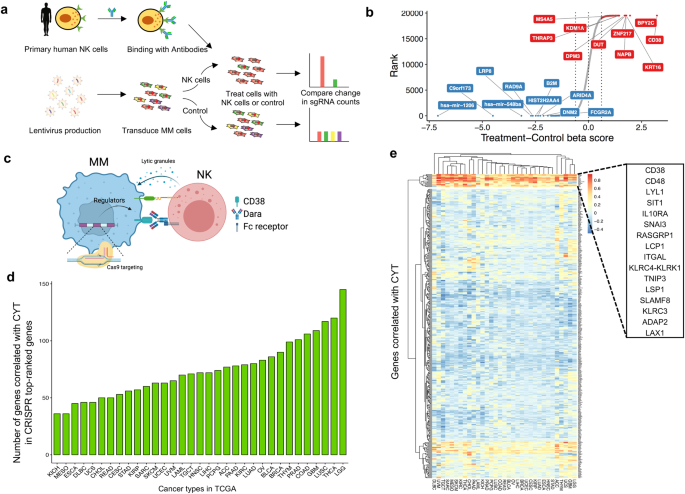
which in turn, leads to reduced ADCC. RESULTS GENOME-WIDE CRISPR SCREEN FOR NK-CELL-MEDIATED CYTOTOXICITY To identify the tumor-intrinsic genes involved in resistance to NK-mediated
cytotoxicity in the presence of Dara in MM cells, we transduced H929 cells with a pooled genome-scale CRISPR knockout (KO) library, selected with puromycin for 7 days, and exposed them to
human primary NK cells at an effector-to-target (E:T) ratio of 1:1 with Dara. This resulted in ~75% tumor cell lysis (Fig. 1a), and the abundance of each sgRNA in the surviving tumor cells
was assessed by next-generation sequencing of genomic DNA (Fig. 1b and Supplementary Data 1), to identify affected genes involved in Dara-mediated ADCC and/or direct NK-cell-mediated
cytotoxicity (Fig. 1c). As expected, the sgRNAs targeting _CD38_, the target of Dara, were listed as the top candidate, indicating the screen performed properly. To identify whether the
candidate genes we identified attenuated cytolytic activity in other cancers, we analyzed the gene expression profiles of 11,409 human tumors from 36 different cancer types in the TCGA
database (Fig. 1d and Supplementary Data 2) and found a set of 16 genes that are correlated with the cytolytic activity in most of the 36 cancer types (Fig. 1e). Loss of these 16 genes
within tumors could play a major role in immune evasion from NK cell-mediated cytotoxicity. KDM6A LOSS LEADS TO CD38 DOWNREGULATION We performed another genome-wide CRISPR KO screen to
identify the genes regulating CD38 expression in H929 cells (Fig. 2a). After puromycin selection, we sorted cells from the bottom 5% of CD38 expression, meaning the enriched genes positively
regulated CD38 (Fig. 2b and Supplementary Data 3). The genes that overlapped in these two screens are implicated in regulating both Dara-mediated ADCC and CD38 expression (Fig. 2c). From
the overlapping genes, we were particularly interested in KDM6A, which belongs to the KDM6 family of histone 3 lysine 27 demethylases. It de-represses genes by removing methylation from
H3K27 and counteracts the activity of EZH2 of the PRC2 complex, which methylates H3K27. The loss or inactivation of KDM6A occurs in several hematological malignancies including MM28, and
patients with _KDM6A_ mutations or deletion have reduced overall survival29. To validate whether KDM6A regulates CD38 expression, we generated _KDM6A_ KO MM cell lines using different sgRNA
sequences and found that loss of _KDM6A_ was indeed associated with CD38 downregulation on the protein and mRNA levels (Fig. 2d,e). Moreover, we confirmed that the cell-surface expression of
CD38, which is important for Dara binding, was significantly decreased (Supplementary Fig. 1a). For the pool CRISPR KO cell lines, there was still KDM6A expression in the KO cells, so we
generated complete KO single clones and found that their CD38 expression was much lower than that in pool KO cells (Fig. 2f). We next sought to confirm whether the downregulation of CD38 was
a specific effect of _KDM6A_ KO. We added back _KDM6A_ in _KDM6A_ KO cells and found that the CD38 level was restored by KDM6A overexpression (Fig. 2g, h and Supplementary Fig. 1b–d). We
did not detect a significant difference in CD38 expression between different media conditions, indicating that CD38 was specifically downregulated by _KDM6A_ KO in a cell-autonomous manner
(Supplementary Fig. 1e, f). These data indicate that loss of _KDM6A_ decreased CD38 expression at the transcriptional and cell surface expression levels in myeloma cells. KDM6A REGULATES
CD38 EXPRESSION VIA H3K27ME3 AND CHROMATIN ACCESSIBILITY Given KDM6A’s function as an H3K27 demethylase, we hypothesized that KDM6A positively regulated the expression of CD38 by decreasing
the level of H3K27me3 on its promoter. We, therefore, performed H3K27me3 ChIP-seq in control and two isogenic _KDM6A_ KO cell lines. The _KDM6A_-KO cells had more H3K27me3 loci than control
cells (Fig. 3a) and significantly more at the promoter region of _CD38_ (Fig. 3b), evidenced by ChIP-qPCR with three different primers (Fig. 3c). Conversely, the re-introduction of _KDM6A_
into _KDM6A_-KO cells decreased the H3K27me3 level at the promoter region of _CD38_ (Supplementary Fig. 1g). Interestingly, the H3K27Ac level, which is recognized as a marker of active
enhancers30, was increased at the same time (Supplementary Fig. 1h). KDM6A facilitates gene expression by removing a suppressive chromatin modification and increasing chromatin
accessibility. We, therefore, performed ATAC-seq to check chromatin accessibility and found a substantially large number of differentially accessible sites between control and _KDM6A_-KO
cells (Supplementary Data 4). There were 898 and 2395 less-accessible sites in two different _KDM6A_-KO cell lines relative to the control, respectively (Fig. 3d). In particular, the peak on
the _CD38_ gene had decreased intensity, leading to the inhibition of _CD38_ transcription (Fig. 3e). To explore the mechanism of acquired resistance in MM cells, we generated a
Dara-resistant MM cell line by continuously exposing MM cells to NK and Dara for several rounds (Supplementary Fig. 2a). In these resistant cells, CD38 expression was significantly decreased
(Fig. 3f and Supplementary Fig. 2b), leading to attenuated sensitivity to ADCC (Supplementary Fig. 2c). Importantly, the CD38 promoter had increased levels of H3K27me3 and decreased levels
of H3K27Ac (Fig. 3g and Supplementary Fig. 2d). KDM6A KO RENDERS MM CELLS RESISTANT TO DARA-MEDIATED ADCC Importantly, we found that _KDM6A_ and _CD38_ mRNA expression was decreased in
Dara-resistant patient samples relative to newly diagnosed patients (Fig. 3h, i). To evaluate whether the decreased CD38 expression caused by _KDM6A_ KO affects Dara-mediated ADCC, we
co-incubated MM cells with Dara and human primary NK cells or PBMC cells. The parental MM cells were profoundly lysed, whereas _KDM6A_-KO cells were not (Fig. 4a and Supplementary Fig.
3a,b). This also occurred with another anti-CD38 mAb, Isatuximab, indicating that ADCC resistance was specific to CD38 expression (Fig. 4b). Importantly, when we re-introduced CD38 into
_KDM6A_-KO cells, their sensitivity to Dara-mediated ADCC was partially restored (Supplementary Fig. 3c,d). Subsequently, we tested the sensitivity of Dara-mediated ADCC in a _KDM6A_-null
xenograft model. After tumor growth, we gave the mice an i.v. injection of human primary NK cells and i.p. injection of Dara. The _KDM6A_-KO engrafted tumors were significantly resistant to
this treatment (Fig. 4c and Supplementary Fig. 3e), associated with shorter survival times (Fig. 4d). NK cells kill cancer cells through the secretion of the pore-forming protein perforin
and granzymes, we next examined whether the resistance of _KDM6A_-KO cells to ADCC was due to attenuated NK activity. We co-incubated MM and primary NK cells with Dara, then collected
culture supernatant for an ELISA detecting perforin and granzyme B (Fig. 4e), and found that _KDM6A_ KO suppressed NK activity by lowering the secretion of perforin and granzyme B (Fig. 4f,
g). We also found that human NK cells produced lower amounts of IFN-γ when co-cultured with _KDM6A_-KO MM cells than with control MM cells in the presence of Dara (Fig. 4h). Taken together,
these data demonstrate that the loss of _KDM6A_ mediates resistance of MM cells to Dara-mediated ADCC through CD38 downregulation and suggests that the KDM6A expression of MM cells regulates
the activity of NK cells. KDM6A REGULATES CD48 EXPRESSION IN MM CELLS Overexpressing CD38 in _KDM6A_-KO MM cells only partially rescued Dara-mediated ADCC (Supplementary Fig. 3c, d), even
when we utilized an anti-SLAMF7 mAb, elotuzumab, _KDM6A_ KO MM cells continued to exhibit resistance to ADCC (Supplementary Fig. 4a, b). These findings imply the presence of an additional
mechanism mediating ADCC resistance by KDM6A beyond the regulation of CD38 expression. Moreover, we found that _KDM6A_ KO MM cells efficiently evade direct primary NK cell and NK cell
lines-mediated cytotoxicity (Fig. 5a and Supplementary Fig. 4c–e). The IFN-γ produced by primary NK cells was inhibited when co-cultured with _KDM6A_-KO cells (Fig. 5b). These data imply
that KDM6A may function as a regulator of NK activity in MM cells. To investigate how KDM6A regulates NK activity, we performed RNA-seq analysis in _KDM6A_-KO cells (Fig. 5c and
Supplementary Data 5). As expected, gene enrichment analysis revealed that PRC2-regulated genes were highly repressed in _KDM6A_-KO cells (Supplementary Fig. 4f). Of note, CD48 was highly
repressed and was also a top hit in the previous CRISPR screens (Fig. 5d). CD48 is a ligand expressed on cancer cells that binds with its receptor 2B4 on NK cells for their activation. In
_KDM6A_-KO cells, CD48 had significantly reduced mRNA and surface expression (Fig. 5e, f) and a higher level of H3K27me3 on its gene (Fig. 5g, h), which corresponded to profoundly reduced
chromatin accessibility in the _CD48_ promoter region (Fig. 5i). We also validated other well-studied NK activating and inhibitory ligands and found there were no significant alterations in
the expression of other evaluated NK cell ligands (Supplementary Fig. 4g, h). Finally, we found that CD48 expression levels in Dara-resistant patient MM samples are lower than in the newly
diagnosed MM (Fig. 5j). In addition, using the TCGA cancer RNA-seq dataset and TIMER31, we found that the expression of CD48 and KDM6A was positively correlated with intertumoral NK cell
abundance in many human cancer types, suggesting that KDM6A and CD48 affect NK-mediated tumor immunity in a variety of human cancers (Supplementary Fig. 4i,j). CD48 MEDIATES ADCC SENSITIVITY
BY REGULATING NK ACTIVITY To establish whether CD48 loss downregulates NK activity leading to ADCC resistance, we generated _CD48_-KO cells using CRISPR, and confirmed the depletion of cell
surface expression of CD48 in these cells (Fig. 6a). Upon exposure to primary NK cells with or without Dara, the lysis of _CD48_-KO MM cells was profoundly inhibited (Fig. 6b,c and
Supplementary Fig. 5a). The effects were similar to those observed in _KDM6A_-KO cells (Fig. 5a and Supplementary Fig. 5b). When we overexpressed CD48 in _KDM6A_-KO cells (Fig. 6d), we found
these cells significantly increased the IFN-γ expression in primary NK cell fractions (Supplementary Fig. 5c, d), as well as secretion of perforin and granzyme B from NK cells
(Supplementary Fig. 5e, f). Importantly, CD48 overexpression significantly restored the sensitivity of _KDM6A_-KO MM cells to Dara-mediated ADCC (Fig. 6e and Supplementary Fig. 5g) and
recovered IFN-γ production, secretion of perforin and granzyme B by NK cells (Fig. 6f-h). Taken together, these data suggest that KDM6A mediates Dara-mediated ADCC sensitivity through both
CD38 upregulation and CD48 upregulation, with associated increased NK activity. EZH2 INHIBITORS INCREASE CD38 AND CD48 EXPRESSION AND ENHANCE DARA-MEDIATED ADCC KDM6A acts to oppose the
EZH2/PRC2 complex by demethylating H3K27me3; conversely, inactivating KDM6A leads to increased H3K27me3 and inhibition of gene transcription (Fig. 7a). Several pre-clinical studies have
shown that EZH2 can be a therapeutic target in MM. Therefore, we hypothesized that inhibiting EZH2 may restore the balance of gene expression by downregulating H3K27me3, resulting in the
upregulation of CD38 and CD48 expression in _KDM6A_-KO cells. Tazemetostat (Taze), an FDA-approved EZH2 inhibitor (EZHi), has already provided meaningful and sustained responses for relapsed
or refractory follicular lymphoma patients32. We found that Taze increased the expression of CD38 and CD48 at the protein (Fig. 7b), mRNA (Fig. 7c, d), and surface expression levels (Fig.
7e), especially in _KDM6A_-KO cells. The expression of CD48 did not increase as much as CD38, suggesting other regulatory mechanisms. For example, KDM6A also interacts with p300, H3K4
methyltransferases, and the SWI/SNF complex, besides functioning as a demethylase. ChIP-Q-PCR data showed that the H3K27me3 level on the _CD38_ and _CD48_ genes was reduced by Taze in MM
cell lines (Fig. 7f,g). Importantly, we validated the above findings with another EZH2 inhibitor, GSK343, which inhibits EZH2 catalytic activity (Supplementary Fig. 6a–g)33. Interestingly,
we found that Taze did not change the CD38 expression level in NK cells (Supplementary Fig. 6h). Finally, we sought to determine whether Taze can re-sensitize the _KDM6A_-KO cells to
Dara-mediated ADCC. We treated MM cells with Taze for 4 days, then co-incubated them with Dara or Isa and human primary NK cells or PBMC. The sensitivity of the _KDM6A_-KO cells to ADCC was
profoundly restored by Taze treatment (Fig. 7h, i and Supplementary Fig. 7a, b). In addition, NK cell activity was also restored, evidenced by increased granzyme B secretion (Fig. 7j). We
also confirmed that Taze increased Dara-mediated killing of MM patient cells (Fig. 7k) and increased CD38 and CD48 expression (Supplementary Fig. 7c, d). Even in a CD38 low-expression cell
line, Taze increased CD38 expression and enhanced ADCC activity (Supplementary Fig. 7e–g). These data suggest that adding an EZH2 inhibitor to the therapeutic regimen would overcome Dara
resistance by upregulating CD38 and NK cell activity. DISCUSSION KDM6A, also known as UTX, is a histone demethylase that removes the trimethylation of H3K27 (H3K27me3), which is catalyzed by
the EZH2-containing polycomb repressive complex 2 (PRC2)34. Loss or inactivation of KDM6A occurs in multiple human cancers, including MM28,29. In many cancers, KDM6A functions as a tumor
suppressor. Nevertheless, overexpression of KDM6A in breast cancer increases the proliferation of cancer cells, suggesting that the function of KDM6A might be tissue-specific35 or that the
genes targeted by KDM6A are different among cell types36. In MM, KDM6A loss leads to an enhanced malignant phenotype and sensitizes the cell to EZH2 inhibition37. However, little is known
about the role of KDM6A in MM immunotherapy. Here, we describe a new mechanism of KDM6A, whereby its loss induces Dara resistance by downregulating CD38 and CD48 on MM cells, thereby
decreasing NK cell activity. Previous studies have shown that different variants of FcγRs expressed on NK cells are associated with anti-tumor activity of Dara20. In addition, Dara can also
mediate NK fratricide by binding to CD38 on the NK cell surface38. Deletion of CD38 in human primary NK cells eliminates Dara-mediated fratricide and enhances the anti-tumor activity of NK
cells39. In recent years, many CRISPR screens have been done to explore the mechanism of tumor cell resistance to NK cell treatment. Different mechanisms of resistance were found in
different approaches and in different cancer types40,41,42. In our study, we focused on the screens for mAb-mediated ADCC, and the mechanisms underlying NK cell resistance identified in our
study are distinct from those findings. The diversity of identified mediators emphasizes the complexity of tumor cell resistance mechanisms against immune cells. Further research and
comparative analyses across different mediators are crucial for a comprehensive understanding of the regulatory networks governing immune evasion in tumor cells. This could potentially
uncover new therapeutic targets and strategies for overcoming resistance in cancer immunotherapy. In this study, we focused on the intra-tumor factors that affect NK cell activity. We found
that KDM6A epigenetically upregulates the expression of CD48 in MM cells, whereas loss of CD48 has a known role in the evasion of NK cell-mediated surveillance in hematological
neoplasms43,44. In recent years, various drugs have been developed to target epigenetic regulators in the treatment of cancers, such as Azacitidine targeting DNMT1 and Panobinostat targeting
pan-histone deacetylase45. Tazemetostat is a potent selective EZH2 inhibitor that was approved by the FDA in 2020 for the treatment of epithelioid sarcoma46 and follicular lymphoma. Several
preclinical studies have found that EZH2 inhibitors have potent anti-MM activity as a monotherapy or in combination with other conventional drugs47. In our current study, we found that the
EZH2 inhibitor can overcome the resistance of MM cells to Dara-induced ADCC triggered by the loss of KDM6A through upregulating CD38 and CD48 expression. At the same time, the EZH2 inhibitor
does not increase the CD38 expression level in NK cells, suggesting it will not enhance Dara-mediated NK fratricide. In summary, our data reveal a molecular mechanism whereby KDM6A mediates
ADCC not only through regulating CD38 but also by modulating NK activity through CD48 regulation, demonstrating the therapeutic potential of EZH2 inhibitor in MM treatment to overcome Dara
resistance and providing the preclinical rationale for a Dara-based combination therapeutic strategy to improve patient outcome in MM (Fig. 8). METHODS The research methods applied in this
study followed the guidelines of the World Medical Association’s Declaration of Helsinki and subsequent revisions, and the study was reviewed and approved by the ethics committee of
Dana-Farber Cancer Institute. CELL LINES H929, MM1.S, U266, and human embryonic kidney (HEK) 293 T cells were purchased from the American Type Culture Collection (ATCC). KMS-11 cells were
purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). All cell lines were verified by short tandem repeat (STR) DNA fingerprinting analysis (Molecular Diagnostic
Laboratory, DFCI) and tested negative for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza). All cells were grown at 37 °C in 5% CO2. MM.1 S, H929, and U266 cells were
maintained in RPMI 1640 medium; HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM). All media were supplemented with 10% fetal bovine serum (FBS), 1X
antibiotic-antimycotic, 1X GlutaMAX, and 1X Hepes. PRIMARY MM CELLS Primary MM cells were obtained from bone marrow samples of patients after informed consent and approved by the
Institutional Review Board of the Dana-Farber Cancer Institute. Mononuclear cells were separated by using Ficoll-Paque PLUS (Cytiva). Primary MM cells were purified by CD138 positive
selection with anti-human CD138 Microbeads (Miltenyi). REAGENTS AND ANTIBODIES Daratumumab and Isatuximab were purchased from the Department of Pharmacy at Dana-Farber Cancer Institute;
Tazemetostat and Elotuzumab were purchased from Selleckchem; GSK343 was purchased from Cell Signaling Technology; Tazemetostat and GSK343 were dissolved in dimethyl sulfoxide (DMSO) and
stored at –20 °C for up to 6 months. For all cell-based experiments, drugs were diluted at least by 1:1000 to ensure that the final DMSO concentration was lower than 0.1%. Human recombinant
IL-2 was purchased from R&D Systems. Antibodies were obtained as follows: KDM6A (no. 33510, Cell Signaling Technology, 1:1000), CD38 (no. 51000, Cell Signaling Technology, 1:1000), CD48
(no. 29499, Cell Signaling Technology, 1:1000), GAPDH (no. 5174, Cell Signaling Technology, 1:1000), H3K27me3 (no. 9733, Cell Signaling Technology, 1:1000), anti-rabbit immunoglobulin G
(IgG), horseradish peroxidase (HRP)-linked Ab (no. 7074, Cell Signaling Technology, 1:2000), FITC anti-human CD38 (no. 356610, Biolegend, 1:20), APC anti-human CD38 (no. 356606, Biolegend,
1:20), FITC mouse IgG1 (no. 400110, Biolegend, 1:20), APC mouse IgG1 (no. 981806, Biolegend, 1:20), APC anti-human CD138 (no. 356506, Biolegend, 1:20), FITC anti-human IFN-γ (no. 502506,
Biolegend, 1:20), APC anti-human CD48 (no. 336714, Biolegend, 1:20), Alexa Fluor 488 anti-human MICA/B (no. 320912, Biolegend, 1:20), APC anti-human CD56 (no. 985906, Biolegend, 1:20), APC
anti-human HLA-A,B,C (no. 311409, Biolegend, 1:20), APC anti-human CD253 (no. 308209, Biolegend, 1:20), FITC anti-human CD319 (no. 331817, Biolegend, 1:20), FITC anti-human CD155 (no.
337627, Biolegend, 1:20), PE anti-human ULBP-2/5/6 (no. FAB1298P, R&D systems, 1:20), BV421 anti-human CD178 (no. 306411, Biolegend, 1:20). TWO CELL TYPE GENOME-WIDE CRISPR-CAS9 SCREEN
The human GeCKOv2 sgRNA library was purchased from Addgene and amplified according to Dr. Zhang’s lab protocol48. To produce lentivirus, HEK293T cells were plated in T150 flasks one day
before transfection at 70% confluency. Two hours before transfection, DMEM media was replaced with 12 ml serum-free OptiMEM media. For each flask, 20 μg of GeCKOv2 A library plasmid, 15 μg
psPAX2 (Addgene #12260), 10 μg pMD2.G (Addgene #12259), and 200 μl PLUS reagent diluted in 4 ml Opti-MEM were combined with 150 μL Lipofectamine 2000 diluted in 4 ml Opti-MEM. The
transfection mixture was left for 20 min and then added dropwise to the cells. 6 h after transfection, the media was replaced by 20 ml fresh complete DMEM media for each flask.
Virus-containing media was collected 72 h post-transfection followed by centrifugation at 800 x g for 10 min to pellet cell debris. Filtration was then performed with a 0.45 μm low protein
binding membrane (Millipore #SE1M003M00). Viral supernatants were concentrated by centrifugation at 4,000 r.c.f. and 4 °C for 35 min in Amicon Ultra-15 filters (Millipore Ultracel-100K).
Concentrated viral supernatants were stored in aliquots at -80 °C. Before cell infection, the virus was first tested to achieve an MOI of 0.3 in H929 cells. A total of 2.1 × 108 H929 cells
(70 wells per 12-well plate, 10 wells for transfection efficiency control) were transduced with the concentrated virus, attaining transduction efficiency of 25% (700 cells per lenti-CRISPRv2
construct). Puromycin was added to cells 24 h after transduction and maintained for 7 days. Subsequently, 4 × 107 cells were collected for baseline genomic DNA analysis. For the screen,
cells were split into two groups of 6 × 107 transduced cells. One group was co-cultured with 6 × 107 primary NK cells (E:T ratio of 1:1) and Daratumumab (1 μg/ml). The other group was
cultured under the same density and conditions but without NK cells and Daratumumab. Cells were co-cultured for 12 h, after which cells were washed with fresh media. The recovery phase was
maintained for another 72 h to get 75% lysis of tumor cells. To evaluate sgRNA enrichment, the surviving cells after co-culture or not were collected. Genomic DNA was extracted from
collected tumor cells (along with the cells collected at the early time point just after puromycin selection) using the Quick-gDNA MidiPrep kit (Zymo Research, cat. no. D3100). To generate
the NGS library, PCR was performed on gDNA using NEBNext® Ultra™ II Q5® Master Mix (New England Biolabs). The sequences of primers, including full barcodes, used for PCR are from Dr. Zhang’s
published protocol49. For each sample, the PCR products were pooled and then purified using the Zymo-Spin V with Reservoir. Purified libraries were quantified by Qubit dsDNA Assay Kit and
sent to Novogene for quenching with 20% PhiX on Illumina Platform PE150. To analyze the enriched sgRNA in our screen, the normalized gRNA count table was loaded into MaGeCK (Model-based
Analysis of Genome-wide CRISPR-Cas9 Knockout)50 by comparing the co-culture and control conditions described above. Top genes were determined based on mean log2 fold change (LFC) for all
gRNAs and false discovery rate (FDR). THE CANCER GENOME ATLAS (TCGA) CORRELATION ANALYSIS 33 RNA-seq datasets from The Cancer Genome Atlas (TCGA) were downloaded through the R package
TCGAbiolinks51. The gene expression in each type of cancer was normalized by DESeq252. The geometric mean of gene GZMA and PRF1 in each dataset was calculated as the expression of cytolytic
activity signature (CYT) first and then identified genes correlated with CYT (Pearson’s r > 0 and _p_ < 0.05). We illustrated the intersection between CYT-correlated genes and 433
CRISPR screen genes in each dataset. The Pearson’s correlation coefficients of these CRISPR screen genes in each dataset were used for clustering and were displayed in the heatmap. CD38
SORTING GENOME-WIDE CRISPR SCREEN The lentivirus library was the same as described above. After cell transduction and puromycin selection, 5% of cells with the lowest expression of CD38 were
sorted for NGS. Data analysis is the same as described above. GENERATION OF STABLE CELL LINES For the generation of CRISPR KO cell lines, oligonucleotides (Supplementary Data 6) targeting
different genes were annealed and subcloned into LentiCRISPRv2 vectors48. Constructs were packaged into lentivirus in HEK293T cells. Target cells were seeded in 12-well plates and spinfected
with the virus for 1.5 hours at 800 x _g_ at 35 °C, supplemented with Polybrene (8 μg/ml). The media was then aspirated, and fresh complete media was added to exclude Polybrene. After 1
day, cells were selected for stable KO using puromycin (0.5 μg/ml). After 7 days, cells were collected for immunoblotting or other experiments. To generate complete KDM6A KO single clone
cells, we delivered indicated sgRNA and cas9-GFP protein ribonucleoprotein complex into MM cells using the Neon Transfection system. One day after transfection, cells were washed, and
single-cell sorted viable and GFP+ cells dispensed into 96-well cell culture plates (1 cell per well). After cells grew up, we transferred the cells to large plates and flasks. Cells were
collected and KO was confirmed by immunoblotting. RE-INTRODUCE INDICATED GENES INTO MM CELLS The pLenti expression vectors were purchased from GeneCopoeia. Viral particles were packaged and
concentrated as described above. MM cells were transduced with the virus and selected for stable overexpression cells using related antibiotics. After 7 days, cells were collected and
identified by immunoblotting. IMMUNOBLOT ANALYSIS Cells were harvested, washed with PBS, and total protein was extracted with RIPA lysis buffer supplemented with protease and phosphatase
inhibitors (no. 78440, Thermo Fisher Scientific). The suspension was incubated for 15 min on ice and vortexed for 5 min. Then, samples were centrifuged at 16,000 x g at 4 °C for 10 min, and
the supernatant was transferred to a new tube. Protein concentration was determined using the BCA protein assay kit (no. 23227, Thermo Fisher Scientific). Samples were mixed with 4X LDS
sample buffer (no. NP0007, Thermo Fisher Scientific) and boiled at 95 °C for 8 min. Equal amounts of protein were run on NuPAGE Bis-Tris gels (Thermo Fisher Scientific) at a constant voltage
and transferred to nitrocellulose membrane by iblot2 Gel Transfer Device (Thermo Fisher Scientific). Then membranes were blocked in 5% nonfat dry milk for 1 hour at room temperature, and
incubated with primary Abs in 5% bovine serum albumin at 4 °C overnight. Blots were then washed three times with 1X Tris Buffered Saline with Tween (TBS-T) before incubation with secondary
Abs for 1 hour. SuperSignal chemiluminescent substrate (Thermo Fisher Scientific) was used for signal detection. For reblotting the membranes, blots were stripped in stripping buffer (no.
46428, Thermo Fisher Scientific) according to the manufacturer’s instruction and re-blocked. QRT-PCR Total RNA was extracted with an RNeasy Mini kit (Qiagen). cDNA was generated by reverse
transcription using the SuperScript VILO cDNA synthesis kit (no. 11754050, Thermo Fisher Scientific). Quantitative real-time PCR was carried out using Taqman Universal PCR master Mix and
related Taqman Assay primers in a QuantStudio6 Flex real-time PCR system. The relative level of each transcript was normalized to control GAPDH expression. The primers for each gene are
listed in Table S6. FLOW CYTOMETRY Cells were collected and washed with PBS and stained with Fixable Viability Dye eFluor 780 at 4 °C for 30 min to exclude dead cells. After the incubation,
cells were washed with PBS and stained with conjugated primary antibodies or isotype control (IgG). Cells were then washed with 2% FBS containing PBS, and resuspended in Flow Staining
Buffer. Cells were acquired in a BD LSRFortessa Flow Cytometer, and data were analyzed by FlowJo software. IN VITRO ADCC ASSAY MM cells were counted and stained with the dye Calcein Am at 37
°C for 30 min and protected from light. Then we added five times complete culture media to remove any free dye remaining in the solution. Cells were pelleted by centrifugation, resuspended
in pre-warmed media, and plated in 96-well U-bottom plates (10,000 target cells per well) with 1 μg/ml Daratumumab. Following this, MM cells were thoroughly washed to remove excess
antibodies before co-culturing them with effector NK cells. Natural killer cells or PBMC from healthy donors were isolated or thawed one day before the ADCC assay and added in at the
indicated E:T ratio. To determine maximum and spontaneous release, target cells were mixed with an equal volume of 1% Triton-X100 in media or an equal volume of media, respectively. After
incubation, plates were centrifuged at 100 x _g_ for 3 min. Supernatants were collected to a new black 96-well plate, and the intensity of fluorescence from the supernatant was detected
using 485 nm excitation and 520 nm emission. The specific killing was calculated using the following formula: 100 x (Experimental release – Spontaneous release)/(Maximum release –
Spontaneous release). GENERATION OF DARA-RESISTANT CELL LINE KMS11 cells were co-cultured with primary NK cells and Dara (1 μg/ml) at E:T ratio of 6:1 in 96-well U-bottom plates. Cells were
incubated at 37 °C for 24 h, followed by washing and resuspending with complete media. Cells were cultured for 1 week, and the co-culture was repeated weekly 10 times (a total of 10 weeks).
Resistance of cells to Dara was determined by ADCC assay. NK CELL CYTOTOXICITY ASSAY The co-culture of MM cells and primary NK cells is as described above of ADCC, but without Dara. Primary
NK cells were pre-cultured with IL-2 one day before. Specific killing was calculated for ADCC. INTRACELLULAR INTERFERON-Γ PRODUCTION BY NK CELLS NK cells were co-cultured for 6 h at a ratio
of 1:1 with MM cells in the presence or absence of Dara. After incubation, cells were washed with PBS and stained with Live/Dead Fixable dead cell stain kit (Near-IR, Thermo Fisher),
followed by Fc receptor blockade. After washing, cells were stained with NK cell marker CD56-APC, fixed with Intracellular Fixation Buffer (eBioscience), permeabilized with Permeabilization
Buffer (eBioscience), and stained with FITC-conjugated anti-human interferon-γ (Biolegend). Cells were acquired in a BD LSRFortessa Flow Cytometer, and analyzed by FlowJo software. ELISA NK
cells were co-cultured for 6 h at a ratio of 1:1 with MM cells in the presence or absence of Dara. After incubation, supernatants were collected and measured using human Granzyme B ELISA kit
(Biolegend) and human Perforin ELISA kit (Abcam), according to the manufacturer’s protocol. HUMAN NK-RECONSTITUTED MURINE MODEL OF HUMAN MULTIPLE MYELOMA The experimental procedures and
protocol (No: 03-043) were approved by the Institutional Animal Care and Use Committee (IACUC; DFCI). All mice were housed in a pathogen-free environment at a DFCI animal facility and were
handled in strict accordance with Good Animal Practice, as defined by the Office of Laboratory Animal Welfare. 2 ×106 H929-luciferase WT or KDM6A KO cells were subcutaneously injected into
the right flank of 6-week-old female NOD/SCID gamma (NSG; NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice lacking functional NK cells (Jackson Labs, #005557-NSG). Tumor burden was serially monitored
by BLI using the IVIS Imaging System and Living Image Software (PerkinElmer, Waltham, MA, USA). After tumor engraftment was confirmed by BLI, the mice were randomly divided into 4 groups (WT
+ NK; WT + NK+Dara; KO + NK; KO + NK+Dara). Dara was given intraperitoneally for a total of 3 administrations (8 mg/kg per mouse weekly) starting on treatment day 1 following randomization.
Human primary NK cells (5 ×106) were injected intravenously the day after the Dara injection once per week for 3 weeks. Tumor sizes were detected by BLI once per week. The mice were
euthanized by carbon dioxide before the tumors exceeded 2.0 cm in any dimension. CHIP-SEQ AND CHIP-QPCR ChIP assays were performed using a Simple Plus Enzymatic Chromatin IP kit (Magnetic
Beads). Purified DNA was delivered to the molecular biology core facilities (MBCF) at Dana-Farber Cancer Institute for NGS. The raw data were saved in files with fastq format. Low-quality
reads and sequencing adapters were removed by Trimmomatic v0.3853, and the qualified reads were mapped to the human genome (hg38) by bowtie254. Peak calling was performed by MACS255 and
visualized in IGV56. The intensity of peaks around the transcription start site (TSS) was calculated and shown by deeptools57. RNA-SEQ ANALYSIS For RNA-seq, total RNA of WT and KDM6A KO
cells was extracted using the RNeasy Mini Kit (Qiagen). Library preparation and sample sequencing were performed in the molecular biology core facilities (MBCF) at Dana-Farber Cancer
Institute. The RNA-Seq data were processed following the VIPER pipeline58. Raw reads were aligned to the human genome (hg38) by STAR59, and the differential gene expression analysis was
performed by limma60. The differentially expressed genes (DEG) were defined by the log of transformed fold change greater than 1 (log2(fold change)>1) and adjusted p value less than 0.05.
Volcano plots were used to display the DEGs with down-regulated genes (blue) and up-regulated genes (pink). Gene set enrichment analysis (GSEA) was performed to identify significantly
enriched pathways. The biologically defined gene sets were obtained from the Molecular Signatures Database (http://software.broadinstitute.org/gsea/msigdb/index.jsp). Genes used for GSEA
analysis were pre-ranked on the basis of log2 fold change of TPM (transcripts per kilobase million) between WT and KDM6A KO cells. ATAC-SEQ ATAC-seq was performed by the Genewiz Corporation.
The raw reads of ATAC-seq were trimmed by Trimmomatic v0.3853 first, and then aligned to human reference genome hg38 by bowtie254. The alignment was filtered by samtools61, screening out
reads with mapping quality less than 30, inconsonant alignment, and secondary alignment. PCR duplicates and mitochondria were also filtered out prior to peak calling by Picard v2.18.2662.
MACS2 v2.1.255 was used for peak calling, and the differential peak analysis was performed by R package Diffbind63. We summarized the peaks across the three samples and compared the
intensity of peaks together. The missing peaks in any sample were assigned zero intensity. Wilcox test was used to compare KO samples and wild-type samples, and the Kruskal-Wallis test was
used to examine the variance of the group. STATISTICS AND REPRODUCIBILITY. NO STATISTICAL METHODS WERE USED TO PREDETERMINE SAMPLE SIZE Two-sided student’s _t_-test or analysis of variance
followed by Dunnett’s test is used to compare differences between the treated group and the relevant control group. Comparisons between three groups or more are done using one-way analysis
of variance (ANOVA) with the Tukey post hoc test. The n number of each experiment is listed in the figure legends. Statistical tests were performed using GraphPad Prism 10.0 (GraphPad
Software Inc.) unless otherwise specified. Values of _p_ < 0.05(*), _p_ < 0.01(**), _p_ < 0.001(***), _p_ < 0.0001(****) are considered significant. Values of _p_ > 0.05 are
indicated as ns. The repeated time of each experiment is indicated in the figure legends. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio
Reporting Summary linked to this article. DATA AVAILABILITY CRISPR screen sequencing data, RNA-seq, ChIP-seq and ATAC-seq data generated in this study are deposited in the National Institute
of Health Gene Expression Omnibus (GEO) database (GSE228771). Source data are provided with this paper. REFERENCES * Palumbo, A. & Anderson, K. Multiple myeloma. _N. Engl. J. Med._ 364,
1046–1060 (2011). Article CAS PubMed Google Scholar * Kuehl, W. M. & Bergsagel, P. L. Multiple myeloma: evolving genetic events and host interactions. _Nat. Rev. Cancer_ 2, 175–187
(2002). Article CAS PubMed Google Scholar * Kumar, S. K. & Anderson, K. C. Immune therapies in multiple myeloma. _Clin. Cancer Res._ 22, 5453–5460 (2016). Article CAS PubMed
Google Scholar * Tai, Y. T. et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow
milieu. _Blood_ 112, 1329–1337 (2008). Article CAS PubMed PubMed Central Google Scholar * Lonial, S. et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in
relapsed or refractory multiple myeloma. _J. Clin. Oncol._ 30, 1953–1959 (2012). Article CAS PubMed Google Scholar * de Weers, M. et al. Daratumumab, a novel therapeutic human CD38
monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. _J. Immunol._ 186, 1840–1848 (2011). Article PubMed Google Scholar * Velasquez, M. P., Bonifant,
C. L. & Gottschalk, S. Redirecting T cells to hematological malignancies with bispecific antibodies. _Blood_ 131, 30–38 (2018). Article CAS PubMed PubMed Central Google Scholar *
Ramadoss, N. S. et al. An anti-B cell maturation antigen bispecific antibody for multiple myeloma. _J. Am. Chem. Soc._ 137, 5288–5291 (2015). Article CAS PubMed Google Scholar * Hipp, S.
et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. _Leukemia_ 31, 2278 (2017). Article CAS PubMed
Google Scholar * Lonial, S. et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. _Lancet Oncol._ 21, 207–221
(2020). Article CAS PubMed Google Scholar * Ikeda, H. et al. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive
multiple myeloma cells in vitro and in vivo. _Clin. Cancer Res._ 15, 4028–4037 (2009). Article CAS PubMed Google Scholar * Raje, N. et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed
or refractory multiple myeloma. _N. Engl. J. Med._ 380, 1726–1737 (2019). Article CAS PubMed PubMed Central Google Scholar * Mikkilineni, L. & Kochenderfer, J. N. CAR T cell
therapies for patients with multiple myeloma. _Nat. Rev. Clin. Oncol._ 18, 71–84 (2021). Article CAS PubMed Google Scholar * McKeage, K. Daratumumab: First global approval. _Drugs_ 76,
275–281 (2016). Article CAS PubMed Google Scholar * Markham, A. Elotuzumab: First global approval. _Drugs_ 76, 397–403 (2016). Article CAS PubMed Google Scholar * Dhillon, S.
Isatuximab: First Approval. _Drugs_ 80, 905–912 (2020). Article CAS PubMed Google Scholar * Lokhorst, H. M. et al. Targeting CD38 with Daratumumab monotherapy in multiple myeloma. _N.
Engl. J. Med._ 373, 1207–1219 (2015). Article CAS PubMed Google Scholar * Mateos, M. V. et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. _N. Engl. J.
Med._ 378, 518–528 (2018). Article CAS PubMed Google Scholar * Palumbo, A. et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. _N. Engl. J. Med._ 375, 754–766 (2016).
Article CAS PubMed Google Scholar * van de Donk, N. & Usmani, S. Z. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. _Front. Immunol._ 9, 2134
(2018). Article PubMed PubMed Central Google Scholar * Nijhof, I. S. et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma.
_Blood_ 128, 959–970 (2016). Article CAS PubMed Google Scholar * Ogiya, D. et al. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: therapeutic
implications. _Blood_ 136, 2334–2345 (2020). Article PubMed PubMed Central Google Scholar * Leo, R. et al. Multiparameter analyses of normal and malignant human plasma cells: CD38++,
CD56+, CD54+, cIg+ is the common phenotype of myeloma cells. _Ann. Hematol._ 64, 132–139 (1992). Article CAS PubMed Google Scholar * Fedele, P. L. et al. IMiDs prime myeloma cells for
daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. _Blood_ 132, 2166–2178 (2018). Article CAS PubMed Google Scholar * Nijhof, I. S. et al. Upregulation of CD38
expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. _Leukemia_ 29, 2039–2049 (2015). Article CAS PubMed Google Scholar *
Garcia-Guerrero, E. et al. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. _Blood_ 129, 3386–3388 (2017). Article CAS PubMed Google Scholar *
Wang, W., Erbe, A. K., Hank, J. A., Morris, Z. S. & Sondel, P. M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. _Front. Immunol._ 6, 368 (2015).
Article PubMed PubMed Central Google Scholar * van Haaften, G. et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. _Nat. Genet._ 41, 521–523 (2009).
Article PubMed PubMed Central Google Scholar * Pawlyn, C. et al. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. _Clin. Cancer Res._ 22, 5783–5794 (2016).
Article CAS PubMed PubMed Central Google Scholar * Zhang, T., Zhang, Z., Dong, Q., Xiong, J. & Zhu, B. Histone H3K27 acetylation is dispensable for enhancer activity in mouse
embryonic stem cells. _Genome Biol_ 21, 45 (2020). Article CAS PubMed PubMed Central Google Scholar * Li, B. et al. Comprehensive analyses of tumor immunity: implications for cancer
immunotherapy. _Genome Biol._ 17, 174 (2016). Article PubMed PubMed Central Google Scholar * Morschhauser, F. et al. Tazemetostat for patients with relapsed or refractory follicular
lymphoma: an open-label, single-arm, multicentre, phase 2 trial. _Lancet Oncol_. 21, 1433–1442 (2020). Article CAS PubMed PubMed Central Google Scholar * Beguelin, W. et al. EZH2 is
required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. _Cancer Cell_ 23, 677–692 (2013). Article CAS PubMed PubMed Central Google Scholar *
Ezponda, T. & Licht, J. D. Molecular pathways: deregulation of histone h3 lysine 27 methylation in cancer-different paths, same destination. _Clin. Cancer Res._ 20, 5001–5008 (2014).
Article CAS PubMed PubMed Central Google Scholar * Kim, J. H. et al. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer
cells. _Cancer Res_ 74, 1705–1717 (2014). Article CAS PubMed PubMed Central Google Scholar * Wang, J. K. et al. The histone demethylase UTX enables RB-dependent cell fate control.
_Genes Dev_ 24, 327–332 (2010). Article PubMed PubMed Central Google Scholar * Ezponda, T. et al. UTX/KDM6A Loss Enhances the Malignant Phenotype of Multiple Myeloma and Sensitizes Cells
to EZH2 inhibition. _Cell Rep_ 21, 628–640 (2017). Article CAS PubMed PubMed Central Google Scholar * Casneuf, T. et al. Effects of daratumumab on natural killer cells and impact on
clinical outcomes in relapsed or refractory multiple myeloma. _Blood Adv._ 1, 2105–2114 (2017). Article CAS PubMed PubMed Central Google Scholar * Naeimi Kararoudi, M. et al. CD38
deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. _Blood_ 136, 2416–2427 (2020). Article PubMed PubMed Central Google
Scholar * Pech M. F. et al. Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. _Elife_ 8, (2019). * Bernareggi, D. et al. CHMP2A
regulates tumor sensitivity to natural killer cell-mediated cytotoxicity. _Nat Commun._ 13, 1899 (2022). Article CAS PubMed PubMed Central Google Scholar * Sheffer, M. et al.
Genome-scale screens identify factors regulating tumor cell responses to natural killer cells. _Nat Genet._ 53, 1196–1206 (2021). Article CAS PubMed Google Scholar * Elias, S. et al.
Immune evasion by oncogenic proteins of acute myeloid leukemia. _Blood_ 123, 1535–1543 (2014). Article CAS PubMed Google Scholar * Chiba, M. et al. Genome-wide CRISPR screens identify
CD48 defining susceptibility to NK cytotoxicity in peripheral T-cell lymphomas. _Blood_ 140, 1951–1963 (2022). Article CAS PubMed PubMed Central Google Scholar * Cheng, Y. et al.
Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. _Signal Transduct Target Ther._ 4, 62 (2019). Article PubMed PubMed Central Google Scholar
* Hoy, S. M. Tazemetostat: First Approval. _Drugs_ 80, 513–521 (2020). Article PubMed Google Scholar * Alzrigat, M., Jernberg-Wiklund, H. & Licht, J. D. Targeting EZH2 in multiple
myeloma-multifaceted anti-tumor activity. _Epigenomes_ 2, 16 (2018). * Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. _Nat.
Methods_ 11, 783–784 (2014). Article CAS PubMed PubMed Central Google Scholar * Joung, J. et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. _Nat.
Protoc._ 12, 828–863 (2017). Article CAS PubMed PubMed Central Google Scholar * Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9
knockout screens. _Genome Biol_. 15, 554 (2014). Article PubMed PubMed Central Google Scholar * Colaprico, A. et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of
TCGA data. _Nucleic Acids Res._ 44, e71 (2016). Article PubMed Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data
with DESeq2. _Genome Biol_. 15, 550 (2014). Article PubMed PubMed Central Google Scholar * Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina
sequence data. _Bioinformatics_ 30, 2114–2120 (2014). Article CAS PubMed PubMed Central Google Scholar * Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2.
_Nat. Methods_ 9, 357–359 (2012). Article CAS PubMed PubMed Central Google Scholar * Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). _Genome Biol_. 9, R137 (2008). Article
PubMed PubMed Central Google Scholar * Robinson, J. T. et al. Integrative genomics viewer. _Nat. Biotechnol._ 29, 24–26 (2011). Article CAS PubMed PubMed Central Google Scholar *
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. _Nucleic Acids Res._ 42, W187–W191 (2014). Article CAS
PubMed PubMed Central Google Scholar * Cornwell, M. et al. VIPER: Visualization Pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. _BMC
Bioinformatics_ 19, 135 (2018). Article PubMed PubMed Central Google Scholar * Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS
PubMed Google Scholar * Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. _Nucleic Acids Res._ 43, e47 (2015). Article
PubMed PubMed Central Google Scholar * Li, H. et al. The Sequence Alignment/Map format and SAMtools. _Bioinformatics_ 25, 2078–2079 (2009). Article PubMed PubMed Central Google Scholar
* Picard2019toolkit. Picard toolkit. _Broad Institute, GitHub repository._ http://broadinstitute.github.io/picard/ (2019). * Stark, R. & Brown, G. DiffBind: Differential binding
analysis of ChIP-Seq peak data. https://bioconductor.org/packages/release/bioc/html/DiffBind.html (2012). Download references ACKNOWLEDGEMENTS We thank Molecular Biology Core Facilities,
Dana-Farber Cancer Institute for assistance with RNA-seq and ChIP-seq; the animal Resources Facility, Dana-Farber Cancer Institute for support with animal studies; the flow cytometry core
facility, Dana-Farber Cancer Institute for the help with cell sorting. This work was supported by the National Institutes of Health grants SPORE-P50100707 (K.C.A.), R01-CA050947 (K.C.A.),
R01-CA178264 (T.H. and K.C.A.), and P01-155258 (K.C.A. and N.M.). This work was also supported by the National Natural Science Foundation of China 82200224 (L.X.) and the Natural Science
Foundation of Shandong Province ZR2021MH072 (L.X). A.G. is a Fellow of the Leukemia & Lymphoma Society and a Scholar of the American Society of Hematology; she is supported by an
Individual Start-UP grant from the Italian Association for Cancer Research (AIRC) (project #27750). This study was also supported by Dr. Miriam and Sheldon G. Adelson Medical Research
Foundation and the Riney Family Myeloma Initiative. AUTHOR INFORMATION Author notes * These authors contributed equally: Jiye Liu, Lijie Xing. AUTHORS AND AFFILIATIONS * Jerome Lipper
Multiple Myeloma Center, Lebow Institute for Myeloma Therapeutics, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, 02215, USA Jiye Liu, Kenneth Wen, Ning Liu,
Yuntong Liu, Keiji Kurata, Eugenio Morelli, Annamaria Gulla, Yu-Tzu Tai, Nikhil Munshi, Paul Richardson, Teru Hideshima & Kenneth C. Anderson * Department of Hematology, Shandong Cancer
Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, Shandong, 250117, China Lijie Xing * Clinical Big Data Research Center, The Seventh
Affiliated Hospital of Sun Yat-Sen University, Shenzhen, Guangdong, 518107, China Jiang Li & Xiaoning Hong * Department of Marine Bio-Pharmacology, College of Food Science and
Technology, Shanghai Ocean University, Shanghai, 201306, China Ning Liu * Center for Functional Cancer Epigenetics, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA,
02215, USA Gongwei Wu * Vertex pharmaceuticals, Boston, MA, 02210, USA Su Wang * Department of Hematology and Oncology, School of Medicine, Tokai University, Isehara, 259-1193, Japan Daisuke
Ogiya * Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, 02215, USA Tian-Yu Song * Broad Institute of Harvard and MIT, Cambridge, MA, 02142, USA Tian-Yu Song *
Department of Oncologic Pathology, Dana-Farber Cancer Institute, Boston, MA, 02215, USA Johany Penailillo & Ruben Carrasco * Department of Cancer Biology, Dana-Farber Cancer Institute,
Boston, MA, 02215, USA Tingjian Wang * Candiolo Cancer Institute, FPO-IRCCS, Candiolo (TO), 10060, Italy Annamaria Gulla * Department of Pathology, Brigham and Women’s Hospital, Harvard
Medical School, Boston, MA, 02215, USA Ruben Carrasco Authors * Jiye Liu View author publications You can also search for this author inPubMed Google Scholar * Lijie Xing View author
publications You can also search for this author inPubMed Google Scholar * Jiang Li View author publications You can also search for this author inPubMed Google Scholar * Kenneth Wen View
author publications You can also search for this author inPubMed Google Scholar * Ning Liu View author publications You can also search for this author inPubMed Google Scholar * Yuntong Liu
View author publications You can also search for this author inPubMed Google Scholar * Gongwei Wu View author publications You can also search for this author inPubMed Google Scholar * Su
Wang View author publications You can also search for this author inPubMed Google Scholar * Daisuke Ogiya View author publications You can also search for this author inPubMed Google Scholar
* Tian-Yu Song View author publications You can also search for this author inPubMed Google Scholar * Keiji Kurata View author publications You can also search for this author inPubMed
Google Scholar * Johany Penailillo View author publications You can also search for this author inPubMed Google Scholar * Eugenio Morelli View author publications You can also search for
this author inPubMed Google Scholar * Tingjian Wang View author publications You can also search for this author inPubMed Google Scholar * Xiaoning Hong View author publications You can also
search for this author inPubMed Google Scholar * Annamaria Gulla View author publications You can also search for this author inPubMed Google Scholar * Yu-Tzu Tai View author publications
You can also search for this author inPubMed Google Scholar * Nikhil Munshi View author publications You can also search for this author inPubMed Google Scholar * Paul Richardson View author
publications You can also search for this author inPubMed Google Scholar * Ruben Carrasco View author publications You can also search for this author inPubMed Google Scholar * Teru
Hideshima View author publications You can also search for this author inPubMed Google Scholar * Kenneth C. Anderson View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS Conceptualization, J.L., L.X., T.H. and K.C.A.; Methodology, J.L., L.X. and T.H.; Software, Jiang Li; Validation, J.L., L.X, N.L. and Y.L.; Formal analysis,
J.L., Jiang Li, S.W. and T-Y.S.; Investigation, J.L., L.X., Jiang Li, K.W., N.L., Y.L., S.W., G.W., D.O., T-Y.S., K.K., J.P., E.M., T.W. and X.H., A.G.; Resources, Y-T.T., N.M. and P.R.,
R.C.; Data Curation, J.L.; Writing-Original Draft, J.L. L.X., and T.H.; Writing-Review & Editing, all co-authors; Visualization, J.L.; Supervision, T.H. and K.C.A; Project
Administration, J.L., T.H. and K.C.A; Funding Acquisition, L.X., N.M., T.H. and K.C.A. CORRESPONDING AUTHOR Correspondence to Kenneth C. Anderson. ETHICS DECLARATIONS COMPETING INTERESTS
K.C.A. serves on advisory boards to Pfizer, AstraZeneca, Janssen, Starton, Window, and Bristol Myers Squibb; and is a Founder of OncoPep, C4 Therapeutics, Dynamic Cell Therapies, and
NextRNA. All the other authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Tony Reiman, Rhonda Voskuhl and the other, anonymous,
reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES
SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5 SUPPLEMENTARY DATA 6 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Liu, J., Xing, L., Li, J.
_et al._ Epigenetic regulation of CD38/CD48 by KDM6A mediates NK cell response in multiple myeloma. _Nat Commun_ 15, 1367 (2024). https://doi.org/10.1038/s41467-024-45561-z Download citation
* Received: 17 July 2023 * Accepted: 29 January 2024 * Published: 14 February 2024 * DOI: https://doi.org/10.1038/s41467-024-45561-z SHARE THIS ARTICLE Anyone you share the following link
with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative







