- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Chimeric antigen receptor (CAR) T cells have transformed the treatment landscape for hematological malignancies. However, CAR T cells are less efficient against solid tumors,
largely due to poor infiltration resulting from the immunosuppressive nature of the tumor microenvironment (TME). Here, we assessed the efficacy of Lewis Y antigen (LeY)-specific CAR T cells
in patient-derived xenograft (PDX) models of prostate cancer. In vitro, LeY CAR T cells directly killed organoids derived from androgen receptor (AR)-positive or AR-null PDXs. In vivo,
although LeY CAR T cells alone did not reduce tumor growth, a single prior dose of carboplatin reduced tumor burden. Carboplatin had a pro-inflammatory effect on the TME that facilitated
early and durable CAR T cell infiltration, including an altered cancer-associated fibroblast phenotype, enhanced extracellular matrix degradation and re-oriented M1 macrophage
differentiation. In a PDX less sensitive to carboplatin, CAR T cell infiltration was dampened; however, a reduction in tumor burden was still observed with increased T cell activation. These
findings indicate that carboplatin improves the efficacy of CAR T cell treatment, with the extent of the response dependent on changes induced within the TME. SIMILAR CONTENT BEING VIEWED
BY OTHERS PSMA-TARGETING TGFΒ-INSENSITIVE ARMORED CAR T CELLS IN METASTATIC CASTRATION-RESISTANT PROSTATE CANCER: A PHASE 1 TRIAL Article 21 March 2022 THE POTENTIAL OF CAR T CELL THERAPY
FOR PROSTATE CANCER Article 08 July 2021 PSCA-CAR T CELL THERAPY IN METASTATIC CASTRATION-RESISTANT PROSTATE CANCER: A PHASE 1 TRIAL Article Open access 12 June 2024 INTRODUCTION
Immune-based therapies have transformed the treatment of many cancers, but not prostate cancer1. Chimeric-antigen receptor (CAR) T cell therapy, in which a patient’s own T cells are
genetically modified to target tumor-associated antigens, has greatly improved clinical outcomes in advanced acute lymphocytic leukemia and B cell lymphoma2,3. However, success in treating
solid tumors has been very limited due to the immunosuppressive tumor microenvironment (TME), which prevents T cell infiltration and activation at the tumor site4. Herein we address the
challenge of how to modify the TME to enhance CAR T cell recruitment and effector function using combination approaches. Target antigens for CAR T cells in prostate cancer include
prostate-specific membrane antigen (PSMA), prostate stem cell antigen (PSCA), KLK2, and six-membrane epithelial antigen of the prostate-1 (STEAP-1), mainly as monotherapies and with limited
success thus far5,6,7,8,9. In this study, we examine the efficacy of CAR T cells directed against the Lewis Y (LeY) glycolipid antigen. LeY is an onco-fetal glycolipid antigen with minimal
expression in normal tissues following birth; however, >50% of human carcinomas reactivate LeY biosynthesis at very high levels10. Studies in the 1990s demonstrated that LeY is
prominently expressed in many high-grade prostate cancers and their metastases11,12. LeY CAR T cells demonstrated high safety and tolerability in a Phase 1a clinical trial in patients with
LeY-positive acute myeloid leukemia13. In addition to ensuring antigen specificity, it is important to consider the immunosuppressive TME, which can interfere with T cell differentiation,
proliferation, and exhaustion in solid tumors14,15. To overcome this issue, novel approaches including the engineering of CARs or therapeutic combinations have been tested in preclinical
cancer models7,16. Promising effects have been observed with checkpoint blockade in various cancer types and are being tested in clinical trials (i.e., ACTRN12622001542785 by our team).
Checkpoint inhibitors effectively block immune checkpoints on T cells and their ligands on tumor cells (i.e. PD-1/L1) and checkpoint blockade allows CAR T cells to penetrate the TME, exert
their actions, and control tumor growth16. Alternatively, accumulating evidence suggests that chemotherapies can increase the immunogenicity of tumor cells and dampened the immunosuppressive
TME. Chemotherapies, including docetaxel, oxaliplatin, and carboplatin in prostate, lung, and ovarian cancer, have been shown to increase tumor antigen presentation, increase immune
checkpoint expression and promote immune cell infiltration and activation5,7,17,18,19,20,21,22. This shift from an immunologically cold to warm TME may prime the tumor to immunotherapies,
including CAR T cells. However, studies on pre- and post-treated ovarian cancer biopsies show different patterns of immune response to chemotherapy, suggesting that TME modulation may be
tumor-specific19. Therefore, whilst there is a strong rationale for modifying the TME with chemotherapy, further work needs to optimize their roles in enhancing immunotherapy and identifying
which tumors will respond to this treatment strategy. In this study, we tested the combination of LeY CAR T cells with either carboplatin, docetaxel, or the checkpoint inhibitor nivolumab
on tumor growth using patient-derived xenografts (PDXs) of prostate cancer. Our data show that carboplatin can synergize with CAR T therapy to facilitate early and persistent CAR T cell
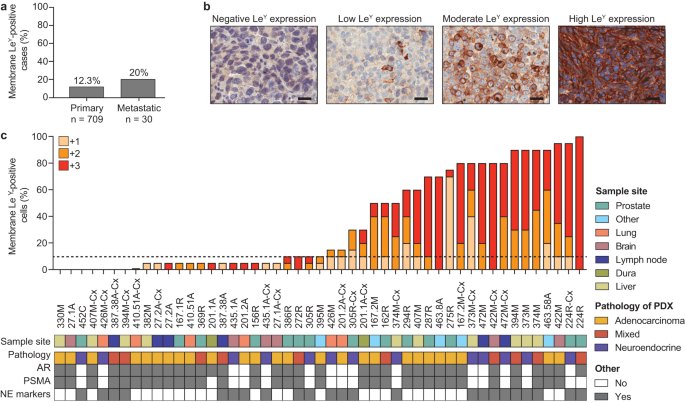
infiltration and activation within select prostate tumors to maximize therapeutic response. RESULTS Using immunohistochemistry, we examined LeY expression in 800 prostate cancer specimens
from 739 patients. As cell surface expression of the target antigen is critical for CAR T cells to form an immune synapse, we quantified both membrane- and cytoplasmic LeY staining in this
unselected patient cohort. 12.6% of tumors had \(\ge\)10% membrane LeY-positive cells (the cut-off for admission into our current Phase I clinical trial in LeY-positive lung cancer); more
specifically, 12.3% (87/709) and 20% (6/30) of patients with localized and metastatic disease respectively demonstrated this level of membrane staining (Fig. 1a). However, in 49 PDXs from
the Monash Urology Research Alliance (MURAL) collection, which are typically derived from more aggressive cases of prostate cancer23, 57% (28/49) had \(\ge\)10% membrane LeY-positive cells,
with far higher expression in many specimens, based on percent cell positivity and staining intensity (Fig. 1b, c). LeY-positive specimens included AR-null tumors as well as AR-positive
tumors (Fig. 1c), and its expression was independent of AR or PSMA (Fig. 1c). This suggested that anti-LeY CAR T cells may provide another treatment option for patients who would not benefit
from AR- or PSMA-directed therapies, or who had progressed following these therapies. To show the on-target action of LeY CAR T cells in vitro, we used organoids derived from PDXs, which
maintain LeY expression (Fig. 2a and Supplementary Fig. 1a, b). We selected six PDXs with different phenotypes and genomic features (Fig. 1c and Supplementary Fig. 1c)23, including three
androgen receptor (AR)-positive adenocarcinomas and three AR-null tumors that express neuroendocrine markers, with a range of LeY expression (Supplementary Fig. 1a, b). Co-culture with LeY
CAR T cells, but not control empty vector T (ev-T) cells, caused morphological destruction of organoids with pronounced propidium iodine uptake, indicating secondary necrosis (Fig. 2b–d).
Importantly, tumor cell death correlated with LeY expression, with strongest PI uptake in organoids with high (\(\ge\) 90%) LeY expression (Fig. 2d, Supplementary Fig. 1a, b). In contrast,
PDX-435.1A-Cx organoids, with 10% LeY expression, showed a weaker response, and PDX-201.1A-Cx organoids, with 5% LeY expression, had no response to LeY CAR T cells (Fig. 2d and Supplementary
Fig. 1a, b). This is consistent with data from cell lines, with lysis of the LeY-positive prostate cancer cells PC-3, DU-145, and 22Rv1, but not the LeY-negative melanoma cell line
MDA-MB435, when co-cultured with LeY CAR T cells (Supplementary Fig. 1d, e). The mechanism of cell death in organoids involved the perforin/granzyme-dependent granule exocytosis pathway as
granzyme B and/or perforin inhibitors markedly reduced cell death in PDX-287R and PDX-472M organoids (Fig. 2e). Since LeY can be targeted on primary prostate tumors in vitro, we next
administered LeY CAR T cells to mice bearing PDX-287R grafts derived from a patient with aggressive adenocarcinoma, as previously described23. PDX-287R has 70% LeY expression by
immunohistochemistry and 95% by flow cytometry (Fig. 1c and Supplementary Fig. 2a). As expected, CAR T cells administered alone did not inhibit tumor growth (Fig. 3a). As immune checkpoint
blockade is often combined with CAR T cells in the setting of solid cancers24, we combined CAR T therapy with nivolumab, an anti-PD-1 antibody that blocks its inhibitory interaction with its
ligand, PD-L1. Despite the tumors expressing PD-L1 (Supplementary Fig. 2b, c), nivolumab had no effect on tumor growth alone or in combination with LeY CAR T cells (Fig. 3b). We next
trialed CAR T cells in combination with a single dose of the taxane-based chemotherapy docetaxel, or the platinum-based chemotherapy carboplatin, given a week prior to CAR T cell infusion.
Docetaxel given alone, or with LeY CAR T cells, had no effect on tumor growth (Fig. 3c). Carboplatin treatment alone or with ev-T cells induced an initial decrease in tumor burden that was
sustained for up to 3 weeks before tumors grew back (Fig. 3d, e and Supplementary Fig. 2d). In contrast, carboplatin with CAR T cells caused tumors to regress to <1% of their starting
volume, with no regrowth for 6 weeks post-treatment (Fig. 3d, e). Following LeY CAR T cell plus carboplatin treatment only scattered tumor cells could be detected in 5 of the 6 residual
grafts (Supplementary Fig. 2e), with phospho-histone H3 (pHH3) staining indicating greatly decreased proliferation compared to tumor cells in other treatment groups (Fig. 3f). Residual tumor
cells lacked or had minimal LeY expression (Supplementary Fig. 2f). However, treatment with carboplatin alone did not alter LeY expression in tumors in vivo, as well as in organoids
following short-term carboplatin treatment in vitro, suggesting that the lack of LeY expression in residual tumors was not due to carboplatin treatment (Supplementary Fig. 2g–i). These data
showed that pre-treatment with carboplatin could negate the poor therapeutic response of CAR T cells alone in PDX-287R. To determine whether this combination therapy is effective in other
PDX lines, we treated PDX-224R-Cx, a primary neuroendocrine cancer responsive to LeY CAR T cells in vitro (Fig. 2d). Consistent with PDX-287R, LeY CAR T cells alone did not alter tumor
growth (Fig. 3g). Carboplatin treatment alone caused an initial regression in tumor burden that was sustained for a week before grafts grew back (Fig. 3g). Treatment with carboplatin plus
LeY CAR T cells significantly decreased tumor burden compared to no T cell control, carboplatin, LeY CAR T cells alone and carboplatin with ev-T cells at the end of the treatment period
(Fig. 3g). However, in contrast to PDX-287R, tumors continued to grow across the experimental period (Fig. 3g), suggesting that the response to carboplatin-CAR T cell therapy may depend on
the effect of carboplatin in individual tumors. To investigate this, we first analyzed T cell recruitment and activation, which is a major barrier to CAR T cell therapy in solid tumors. We
assessed the localization of recruited LeY CAR T cells in PDX-287R grafts by staining for the human T cell marker CD3. There was significant enrichment of LeY CAR T cells at the tumor site 6
weeks after treatment with carboplatin, compared to all other treatment groups and controls (Fig. 4a, b), demonstrating that the carboplatin-induced long-term CAR T retention at the graft
sites. To determine if CAR T cells homed directly to the tumor, or if infiltration was secondary to tumor shrinkage, we assessed LeY CAR T cell infiltration 48 h after infusion. Remarkably,
we found high numbers of LeY CAR T cells in grafts 48 h after infusion following pre-treatment with carboplatin compared to grafts treated with CAR T cells alone (Fig. 4c, d), with
concurrent accumulation of mouse F4/80+ macrophages (Fig. 4e, f). Using flow cytometry, we showed that the presence of CAR T cells in the tumor was accompanied by a decrease in CAR T cells
in the spleen and peripheral blood, when compared to CAR T cells administered alone (Fig. 4g). Elevated expression of T cell activation markers CD25 and CD137, and the production of
pro-inflammatory Th1 cytokines IL-2, TNF-α, and IFN-γ, at the tumor site were also consistent with heightened T cell activation when administered after carboplatin (Fig. 4h, i and
Supplementary Fig. 3a–d). In contrast to PDX-287R, similar analysis on PDX-224R-Cx grafts 48 h post-CAR T cell infusion demonstrated that carboplatin failed to increase the infiltration of
CAR T cells into PDX-224R-Cx tumors (Fig. 4j). However, the limited CAR T cells that were present in the tumors were activated following carboplatin treatment (Fig. 4k, l and Supplementary
Fig. 3e–h). Therefore, while TME-modulation with carboplatin increased the activation of LeY CAR T cells in PDX-224R-Cx, the response was restricted by a lack of CAR T cell infiltration into
the tumor. We next interrogated changes in the TME using flow cytometry seven days after carboplatin treatment, concurrent to the time of CAR T cell infusion. Carboplatin-treated PDX-287R
grafts had a significant decrease in the proportion of EpCAM+ tumor epithelial cells 1 week following treatment compared to vehicle-treated grafts (Fig. 5a, b). This was accompanied with a
significant increase in hFAS-positive tumor cells (Fig. 5c and Supplementary Fig. 4a), a death receptor associated with extrinsic apoptosis. In contrast, despite PDX-224R-Cx initially
responding to carboplatin treatment in vivo (Fig. 3g), by 1 week after treatment there was no change in the proportion of tumor cells or hFAS expression (Fig. 5d–f and Supplementary Fig.
4b). To investigate the response of tumor cells to carboplatin treatment in more detail, RNA-sequencing was performed on FACs-isolated tumor cells from PDX-287R at 1 week post-treatment.
Carboplatin resulted in the differential expression of 2017 genes in tumor cells, with 982 upregulated and 1036 downregulated genes (Fig. 5g and Supplementary Data 1). Consistent with the
increase in FAS expression observed by flow cytometry, gene set enrichment analysis showed an enrichment for apoptotic pathways, with significant increased expression of the pro-apoptotic
genes _FAS, BAX, BBC3, IFI6_, and _JUN_ (Fig. 5h and Supplementary Fig. 4c). Gene set enrichment analysis also revealed enrichment of interferon response, JAK/STAT signaling, p53, and
chemokine signaling pathways (Supplementary Fig. 4c). Previously, carboplatin has been shown to activate the cGAS/STING pathway, resulting in an increase in chemokine expression by lung
tumor cells22. Here, we also found an increase in STING signaling, with an enrichment for the cytosolic DNA sensing pathway and increased expression of _STING1, STAT1_, and _STAT2_ (Fig. 5i
and Supplementary Fig. 4c). There was also a significant increase in the expression of genes involved in T cell chemotaxis, _CXCL10, CXCL11, CCL20_ (Fig. 5j and Supplementary Data 1). This
increase in chemokine expression may contribute to early recruitment of CAR T cells into the tumor. Collectively, carboplatin-induced cell death in tumor cells likely initiated a
pro-inflammatory phenotype in prostate cancer PDXs. At 1 week post-treatment, flow cytometry analysis also showed that carboplatin caused an influx of CD45+ immune cells into PDX-287R grafts
(Fig. 5a and Supplementary Fig. 4d). The majority of immune cells in both control and treated grafts were F4/80+ cells of the myeloid monocytic lineage, and RNA seq of isolated immune cells
demonstrated no change in macrophage phenotype between control and treated grafts (Supplementary Fig. 4e–h and Supplementary Data 2). In contrast, in PDX-224R-Cx grafts there was no change
in CD45+ immune cell infiltration following carboplatin treatment (Fig. 5d and Supplementary Fig. 4i). PDX-287R grafts treated with docetaxel also showed no change in tumor cell proportion
and FAS expression at 1 week post-treatment, and a reduction in immune cell infiltration was observed (Supplementary Fig. 4j–n). Therefore, a response to combination therapy with CAR T cells
may depend on initial sensitivity to the chemotherapeutic agent, with tumor cell death at the time of CAR T cell infusion required to promote T cell recruitment into the tumor. To further
understand the long-term changes following carboplatin treatment in PDX-287R that may facilitate CAR T cell retention and the sustained response to LeY CAR T cells, we used scRNA-seq to
transcriptionally define immune and non-immune cell populations and anti-tumor innate immune signaling within the TME 3 weeks post-carboplatin treatment, when the response to carboplatin was
maximal. Consistent with the previous experiment, a single dose of carboplatin initially induced a decrease in tumor burden before tumors grew back (Supplementary Fig. 5a). Indeed, by 3
weeks post-carboplatin treatment 97% (28/29) of grafts from across the two experiments had regressed from starting volume (Supplementary Fig. 5b). Given the tumor mass was considerably
reduced by this timepoint, we expected a substantial reduction in tumor cells. This was demonstrated by scRNA-seq, where epithelial cells comprised only 2.1% of the cellular content of the
graft, compared to 86.1% in vehicle control, and confirmed histologically (Fig. 6a, Supplementary Fig. 5c, and Supplementary Data 3). Unlike the 1 week time point, there was no significant
difference in the expression of pro-apoptotic genes; however, _BAX_ and _BAD_ were still upregulated suggesting ongoing apoptosis (Supplementary Fig. 5d and Supplementary Data 4). As the
epithelial cell content was reduced in carboplatin-treated PDX tumors, there was a proportionate increase in mouse stroma (97.9%) compared to vehicle control (13.9%; Fig. 6a, Supplementary
Fig. 5c, and Supplementary Data 3). This was also accompanied by changes in the relative populations of stromal cell types, with an increase in macrophages and monocytes, and fewer
fibroblasts, endothelial and perivascular cells (Fig. 6b and Supplementary Data 3). UMAP (Uniform Manifold Approximation and Projection) density-based clustering analysis separated
PDX-287R-associated mouse stromal cells, showing distinct clusters of stromal cell types with differential gene expression patterns between cells from carboplatin-treated and vehicle grafts
(Fig. 6c). The gene expression profile of each cell type was cross-referenced to known cell population markers25,26,27,28,29,30 to identify the cell types represented by each cluster in our
analysis (Supplementary Fig. 5e–g). In contrast to the epithelial cells, carboplatin treatment led to increased expression of pro-survival factors (_Bcl2, Mcl1, Ier3, Ier5, Hmox1_) in
stromal cell populations, thus preserving their viability and potentially providing a means of stabilizing and prolonging the anti-tumor response (Supplementary Fig. 5d, Supplementary Data
5–7). In contrast to 1 week post-treatment, gene set enrichment analyses demonstrated that by 3 weeks following carboplatin treatment macrophages had undergone polarization to the
pro-inflammatory M1, but not M2, phenotype, with upregulated of _Cd68_, _Cd86_, and _MHCII (H2-Aa, H2-Eb1_, and _H2-Ab1)_ (Fig. 6d, e and Supplementary Data 5). Consistent with their M1
differentiation, the macrophages upregulated expression of the chemotactic factors _Ccl5_ and _Cxcl10_, responsible for recruiting T lymphocytes (in this instance CAR T cells) into the tumor
stroma (Fig. 6f). The transcriptional profile of cancer-associated fibroblasts (CAFs) was also substantially altered 3 weeks after a single dose of carboplatin. The proportion of CAFs in
carboplatin-treated grafts was markedly reduced (Fig. 6b, c), and there was a shift from a myofibroblast (myCAF) to a pro-inflammatory phenotype (iCAF) (Fig. 6g and Supplementary Data 6).
This involved the downregulation of genes encoding the immunosuppressive factors _Tgfß-1_ and _Vegfa_, as well as the _Loxl_ gene family, responsible for stabilizing the extracellular matrix
(ECM; Fig. 6h and Supplementary Data 6). Following carboplatin treatment, CAFs increased their expression of numerous MMP genes (_Mmp2, Mmp3, Mmp13, Mmp14, Mmp27_); proteases involved in
the degradation of the tumor-supporting matrix (Fig. 6h and Supplementary Data 6). Carboplatin-treated CAFs also upregulated the chemoattractant _Ccl2_, which has been associated with
promoting monocyte and macrophage infiltration of the tumor site, and _Irf1_ and _IL-1ß_, which enhance M1 polarization in macrophages (Fig. 6h and Supplementary Data 6). Thus, the
carboplatin-induced CAF phenotype orchestrates a coordinated anti-tumorigenic response, which facilitates the polarization of macrophages to a pro-inflammatory M1 phenotype, enables
infiltration of phagocytes to the tumor site, and alters the ECM to promote immune cell trafficking. Taken together, carboplatin induces multiple changes that could overcome the
immunosuppressive TME. CAR T cells gain entry to the tumor site through the process of transendothelial migration, and tumor-associated high endothelial venule (TA-HEV) cells have been
described to facilitate this trafficking30. Three weeks post-carboplatin treatment there was a reduction in the proportion of endothelial cells in the PDXs (Fig. 6b). However, multiple
phenotypic changes were observed. Firstly, endothelial cells showed decreased expression of mouse _Angpt2_ and _Egln1_ genes, which typically promote tumor cell metastasis and the
dysregulation of vascular stability (Fig. 6i and Supplementary Data 7). Furthermore, carboplatin-treated endothelial cells had increased expression of _Sele_ and _Selp_, which encode key
selectins involved in the initial “capture and rolling” of leukocytes along the endothelial layer (Fig. 6i and Supplementary Data 7). Downstream in the signaling pathway, carboplatin-treated
endothelial cells also upregulated endothelial adhesion molecules _Icam1_, as well as _Vcam1_ gene expression, both of which may enhance that typify the TA-HEV phenotype and could be
expected to enhance CAR T cells traversal through the endothelial layer (Fig. 6i and Supplementary Data 7). Collectively, we describe a cascade of anti-tumorigenic alterations in cell
content and gene expression in response to low-dose carboplatin in PDX-287R that demonstrate how carboplatin effectively primes the TME and promotes LeY CAR T efficacy (Fig. 7). In the
absence of carboplatin sensitivity, including upregulation of hFAS and immune cell infiltration, CAR T cell entry to the tumor is limited and the response to LeY CAR T cell treatment is
dampened. DISCUSSION Collectively, these studies demonstrate that LeY CAR T cells are effective at targeting prostate cancer cells when co-cultured in vitro and can eliminate a prostatic PDX
tumor in vivo when preceded by a single dose of carboplatin. Mechanistically, carboplatin induced a cascade of events following the initial apoptosis of tumor cells, resulting in a
pro-inflammatory, anti-tumor immune response that enables CAR T cells to enter the tumor site and exert cytotoxic effector functions on LeY-expressing tumor cells. Failure of carboplatin to
induce apoptosis at the time of CAR T cell infusion and decreased T cell recruitment into the tumor dampens, but does not abolish, the response to treatment. Therefore, carboplatin improves
the efficacy of CAR T cell treatment, with the extent of the response dependent on changes induced within the tumor. Trafficking of CAR T cells to the tumor is a major barrier to CAR T cell
therapy in all cold, immune excluded solid tumors, and our data further supports this notion. In PDX-287R, carboplatin treatment initially induced apoptosis within the tumor cells,
accompanied by a pro-inflammatory response, increased cGAS-STING signaling and increased expression of chemokines, including CXCR3 ligands for T cell chemotaxis. At this time, there was an
influx of mouse myeloid cells into the tumor, and enhanced recruitment and activation of human CAR T cells following carboplatin treatment. In contrast, carboplatin failed to increase tumor
cell apoptosis, myeloid cell recruitment and T cell infiltration in PDX-224R-Cx. Interestingly, the T cells present within the PDX-224R-Cx tumors still showed increased activation following
carboplatin, presumably accounting for the decrease in tumor burden in carboplatin-LeY CAR T cell treated grafts compared to treatment with LeY CAR T cells alone. Collectively, this
demonstrates that pro-inflammatory changes induced by carboplatin are mandatory to enable the CAR T cells to gain unfettered access to the LeY-positive tumor cells and eliminate the tumor.
Whilst tumor cell sensitivity to carboplatin is clearly a feature, the transcriptomic changes within the TME following carboplatin treatment strongly suggest that pro-inflammatory effects
are not solely dependent on cancer cell death. A single dose of carboplatin induced a cascade of events to sustain a pro-inflammatory microenvironment and T cell persistence. The increase in
macrophages initially observed at 1 week post-treatment was sustained, with a shift to the M1 pro-inflammatory phenotype by 3 weeks post-treatment. Indeed, carboplatin is known to activate
mouse macrophages31,32, and consequently, the increased production of macrophage-derived factors, _Ccl5_ and _Cxcl10_, likely mediated the recruitment of T cells through known mechanisms,
including TCR3 receptor recruitment33. The evidence from our animal studies strongly suggests that the myeloid compartment plays a key role in the mechanism of action of carboplatin. Whether
the response will be similar in humans is unknown, but it is of high relevance to the clinical translation of this combination therapy. In addition to the activation of macrophages,
carboplatin induced a range of downstream modifications to the TME, including the induction of an iCAF phenotype, involving the downregulation of immunosuppressive genes such as _Tgfβ-1_ and
_Vegfa_, and decreased expression of _LOXL_ genes and increased expression of _MMP_ genes, leading to ECM degradation. We also demonstrated the presence of tumor-associated high endothelial
venule (TA-HEV) cells that are known to effectively traffic lymphocytes to tumors30. Collectively, this cascade of dynamic alterations to the TME (Fig. 7), observed after carboplatin
treatment, resulted in the induction of a pro-inflammatory microenvironment and provides a mechanism by which human T cell efficacy is mediated in this immune-compromised setting. The
chemotherapeutic agents docetaxel and cyclophosphamide have been shown to induce immunogenic cell death, dampen the suppressive TME, and improve the efficacy of immunotherapy, including CAR
T cells and checkpoint blockade, in preclinical studies for prostate cancer5,7,21. Furthermore, docetaxel is currently in clinical trials in combination with immunotherapy for
chemotherapy-naïve patients with advanced prostate cancer, where it is well-tolerated and showing anti-tumor activity34. Here, we demonstrate that the response to chemotherapies as a
modulating agent is likely dependent on the response of individual tumors, with TME modifications at the transcriptional level likely playing a major role in CAR T cell infiltration and
tumor response. In contrast to carboplatin, docetaxel failed to induce apoptosis and myeloid cell infiltration in PDX-287R at 1 week post-treatment, and had no effect on tumor growth when
combined with CAR T cells. It is likely that a multimodal approach is required to enhance the efficacy of immunotherapy in prostate cancer, and a pro-inflammatory gene signature induced by
agents such as carboplatin may be helpful in identifying patients that may benefit from CAR T cell therapy. Interestingly, PDX-287R has a mutation in the mismatch repair gene mutS homologue
2 (_MSH2_), which may be associated with response to immune checkpoint inhibitors in a range of solid tumors35. This may have contributed to the response seen here and needs to be
investigated in other prostate tumors with mismatch repair deficiency. The sequencing and timing of combination therapies may also influence response, and future studies are required to
optimize their roles in enhancing CAR T cell therapy, for example, the use of real-time analyses to provide more dynamic insights into the spectrum of responses across time. In this study,
the action of carboplatin was assessed in immunosuppressed NSG mice, which lack T cells, B cells, and natural killer cells. This model allows us to compare the response of individual patient
tumors to treatment, and thus it is more reflective of tumor heterogeneity seen in the clinic. In addition, a potentially unappreciated benefit of this immunosuppressed model is that it
allows the focus to be on the interaction between the tumor and human CAR T cells without a cognate effector immune response being induced through the murine lymphoid compartment. This lack
of lymphoid cells was advantageous as it did not obscure the major shift we saw in the phenotypes of host myeloid cells, fibroblasts, ECM, and endothelial cells in response to carboplatin.
This is likely to be a critical facet of the mechanism of action and is highly relevant to the human setting. However, this model is limited by the lack of crosstalk between immune cells
within the TME, including the dynamic interaction between immune activation and suppression, and future studies need to be extended into an immunocompetent syngeneic setting. In this study,
we examined the effect of bulk transduced T cells using IL-2; however, future studies should also consider the effectiveness of selected CAR T cell products with defined subsets of T cells
that possess increased memory capability36. Evidence is emerging to show that maintaining CAR T cells in an early, less-differentiated state in vitro results in superior persistence,
proliferation, and anti-tumor effects in vivo36,37. Although effector memory T cells have strong cytolytic capacity, less differentiated T cells, such as stem cell memory T cells, are
critical for in vivo expansion and long-term persistence37,38. LeY is not a predicted target antigen for prostate cancer treatment. Unlike other cell-surface prostate adenocarcinoma targets,
such as prostate stem cell antigen (PSCA), prostate-specific membrane antigen (PSMA), and six-transmembrane epithelial antigen of the prostate (STEAP-1)7,39, LeY is expressed on a broad
spectrum of tumors. In general, expression of PSCA, PSMA and STEAP-1 is limited to AR-positive adenocarcinoma tumors and is rarely detected in AR-null or neuroendocrine prostate cancer,
limiting their clinical application. Here, we demonstrate that LeY expression is detected in both adenocarcinoma and neuroendocrine prostate cancer, and is independent of AR or PSMA, and
thus LeY CAR T cells could be applicable for the treatment of a diverse range of prostate tumors. Here, PDX-224R, a de novo neuroendocrine tumor, had a weaker response to combination therapy
compared to PDX-287R. However, this is more likely dependent on chemotherapy response given that both adenocarcinoma and neuroendocrine tumors were sensitive to LeY CAR T cells in vitro. A
larger panel of PDXs is required to determine whether this response is phenotype dependent. This is clinically relevant as LeY CAR T cells may provide another treatment option for patients
who would not benefit from AR- or PSMA-directed therapies, or who had progressed following these therapies. Overall, carboplatin induces a cascade of changes within the tumor to increase the
efficacy of CAR T cell treatment, with the extent of the response dependent on TME modifications to promote T cell infiltration, activation and persistence. These data provide preclinical
proof-of-concept evidence for the use of carboplatin as a modulating agent for CAR T cell immunotherapy in the treatment of prostate cancer. METHODS PATIENT-DERIVED XENOGRAFTS
Serially-transplantable PDXs were established and characterized by MURAL, as previously described23. Original specimens of localized prostate cancer tissue were collected from patients
undergoing radical prostatectomy or transurethral resection of the prostate, and metastatic prostate cancer tissue was obtained from biopsy, palliative surgery, or rapid autopsy through the
CASCADE program40. Informed, written consent was obtained from patients prior to tissue collection according to human ethics approval from the Cabrini Institute (03-14-04-08), Monash
University (1636), and Peter MacCallum Cancer Centre (15/98, 97_27), and patient details, including age and treatment history, have been previously published23. All PDX experiments were
performed in 6–8-week-old male non-obese diabetic severe-combined immune-deficient (NSG) mice (RRID:IMSR_JAX:005557)41. NSG mice were purpose bred at Monash Animal Research Laboratories
(Monash University, Monash Breeding Colony approval number MMCA 209/25BC and 15160). All animal care and procedures for the establishment and maintenance of PDXs were performed in accordance
with Monash University animal ethics approvals (MARP/2014/085, MARP/2014/119, MARP/2016/016, 17963, 28911). All mice were bred and housed in a PC2-specific pathogen-free facility under
controlled temperature (22 °C) and lighting (12:12 h light-dark cycle), and were fed a chow diet _ad libitum_. Experimental mice and control mice were co-housed. NSG mice either had a 5 mm
testosterone pellet implanted subcutaneously to supplement host testosterone levels, or were castrated to mimic androgen deprivation. Routine validation of PDX authenticity included DNA and
RNA-sequencing, STR profiling and pathological assessment including biomarker expression for androgen receptor (AR), prostate specimen membrane antigen (PSMA) and the neuroendocrine markers
CD56, synaptophysin and chromogranin A, as previously described23. Genomic alterations in PDXs, determined by targeted DNA sequencing, was previously published23. IMMUNOHISTOCHEMISTRY FOR
LEWIS Y LeY expression was analyzed in 709 primary prostate cancer specimens on the Case PSA Progression tissue microarray (obtained from the Prostate Cancer Biorepository Network) and 91
metastatic prostate cancer specimens from 32 patients in our own clinical collection. LeY expression was also analyzed in 49 PDXs obtained from MURAL derived from 14 high-risk localized and
35 metastatic tumors. Detection of the LeY antigen was performed by immunohistochemistry on formalin-fixed tissue using a humanized monoclonal IgG1 antibody m3S193 (1.9 µg/ml; provided by
Ludwig Institute for Cancer Research) on the Dako Autostainer Link 48 with EnVison FLEX, High pH Link visualization system reagents42. The staining protocol was adapted and validated from a
previously published study43. Briefly, antigen retrieval was performed at 10 oC for 20 min, peroxidase blocking for 10 min, antibody or negative control reagent for 30 min at 1.9 µg/ml,
EnVision Flex HRP polymer for 30 min, DAB for 5 min and finally sections were counterstained using Mayer’s Hematoxylin. Normal human lung, NSCLC squamous, and adenocarcinoma control tissues
were used for staining quality control throughout the study. ESTABLISHMENT OF ORGANOIDS FROM PDX TISSUES Organoids were established from PDX tissues as previously described23,44. Fresh or
frozen-thawed single PDX cells were seeded in growth factor reduced, phenol red-free, ldEV-free Matrigel (Corning), with 1–1.5 × 105 cells seeded per 20 µl of Matrigel in 48 well plates.
Organoids were cultured in organoid media, consisting of advanced DMEM/F-12 media (Thermo Fisher) containing 1% penicillin-streptomycin, 2 mM Glutamax, 1 nM DHT, 1.25 mM _N_-acetylcysteine,
50 ng/ml EGF, 500 nM A83-01, 10 mM nicotinamide, 10 μM SB202190 (Sigma), 2% B27 (Life Technologies), 100 ng/ml noggin (Peprotech), 10 ng/ml FGF10 (VWR), 5 ng/ml FGF2, 1 μM prostaglandin E2
(Tocris), and 10% R-spondin 1 conditioned media. 10 μM Y-27632 dihydrochloride (Selleck Chemicals) was added to culture medium during organoid establishment and the first subsequent passage.
To assess LeY expression, organoids were fixed in 10% formalin for at least 1 h, and then embedded in paraffin before performing immunohistochemistry for LeY, as described above. CAR T CELL
PRODUCTION The anti-LeY scFv-CD3ζ-CD28 CAR construct and retroviral packaging cell line PG13-LeY CAR was used in this study as described previously10. Human PBMCs were isolated from normal
donor buffy coats (Australian Red Cross Blood Service) by density gradient centrifugation (Ficoll-Paque, GE Healthcare Life Science). PBMCs were stimulated with anti-human CD3 (OKT3 30
ng/ml, Miltenyi) and 600 units/ml IL-2 (Peprotech) for 48 h and were transduced on day 3 and 4 with viral supernatant produced by PG13-LeY CAR cells on a Retronectin (Takara Bio, Otsu,
Japan) coated 6-well plates as per the manufacturer’s instructions. After 4 h incubation at 37 °C, viral supernatant was removed, and 2.5 × 106 T cells in 5 ml of fresh retroviral
supernatant with IL-2 (600 units/ ml) were added. 24 h after the second transduction, T cells were transferred to fresh RPMI 1640 (Gibco) supplemented with 10% FBS, penicillin/streptomycin,
Glutamax, and 600 units/ml IL-2. The media was changed every two days until day 13. In the LeY-CAR construct, a CD34 epitope was embedded for purification. During the CAR-T cell production,
the transduced T cells were purified using the EasySep™ Human CD34 Positive Selection Kit (STEMCELL Technologies) on day 4–5 post transduction. The purified cells were further expanded for
infusion. The percentage of CAR+ cells in the final product was no <98%. The gating strategy for CAR T cells is shown in Supplementary Fig. 6a. IN VITRO CAR T CELL KILLING ASSAY Once
organoids had reached approximately 50 μM in size, organoids were recovered from Matrigel using Cell Recovery Solution (Corning, USA #354253) for 1 h on ice. 50–100 organoids were plated in
10 µl of Matrigel to form a Matrigel dome in µ-Plate 96 well ibiTreat (ibidi GMbH, Germany #89626) with organoid media. In all, 3 × 104 LeY CAR T cells or control ev-T cells were added to
triplicate organoid wells with 300 units/ml IL-2. Live CAR T/ev-T cells can infiltrate into the dome, but the dead ones are excluded. Propidium iodide (PI, Sigma-Aldrich) was used as an
indicator for cell death (2 μg/ml) and 1% Triton-X100 was used to obtain maximum cell death (positive control). Live cell images were captured by IX83 widefield live-imaging microscope
(Olympus) over a 48 h period. Briefly, 10 fields were imaged per well at ×10 magnification at one time point and 25 time points were taken with 2 h interval. Images were analyzed using
Fiji45 and Bio-formats46. Approximately 50–100 organoids were analyzed individually per well and the MFI of PI of each organoid at each time point was measured. Organoids were gated
frame-by-frame to determine the PI uptake by dead organoid cells. For mechanism study, granzyme B inhibitor Compound 20 (10 μM) and perforin inhibitor SN34960 (20 μM) were used during the
assay. ANALYSIS OF LEY EXPRESSION ON TUMOR CELL LINES To analyze the LeY expression on different prostate cancer cell lines, flow cytometry was used (Supplementary Fig. 6b). Cells were
stained with Live/Dead Near-IR fixable viability dye (Thermo Fisher) for 10 min at 4 °C, then followed by a 30-min surface staining with a murine monoclonal antibody against human LeY (clone
3S193) conjugated with Alex Fluor 488. Cells were washed and fixed in 2% paraformaldehyde then resuspended in FACS buffer before acquisition on a LSR Fortessa (Becton Dickinson). Flow
cytometry data was then analyzed using FlowJo v10.8 software. 51CHROMIUM RELEASE ASSAY IN TUMOR CELL LINES Target cells (LeY+ cell lines DU-145, PC-3, and 22Rv1 and LeY- cell line MDA-MB435)
were labeled with 50 μCi 51Chromium (51Cr, PerkinElmer) for 1 h at 37 °C. The labeled cells were washed and subsequently co-cultured with LeY-CAR-T cells or empty vector-transduced T cells
in triplicate wells at effector to target ratios ranging from 50:1 to 6.25:1. Target cells alone (spontaneous release) and target cells with 10% Triton X-100 (maximum release) served as
controls. After 4 h coculture, supernatants were collected, and the amount of 51Cr released (experimental release) was detected using a gamma counter (Wallac Wizard 1470, PerkinElmer). The %
lysis was calculated by [(experimental release – spontaneous release)/(maximum release – spontaneous release)] × 100. PDX EXPERIMENTS All animal care and procedures were performed in
accordance with Monash University animal ethics approval 20374 and Peter MacCallum Cancer Centre animal ethics approval E647. To assess the long-term in vivo response to LeY CAR T cells,
tissue from PDX-287R and PDX-224R-Cx was established subcutaneously (1 graft/mouse) in male NSG host mice (6–8 weeks old) until tumor volume reached approximately 50–100 mm3. Mice received a
single dose of irradiation (0.5 Gy) for lymphodepletion on day 0, before receiving three doses of 1 × 107 adoptively transferred LeY CAR T cells or empty vector T cells in PBS by
intravenous injection on days 1, 2, and 3. Mice also received eight doses of 50000 IU of IL-2 by intraperitoneal injection on days 1–5, 7, 9, and 11. A subset of host mice were pre-treated
with 200 µg/dose nivolumab (1 dose every 5 days by intraperitoneal injection starting 5 days before day 1 of CAR T cell transfer, 4 doses in total, dissolved in PBS), 10 mg/kg docetaxel (1
dose 7 days before day 1 of CAR T cell transfer, dissolved in 90% PBS with 5% Tween 80 and 5% ethanol), or 50 mg/kg carboplatin (1 dose 7 days before day 1 of CAR T cell transfer, dissolved
in water). The dose and dosing regimen of nivolumab has been used previously in NSG mice and is an equivalent safe dose for humans47,48. The doses of carboplatin and docetaxel are lower
doses than in clinical practice to investigate the modulatory effects of these agents. IL-2 was purchased from Peprotech (Cranbury, New Jersey, USA), and nivolumab, docetaxel and carboplatin
were purchased from Selleck Chemicals (Houston, Texas, USA). Between 5–21 mice were present per treatment group across 1–3 independent experiments (see figure legend). Tumors were measured
three times a week for up to 6 weeks post-adoptive transfer of CAR T cells using calipers. Tumor volume was determined from caliper measurements by length × width × height × 0.52, as
previously described49, and animals were culled when tumor volume reached the maximum ethical limit of 1000 mm3. At tumor collection, animals were placed under isoflurane for blood
collection by cardiac puncture and then humanely killed by cervical dislocation. The investigators involved in treating mice were not blinded to the treatment groups but were blinded to
tumor measurement data. Harvested tumor tissue was formalin-fixed for histological analysis. For early time point experiments, mouse PDXs were established as described above. Mice were
treated with a single dose of 50 mg/kg carboplatin 7 days before the CAR T cell infusion and were irradiated 1 day before infusion. CAR T cells were given on day 1 and day 2 with 50,000
IU/dose IL-2. On day 3, 48 h after the first CAR T cell infusion mice were placed under isoflurane for blood collection by cardiac puncture, and then humanely killed by cervical dislocation.
Blood, spleen, and tumors were collected for further analysis. For mechanistic single-cell RNA-sequencing studies, mice were treated with a single dose of 50 mg/kg carboplatin or vehicle
control and tumor tissues were harvested on day 21. IMMUNOHISTOCHEMISTRY ON PDX TISSUE Immunohistochemistry for phospho-histone H3 was performed on three representative sections per graft
using the Leica BOND-MAXTM automated system (Leica Biosystems, Mount Waverley, Victoria, Australia). The primary antibody was a rabbit antibody against phospho-histone H3 (Ser10) Antibody
(#9701), Research Resource Identifier (RRID): AB_331535 (Cell Signaling Technology) used at 8 µg/ml. The number of phospho-histone H3-positive cells were counted using Aperio ImageScope
Software (Leica Biosystems), with three sections analyzed per graft, and expressed as a percentage of the total number of cells counted. Immunohistochemistry for human CD3, human CD8, and
mouse F4/80 was performed manually. FFPE slides were dewaxed in xylene and antigens were retrieved at 125 °C for 10 min in sodium citrate pH6 buffer (for CD3), Tris EDTA pH9 buffer (for
CD8), or Tris EDTA pH8 buffer (for F4/80). Endogenous peroxidases were inactivated with 3% H2O2 (Merck), and the slides were blocked in PKI blocking buffer (Akoya Biosciences). Primary CD3
antibody (6.7 µg/ml; clone SP7; Abcam #ab16669; RRID AB_443425), CD8 antibody (1:100; clone 4B11; Invitrogen #MA1-80231; RRID AB_929437) or F4/80 antibody (1: 100; polyclone, Abcam #100790;
RRID AB_10675322) was incubated 30 min at room temperature, and the secondary anti-mouse ImmPress (Vector Laboratories, MP-7402) or anti-rabbit ImmPress (Vector Laboratories, MP-7401) was
added for 30 min at RT, followed by Dako Liquid DAB for 15 min. The slides were then counter-stained by Jung Autostainer (Leica) and scanned using the Olympus VS120 microscope at 20×
magnification. FLOW CYTOMETRIC ANALYSIS OF CAR T CELLS To analyze CAR T cells in blood and spleen in vivo, flow cytometry was used for absolute cell counting and cell phenotyping. Blood and
smashed spleen were treated with ACK buffer to remove the red blood cells. Cells were stained with Live/Dead Near-IR fixable viability dye (Thermo Fisher) for 10 min at 4 °C, then followed
by 30-min surface staining for CD3 (1:100; BD Biosciences #564001), CD4 (BioLegend #317438), CD8 (1:100; BD Biosciences # 612889), CAR (as indicated by Flag-tag, 1:200; BioLegend #637310),
and CD137 (1:100; BioLegend #309814), CD25 (1:200; BioLegend #302629). Before analyzing the cells on Symphony (BD Biosciences), cells were mixed with CountBright Absolute Counting Beads
(ThermoFisher) for absolute cell count according to the manufacture’s instruction. Flow cytometry data was then analyzed using FlowJo v10.8 software. The gating strategy is shown in
Supplementary Fig. 6a. ANALYSIS OF TUMOR-INFILTRATING T CELLS To analyze CAR T cells infiltrating tumors by flow cytometry, mice were euthanized 48 h-post first infusion, and tumors were
digested using RPMI-1640 (Gibco) supplemented with 1 mg/ml collagenase type IV (Sigma-Aldrich) and 0.02 mg/ml DNase (Sigma-Aldrich). Tumors were incubated for 1 h at 37 °C, rendered to
single-cell suspension, and then filtered twice (through 70-μm filter). Cells were then analyzed by flow cytometry directly for phenotyping and absolute cell counting using CountBright
Absolute Counting Beads (ThermoFisher). For analysis of cytokines, single-cell suspensions were incubated on anti-idiotype antibody (0.5 μg/ml) or anti-CD3 antibody (1:100; clone OKT3, 0.5
μg/ml) coated-plate overnight with 25 units/ml IL-2 and were treated with GolgiStop (BD Biosciences) for 6 h before intracellular staining for IL-2 (1:50; BioLegend #500348), TNF-α (1:100;
BioLegend #502912) and IFN-γ (1:100; BioLegend #502546). The gating strategy is shown in Supplementary Fig. 6a. ANALYSIS OF PD-L1 ON ORGANOIDS To investigate CAR T cell-induced PD-L1
expression on organoids, cells from PDX-287R organoids were prepared on Matrigel with organoid growth media in 24-well plate as described above. LeY CAR T cells were co-cultured with
organoids for 24 h, then the supernatant was collected as CAR T cell-conditioned media. The fresh organoids or prostate tumor line DU-145 cells (as positive control) were incubated with
either conditioned media or fresh media containing 150 ng/ml human IFN-γ (PeproTech) for 24 h. The cells were trypsinized and resuspended in single-cell suspension for flow cytometry
analysis using anti-PD-L1 antibody (1:100; BioLegend #393608; Supplementary Fig. 6c, d). DISSOCIATION OF PDXS FOR SINGLE-CELL ANALYSIS PDXs were harvested from host mice and cut into 2 × 2
mm pieces using a scalpel. Tumor pieces were digested in RPMI-1640, containing 0.65 U/ml Liberase TM (Roche) and 0.2 mg/ml DNase I (Roche), for 1 hour at 37 °C, following by lysis of red
blood cells (RBCs) using RBC Lysis Buffer (Sigma) for 1 minute. Cells were then resuspended in PBS, 1 mM CaCl2, with 2% FBS and underwent negative selection for viable cells using the Easy
Sep Dead Cell Removal kit (Miltenyi), according to the manufacturer’s protocol. Viable cells were passed through a 30 µM cell strainer (Miltenyi) to remove cell clumps, and then counted
using Trypan blue. Samples with cell viability >80% were resuspended in PBS with 2% BSA and proceeded to single-cell analysis. FLOW CYTOMETRIC ANALYSIS AND FLUORESCENCE-ACTIVATED CELL
SORTING (FACS) OF PDXS AND ORGANOIDS ANALYSIS OF LEWIS Y EXPRESSION ON PDX AND ORGANOIDS PDX-287R organoids were prepared on Matrigel with organoid growth media in 24-well plate as described
above. Organoids were treated with 10 µM carboplatin for 24 or 72 h in triplicate. PDX tissue or treated organoids cultures were digested into single-cell suspensions as above. Cells were
incubated with pre-titrated anti-human Lewis Y-Alexa Fluorochrome 647 (1:50–1:100; murine IgG3 monoclonal antibody, close 3S193) and IgG3-APC isotype control antibodies. Cells were incubated
on ice in the dark for 15 min, washed with FACS buffer, and centrifuged for 5 min at 200 g to collect cells. Cells were filtered into 5 ml round-bottom tubes with strainer cap (Falcon®)
prior to analysis. 100 ng/ml of propidium iodide (Sigma-Aldrich) was used to exclude dead cells. Samples were analyzed on the BD® LSR II Flow Cytometer. At least 2 × 103 live events were
recorded for each file for downstream analysis using FlowJoTM v10.8.1. The gating strategy is shown in Supplementary Fig. 6c. ANALYSIS OF PDXS FOLLOWING CARBOPLATIN TREATMENT PDX-287R and
PDX-224R mice were established in host mice before being treated with a single dose of 50 mg/kg carboplatin or vehicle control. The tumor was harvested 7 days after treatment and digested
into single-cell suspensions as above. Digested cells were incubated with pre-titrated antibodies diluted in staining buffer (1 x PBS with 10% FCS and 5 mM EDTA). Cells were pre-blocked for
5 min on ice with purified rat anti-mouse CD16/CD32 (25 mg/ml; clone 2.4G2, Cat #553141, BD Biosciences, USA) to block murine Fc receptors. Cells were then stained with the following
antibodies (all antibodies are purchased from BioLegend unless otherwise stated); anti-human EpCAM-PECy7 (1:200; clone 9C4), anti-human CD95-BUV395 (4 mg/ml; clone DX2, BD Biosciences),
anti-mouse MHC I-Ak-PE (0.2 mg/ml; clone 10-3.6), anti-mouse F4/80-APC (1 mg/ml; clone BM8), anti-mouse CD45-APCCy7 (1 mg/ml; clone 30-F11), anti-mouse ICAM1-BV421 (2 mg/ml; clone
YN1/1.7.4), anti-mouse CD31-BV510 (2 mg/ml; clone 390, BD Biosciences) or isotype controls; mouse IgG1k-BUV395 (4 mg/ml; clone X40, BD Biosciences), Rat IgG2bk-BV421 (2 mg/ml; clone
RTK4530). Cells were incubated on ice in the dark for 15 min, washed with FACS buffer, and centrifuged for 5 min at 200 g to collect cells. Cells were filtered into 5 ml round-bottom tubes
with strainer cap (Falcon®) prior to analysis. In all, 100 ng/ml of propidium iodide (Sigma-Aldrich) was used to exclude dead cells. Samples were analyzed on the BD® LSR II Flow Cytometer.
At least 2 × 103 live events were recorded for each file for downstream analysis with FlowJoTM v10.8.1. For FACS, samples were sorted on the BD FACSAria™ Fusion Flow Cytometer at no more
than 5 × 103 cells per second through a 100-micron nozzle at 20 psi. The following gating strategy was used to purified cell subsets; debris was excluded via FSC and SSC gate, PI was used to
excluded dead cells and single cells were gated based on FSC-H and FSC-A parameters (Supplementary Fig. 6e). FACS purification of; tumor cells (EpCAM+ CD45-), immune cells (EpCAM- CD45+),
stromal cells (EpCAM-, CD45-, CD31-) and endothelial cells (EpCAM-, CD45-, CD31+). Samples were collected in 30% (v/v) FCS in RPMI, recovered by centrifugation, counted and analyzed for
purity. Populations were sorted to greater than 90% purity. RNA ISOLATION Total RNA from FACS purified subsets were isolated using the RNAqueous Total RNA Isolation Kit (Ambion) according to
the manufacturer’s instruction. Total RNA was quantified in a Nanodrop ND-1000 spectrophotometer, checked for purity and integrity in a Bioanalyzer-2100 device (Agilent Technologies). BULK
RNASEQ ANALYSIS MULTIPLEX RNA-SEQUENCING Samples with a RIN (RNA Integrity Number) \(\ge\) 6, as determined by the Bioanalyzer-2100, were incorporated into the RNA library for associated
downstream Multiplex RNA-sequencing and analysis. The 3’ end of poly(A) primed transcripts were initially hybridized with a custom primer, 8 bp sample index and 10 bp unique molecular
identifier (UMI). Subsequently, during first complementary DNA (cDNA) strand synthesis, a modified reverse transcription reaction facilitated the addition of a template switching sequence to
the 5′ end of the RNA transcripts. The amplified cDNA libraries were subsequently tagmented utilizing PCR transposase. Indexed complementary DNA (cDNA) was selected for and amplified
utilizing Illumina P5 (5′ AAT GAT ACG GCG ACC ACC GA 3′) and P7 (5′ CAA GCA GAA GAC GGC ATA CGA GAT 3′)50. Multiplex RNA-sequencing was performed on the Illumina NextSeq550 by the Medical
Genomics Facility (Monash Health Translation Precinct). MULTIPLEX RNA-SEQUENCING ANALYSIS The raw Fastq files were quality checked using FastQC and low-quality reads were trimmed using
Cutadapt v1.7.1. Trimmed reads were aligned to both human hg38 and mouse mm39 reference genomes using STAR aligner v2.7.5b. XenofilteR v1.6 was used to select Human and mouse specific reads
and counts matrix were generated using HTSeq v0.11.2. EdgeR v3.28 was used for differential expression analysis. The normalized log-transformed counts per million (CPM) was used to calculate
single-sample gene set enrichment analysis score against specific MsigDB signature gene sets using R v4.2.0 package. Differentially expressed genes between treated vs untreated and its
significance (adjusted P-value < 0.05) were calculated using Wald’s T-test method implemented in the DESeq2 R package. SINGLE-CELL RNA TRANSCRIPTOME ANALYSIS (SCRNA-SEQ) SINGLE-CELL
RNA-SEQUENCING scRNA-Seq for dissociated PDXs was performed using the 10X Genomics Chromium Single Cell 3′ Library & Gel bead Kit V3.0, according to the manufacturers protocol (CG000183
Rev C). Briefly, ~5,000 PDX cells were used as input per sample. Cell encapsulation in microfluidic droplets yielded ~4000 recovered single-cell transcriptomes per sample. After reverse
transcription, barcoded-cDNA was purified using SILANE Dynabeads followed by 11 cycles of PCR-amplification. SPRIselect purification was performed on an Agilent Bioanalyzer High Sensitivity
chip to quantitate the fragment size and concentration of the amplified cDNA. Libraries were sequenced on an Illumina NovaSeq6000 with 151 bp paired-end reads. SINGLE-CELL RNA-SEQ ANALYSIS
XenoCell v1.0 was used to align transcripts to the GRCh38 human reference genome and mm10 mouse genome51, mouse-specific cellular barcodes were selected containing a minimum of 90% of
host-specific reads. Extracted mouse cells were then processed using Alevin tool (Salmon Software v1.3.0) to obtain unique molecular identifiers (UMIs; Supplementary Data 8) and generate a
cell by gene count matrix52, which was imported into Seurat (v3.2.0) for downstream analysis53. We excluded outlier cells that expressed in the range of <200–800 genes depending on the
sample type and had unusual gene count, transcript count, and mitochondrial gene fraction, according to sample-specific thresholds (Supplementary Data 8). We also excluded genes expressed in
fewer than 50 cells. Epithelial tumor cells (vehicle sample) identified were further down sampled to 100 cells, similar to the amount of carboplatin sample for all downstream analysis. For
the carboplatin-treated sample, we subsampled to 500 cells, similar to the amount of the vehicle sample. The SCTransform function from Seurat was then used to log-normalize and scale counts
to 10,000 transcripts per cell and detect highly variable features. Principal component analysis (PCA) was performed using the top 3000 most highly variable features and used to cluster
cells by Uniform Manifold Approximation and Projection (UMAP). Cell clusters were identified using Seurat FindClusters function. The ClusterTree (v0.4.3) R package was used to determine the
optimal resolution and number of clusters for each sample54. Expression of selected genes signatures were calculated per-cell using the AddModuleScore function from Seurat. Differentially
expressed markers were identified using Seurat FindMarkers function and significant genes were selected based on the FDR < 0.05. Gene set enrichment analysis was performed using escape
(v1.2.0) enrichIT function in R (v4.1.0)55. Cell type annotation was performed by a differential gene expression analysis with a ROC test using FindAllMarkers function. Markers were ranked
based on Log2Fold change and the ROC power score. Cross reference of the markers and literature was done to annotate each cell type25. Previously identified gene expression markers were used
to define the phenotypes of cells present in the PDX graft. These signature genes were discerned by cross-examining multiple primary research papers detailing markers of M1/M2
polarization26,27,28, the distinctions between an iCAF and myCAF phenotype25,29, and specialization of the endothelium30. STATISTICAL ANALYSIS Linear mixed model analyses were conducted
using SPSS Statistics (Version 27; International Business Machines Corporation, Armonk, New York, United States of America). All other statistical analyses were performed in Prism v9
software (GraphPad). A _p_-value ≤ 0.05 was considered statically significant. Unless otherwise state, data analyses were conducted using unpaired Student’s _t_ test to compare two data sets
or using one-way/two-way ANOVA when analyzing multiple sets of data. Data were presented as mean ± standard error of the mean (SEM). Statistically significant (FDR < 0.05) gene sets were
calculated using escape package (v1.2.0) in R (v4.1.0). REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this
article. DATA AVAILABILITY The bulk RNA-sequencing, and single-cell RNA-sequencing data that support the findings of this study have been deposited in the NCBI dbGaP repository under
accession number: phs003369.v1.p1. Genomic alterations in PDXs, determined by targeted DNA sequencing, was previously published. The curated set of genomic alterations was based on data
downloaded from the cBioPortal resource Prostate Adenocarcinoma [https://www.cbioportal.org/study/summary?id=prad_p1000 and https://www.cbioportal.org/study/summary?id=prad_su2c_2015]. The
remaining data are available within the Article or its Supplementary Information. Source data are available as Source Data File. To request access to MURAL PDXs and/or biospecimens,
researchers should contact Dr. Melissa Papargiris, MURAL Project Manager ([email protected]) to initiate an Expression of Interest. Researchers would need to provide evidence of
institutional approval to experiment with human PDX tumors and research would be conducted under the conditions of a Materials Transfer Agreement. Source data are provided with this paper.
REFERENCES * Gorchakov, A. A., Kulemzin, S. V., Kochneva, G. V. & Taranin, A. V. Challenges and prospects of chimeric antigen receptor T-cell therapy for metastatic prostate cancer.
_Eur. Urol._ 77, 299–308 (2020). CAS PubMed Google Scholar * Park, J. H. et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. _N. Engl. J. Med._ 378, 449–459
(2018). CAS PubMed PubMed Central Google Scholar * Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. _N. Engl. J. Med._ 371, 1507–1517 (2014).
PubMed PubMed Central Google Scholar * Mardiana, S., Solomon, B. J., Darcy, P. K. & Beavis, P. A. Supercharging adoptive T cell therapy to overcome solid tumor-induced
immunosuppression. _Sci. Transl. Med_. 11, https://doi.org/10.1126/scitranslmed.aaw2293 (2019). * Alzubi, J. et al. PSMA-directed CAR T cells combined with low-dose docetaxel treatment
induce tumor regression in a prostate cancer xenograft model. _Mol. Ther. Oncolytics_ 18, 226–235 (2020). CAS PubMed PubMed Central Google Scholar * Schepisi, G. et al. CAR-T cell
therapy: a potential new strategy against prostate cancer. _J. Immunother. Cancer_ 7, 258 (2019). PubMed PubMed Central Google Scholar * Murad, J. P. et al. Pre-conditioning modifies the
TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. _Mol. Ther._ https://doi.org/10.1016/j.ymthe.2021.02.024 (2021). * Tschernia, N. P., Norberg, S. M. &
Gulley, J. L. CAR T cells reach clinical milestone in prostate cancer. _Nat. Med._ 28, 635–636 (2022). CAS PubMed Google Scholar * Jin, Y. et al. Development of STEAP1 targeting chimeric
antigen receptor for adoptive cell therapy against cancer. _Mol. Ther. - Oncolytics_ 26, 189–206 (2022). CAS PubMed PubMed Central Google Scholar * Westwood, J. A. et al. Adoptive
transfer of T cells modified with a humanized chimeric receptor gene inhibits growth of Lewis-Y-expressing tumors in mice. _Proc. Natl Acad. Sci. USA_ 102, 19051–19056 (2005). CAS PubMed
PubMed Central ADS Google Scholar * Myers, R. B., Srivastava, S. & Grizzle, W. E. Lewis Y antigen as detected by the monoclonal antibody BR96 is expressed strongly in prostatic
adenocarcinoma. _J. Urol._ 153, 1572–1574 (1995). CAS PubMed Google Scholar * Culig, Z. et al. Expression of Lewis carbohydrate antigens in metastatic lesions from human prostatic
carcinoma. _Prostate_ 36, 162–167 (1998). CAS PubMed Google Scholar * Ritchie, D. S. et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute
myeloid leukemia. _Mol. Ther._ 21, 2122–2129 (2013). CAS PubMed PubMed Central Google Scholar * Porter, D. L. et al. Chimeric antigen receptor T cells persist and induce sustained
remissions in relapsed refractory chronic lymphocytic leukemia. _Sci. Transl. Med._ 7, 303ra139 (2015). PubMed PubMed Central Google Scholar * Savoldo, B. et al. CD28 costimulation
improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. _J. Clin. Invest._ 121, 1822–1826 (2011). CAS PubMed PubMed Central Google Scholar
* Klampatsa, A. et al. Analysis and augmentation of the immunologic bystander effects of CAR T cell therapy in a syngeneic mouse cancer model. _Mol. Ther. Oncolytics_ 18, 360–371 (2020). CAS
PubMed PubMed Central Google Scholar * Xin, M. et al. Oxaliplatin facilitates tumor-infiltration of T cells and natural-killer cells for enhanced tumor immunotherapy in lung cancer
model. _Anticancer Drugs_ 33, 117–123 (2022). CAS PubMed Google Scholar * Srivastava, S. et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves
antitumor efficacy when combined with checkpoint blockade. _Cancer Cell_ 39, 193–208.e110 (2021). CAS PubMed Google Scholar * Lo, C. S. et al. Neoadjuvant chemotherapy of ovarian cancer
results in three patterns of tumor-infiltrating lymphocyte response with distinct implications for immunotherapy. _Clin. Cancer Res._ 23, 925–934 (2017). CAS PubMed ADS Google Scholar *
Böhm, S. et al. Neoadjuvant chemotherapy modulates the immune microenvironment in metastases of tubo-ovarian high-grade serous carcinoma. _Clin. Cancer Res._ 22, 3025–3036 (2016). PubMed
Google Scholar * Ma, Z. et al. Docetaxel remodels prostate cancer immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. _Theranostics_ 12, 4965–4979 (2022). CAS
PubMed PubMed Central Google Scholar * Zhou, L. et al. Low-dose carboplatin reprograms tumor immune microenvironment through STING signaling pathway and synergizes with PD-1 inhibitors in
lung cancer. _Cancer Lett._ 500, 163–171 (2021). CAS PubMed Google Scholar * Risbridger, G. P. et al. The MURAL collection of prostate cancer patient-derived xenografts enables discovery
through preclinical models of uro-oncology. _Nat. Commun._ 12, 5049 (2021). CAS PubMed PubMed Central ADS Google Scholar * Giuffrida, L. et al. IL-15 preconditioning augments CAR T
cell responses to checkpoint blockade for improved treatment of solid tumors. _Mol. Ther._ 28, 2379–2393 (2020). CAS PubMed PubMed Central Google Scholar * Elyada, E. et al.
Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. _Cancer Discov._ 9, 1102–1123 (2019). CAS PubMed PubMed
Central Google Scholar * Jayasingam, S. D. et al. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and
challenges in routine clinical practice. _Front. Oncol._ 9, 1512 (2019). PubMed Google Scholar * Orecchioni, M., Ghosheh, Y., Pramod, A. B. & Ley, K. Macrophage polarization: different
gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. _Front. Immunol._ 10, 1084 (2019). CAS PubMed PubMed Central Google Scholar *
Jablonski, K. A. et al. Novel markers to delineate murine M1 and M2 macrophages. _PLoS ONE_ 10, e0145342 (2015). PubMed PubMed Central Google Scholar * Sebastian, A. et al. Single-cell
transcriptomic analysis of tumor-derived fibroblasts and normal tissue-resident fibroblasts reveals fibroblast heterogeneity in breast cancer. _Cancers (Basel)_
https://doi.org/10.3390/cancers12051307 (2020). * Asrir, A. et al. Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to PD-1 plus CTLA-4
combination immunotherapy. _Cancer Cell_ 40, 318–334.e319 (2022). CAS PubMed Google Scholar * Palma, J. P., Aggarwal, S. K. & Jiwa, A. Murine macrophage activation after cisplatin or
carboplatin treatment. _Anticancer Drugs_ 3, 665–676 (1992). CAS PubMed Google Scholar * Palma, J. P. & Aggarwal, S. K. Cisplatin and carboplatin mediated release of cytolytic factors
in murine peritoneal macrophages in vitro. _Anticancer Drugs_ 5, 615–622 (1994). CAS PubMed Google Scholar * House, I. G. et al. Macrophage-derived CXCL9 and CXCL10 are required for
antitumor immune responses following immune checkpoint blockade. _Clin. Cancer Res._ 26, 487–504 (2020). CAS PubMed ADS Google Scholar * Yu, E. Y. et al. Pembrolizumab plus docetaxel and
prednisone in patients with metastatic castration-resistant prostate cancer: long-term results from the phase 1b/2 KEYNOTE-365 cohort B study. _Eur. Urol._ 82, 22–30 (2022). CAS PubMed
Google Scholar * Zhao, P., Li, L., Jiang, X. & Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. _J. Hematol.
Oncol._ 12, 54 (2019). PubMed PubMed Central Google Scholar * Meyran, D. et al. Early-phenotype CAR-T cells for the treatment of pediatric cancers. _Ann. Oncol._ 32, 1366–1380 (2021). CAS
PubMed Google Scholar * Meyran, D. et al. TSTEM-like CAR-T cells exhibit improved persistence and tumor control compared with conventional CAR-T cells in preclinical models. _Sci.
Transl. Med._ 15, eabk1900 (2023). CAS PubMed Google Scholar * Xu, Y. et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by
IL-7 and IL-15. _Blood_ 123, 3750–3759 (2014). CAS PubMed PubMed Central Google Scholar * Dorff, T. B. et al. Novel redirected T-cell immunotherapies for advanced prostate cancer. _Clin.
Cancer Res._ https://doi.org/10.1158/1078-0432.CCR-21-1483 (2021). * Alsop, K. et al. A community-based model of rapid autopsy in end-stage cancer patients. _Nat. Biotechnol._ 34, 1010–1014
(2016). CAS PubMed Google Scholar * Shultz, L. D. et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem
cells. _J. Immunol._ 174, 6477–6489 (2005). CAS PubMed Google Scholar * Scott, A. M. et al. Construction, production, and characterization of humanized anti-Lewis Y monoclonal antibody
3S193 for targeted immunotherapy of solid tumors. _Cancer Res._ 60, 3254–3261 (2000). CAS PubMed Google Scholar * Westwood, J. A. et al. The Lewis-Y carbohydrate antigen is expressed by
many human tumors and can serve as a target for genetically redirected T cells despite the presence of soluble antigen in serum. _J. Immunother._ 32, 292–301 (2009). CAS PubMed Google
Scholar * Choo, N. et al. High-throughput imaging assay for drug screening of 3D prostate cancer organoids. _SLAS Discov._ 26, 1107–1124 (2021). CAS PubMed PubMed Central Google Scholar
* Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. _Nat. Methods_ 9, 676–682 (2012). CAS PubMed Google Scholar * Linkert, M. et al. Metadata matters:
access to image data in the real world. _J. Cell Biol._ 189, 777–782 (2010). CAS PubMed PubMed Central Google Scholar * Agrawal, S., Feng, Y., Roy, A., Kollia, G. & Lestini, B.
Nivolumab dose selection: challenges, opportunities, and lessons learned for cancer immunotherapy. _J. Immunother. Cancer_ 4, 72 (2016). PubMed PubMed Central Google Scholar * Meraz, I.
M. et al. An improved patient-derived xenograft humanized mouse model for evaluation of lung cancer immune responses. _Cancer Immunol. Res._ 7, 1267–1279 (2019). CAS PubMed PubMed Central
Google Scholar * Tomayko, M. M. & Reynolds, C. P. Determination of subcutaneous tumor size in athymic (nude) mice. _Cancer Chemother. Pharmacol._ 24, 148–154 (1989). CAS PubMed
Google Scholar * Grubman, A. et al. Transcriptional signature in microglia associated with Abeta plaque phagocytosis. _Nat. Commun._ 12, 3015 (2021). PubMed PubMed Central ADS Google
Scholar * Cheloni, S., Hillje, R., Luzi, L., Pelicci, P. G. & Gatti, E. XenoCell: classification of cellular barcodes in single cell experiments from xenograft samples. _bioRxiv_
https://doi.org/10.1101/679183 (2019). * Srivastava, A., Malik, L., Smith, T., Sudbery, I. & Patro, R. Alevin efficiently estimates accurate gene abundances from dscRNA-seq data. _Genome
Biol._ 20, 65 (2019). PubMed PubMed Central Google Scholar * Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across
different conditions, technologies, and species. _Nat. Biotechnol._ 36, 411–420 (2018). CAS PubMed PubMed Central Google Scholar * Zappia, L. & Oshlack, A. Clustering trees: a
visualization for evaluating clusterings at multiple resolutions. _Gigascience_ https://doi.org/10.1093/gigascience/giy083 (2018). * Borcherding, N. et al. Mapping the immune environment in
clear cell renal carcinoma by single-cell genomics. _Commun. Biol._ 4, 122 (2021). CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We also acknowledge the
members of the Prostate Cancer Research program, the patients, families, and consumers who support our research, and the members of the Melbourne Urological Research Alliance (MURAL). We
wish to thank the Prostate Cancer Biorepository Network (PCBN) for access to the tissue microarray, whose work is supported by the Department of Defense Prostate Cancer Research Program, DOD
Award No W81XWH-18-2-0013, W81XWH-18-2-0015, W81XWH-18-2-0016, W81XWH-18-2-0017, W81XWH-18-2-0018, and W81XWH-18-2-0019 PCRP PCBN. We acknowledge the association with the Bristol-Myers
Squibb (BMS) study #CA209-7TK and thank BMS for providing access to the LeY CAR T cell construct. We also thank Andrew Scott (Oliver Newton-John Cancer Research Institute) for access to the
anti-LeY antibody. This work was supported by Movember and MRFF (Upfront PSMA Prostate Cancer Research Alliance). Fellowship and grant support was obtained from the National Health and
Medical Research Council (GPR, APP1102752: PKD, APP1136680; JAT, APP1102752; RAT, LHP, Ideas grant, APP2011391), Cancer Council Victoria (JAT, LHP, TP834128), Department of Health and Human
Services acting through the Victorian Cancer Agency (RAT; MCRF15023), Prostate Cancer Foundation of Australia (LHP, PIRA YI-0322); and Monash University (LHP, Bridging Fellowship; LHP,
Platform Access Grant PAG2-9067770177). Infrastructure support was obtained from the EJ Whitten Foundation, Movember Foundation (Global Action Plan 1), the Peter and Lyndy White Foundation
and TissuPath Pathology. This research was supported by the Monash University Histology Platform, Monash University Animal Research Laboratories, Monash Biomedicine Discovery Institute
Organoid Program. This research was funded by a Prostate Cancer Research Alliance (PCRA) funded by Movember and the Medical Research Future Fund (MRFF). ProsTIC is additionally supported by
the Prostate Cancer Foundation (PCF). AUTHOR INFORMATION Author notes * These authors contributed equally: L. H. Porter, J. J. Zhu. * These authors jointly supervised this work: R. A.
Taylor, G. P. Risbridger. AUTHORS AND AFFILIATIONS * Prostate Cancer Research Group, Monash Biomedicine Discovery Institute, Cancer Program, Department of Anatomy and Developmental Biology,
Monash University, Clayton, VIC, 3800, Australia L. H. Porter, N. L. Lister & G. P. Risbridger * Cancer Immunology Program, Cancer Research Division, Peter MacCallum Cancer Centre,
Melbourne, VIC, 3000, Australia J. J. Zhu, P. J. Neeson, P. K. Darcy, J. A. Trapani, R. A. Taylor & G. P. Risbridger * Sir Peter MacCallum Department of Oncology, The University of
Melbourne, Parkville, VIC, 3010, Australia J. J. Zhu, S. Keerthikumar, D. L. Goode, R. Quezada Urban, A. Azad, M. S. Hofman, P. J. Neeson, P. K. Darcy, J. A. Trapani, R. A. Taylor & G.
P. Risbridger * Prostate Cancer Research Group, Monash Biomedicine Discovery Institute, Cancer Program, Department of Physiology, Monash University, Clayton, VIC, 3800, Australia S. G.
Harrison & R. A. Taylor * Cancer Research Division, Peter MacCallum Cancer Centre, Melbourne, VIC, 3000, Australia S. Keerthikumar, D. L. Goode & R. Quezada Urban * Computational
Cancer Biology Program, Peter MacCallum Cancer Centre, Melbourne, VIC, 3000, Australia S. Keerthikumar, D. L. Goode & R. Quezada Urban * Department of Pathology, Peter MacCallum Cancer
Centre, Melbourne, Victoria, Australia D. J. Byrne * Department of Medical Oncology, Peter MacCallum Cancer Centre, Melbourne, VIC, 3000, Australia A. Azad * Queensland Bladder Cancer
Initiative, School of Biomedical Science, Faculty of Health, Queensland University of Technology, Brisbane, QLD, 4102, Australia I. Vela * Australian Prostate Cancer Research Center, School
of Biomedical Science, Faculty of Health, Queensland University of Technology, Brisbane, QLD, 4102, Australia I. Vela * Department of Urology, Princess Alexandra Hospital, Brisbane, QLD,
4102, Australia I. Vela * Molecular Imaging and Therapeutic Nuclear Medicine, Peter MacCallum Cancer Centre, Melbourne, VIC, 3000, Australia M. S. Hofman * Prostate Cancer Theranostics and
Imaging Centre of Excellence (ProsTIC), Peter MacCallum Cancer Centre, Melbourne, VIC, 3000, Australia M. S. Hofman, R. A. Taylor & G. P. Risbridger Authors * L. H. Porter View author
publications You can also search for this author inPubMed Google Scholar * J. J. Zhu View author publications You can also search for this author inPubMed Google Scholar * N. L. Lister View
author publications You can also search for this author inPubMed Google Scholar * S. G. Harrison View author publications You can also search for this author inPubMed Google Scholar * S.
Keerthikumar View author publications You can also search for this author inPubMed Google Scholar * D. L. Goode View author publications You can also search for this author inPubMed Google
Scholar * R. Quezada Urban View author publications You can also search for this author inPubMed Google Scholar * D. J. Byrne View author publications You can also search for this author
inPubMed Google Scholar * A. Azad View author publications You can also search for this author inPubMed Google Scholar * I. Vela View author publications You can also search for this author
inPubMed Google Scholar * M. S. Hofman View author publications You can also search for this author inPubMed Google Scholar * P. J. Neeson View author publications You can also search for
this author inPubMed Google Scholar * P. K. Darcy View author publications You can also search for this author inPubMed Google Scholar * J. A. Trapani View author publications You can also
search for this author inPubMed Google Scholar * R. A. Taylor View author publications You can also search for this author inPubMed Google Scholar * G. P. Risbridger View author publications
You can also search for this author inPubMed Google Scholar CONTRIBUTIONS G.P.R. and R.A.T. had full access to all the data in the study and takes responsibility for the integrity of the
data and the accuracy of the data analysis. Concept and design: G.P.R., R.A.T., J.A.T., P.K.D., and P.J.N. Acquisition of data: L.H.P., J.J.Z., N.L.L., S.G.H., S.K., D.L.G., R.Q.U., and
D.J.B. Analysis and interpretation of data: L.H.P., J.J.Z., N.L.L., S.G.H., S.K., D.L.G., R.Q.U., D.J.B., G.P.R., R.A.T., J.A.T., P.K.D., and P.J.N. Drafting of the manuscript: L.H.P.,
J.J.Z., N.L.L., S.G.H., S.K., R.Q.U., G.P.R., R.A.T., J.A.T., P.K.D., and P.J.N. Critical revision of the manuscript for important intellectual content: L.H.P., J.J.Z., N.L.L., S.G.H.,
D.J.B., A.A., I.V., M.S.H, P.J.N., P.K.D., J.A.T., R.A.T., and G.P.R. Statistical analysis: L.H.P., J.J.Z., N.L.L., S.G.H., S.K., and R.Q.U. Obtaining funding: G.P.R., R.A.T., J.A.T., and
L.H.P. Supervision: G.P.R., R.A.T., J.A.T., P.K.D., and P.J.N. Other: none. CORRESPONDING AUTHORS Correspondence to R. A. Taylor or G. P. Risbridger. ETHICS DECLARATIONS COMPETING INTERESTS
G.P.R. and R.A.T. (Research collaborations: Pfizer, Astellas, Zenith Epigenetics, AstraZeneca); P.K.D. (Research funding from Myeloid Therapeutics, Prescient Therapeutics, and Bristol Myers
Squibb); A.A. (Speakers Bureau: Astellas, Janssen, Novartis, Amgen, Ipsen, Bristol Myers Squibb; Merck Serono, Bayer; Honoraria: Astellas, Novartis, Sanofi, AstraZeneca, Tolmar, Telix, Merck
Serono, Janssen, Bristol Myers Squibb, Ipsen, Bayer, Pfizer, Amgen, Noxopharm, Merck Sharpe Dome; Scientific Advisory Board: Astellas, Novartis, Sanofi, AstraZeneca, Tolmar, Pfizer, Telix,
Merck Serono, Janssen, Bristol Myers Squibb, Ipsen, Bayer, Merck Sharpe Dome, Amgen, Noxopharm; Travel + Accommodation: Astellas, Merck Serono, Amgen, Novartis, Janssen, Tolmar, Pfizer;
Investigator Research Funding: Astellas, Merck Serono, AstraZeneca; Institutional Research Funding: Bristol Myers Squibb, AstraZeneca, Aptevo Therapeutics, Glaxo Smith Kline, Pfizer,
MedImmune, Astellas, SYNthorx, Bionomics, Sanofi Aventis, Novartis, Ipsen); M.S.H. (personal fees for lectures or advisory boards: Jannsen, Mundipharma, Astellas, Merck/MSD, Astra Zeneca,
Point Biopharma; Research support paid to institution: Endocyte and Advanced Accelerator Applications, both Novartis companies); I.V. (Honoraria: Astellas, Abbvie, Tolmar, Janssen;
Scientific Advisory Board: Astellas, AstraZeneca, Janssen, Bayer); P.J.N. (Research funding from BMS, Roche Genentech, MSD, Prescient Therapeutics, CRISPR Therapeutics, Allergan, Compugen);
All other authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Yuzhuo Wang and the other, anonymous, reviewer(s) for their contribution
to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published
maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SUPPLEMENTARY DATA 1-8 SOURCE DATA SOURCE DATA FILE RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Porter, L.H., Zhu,
J.J., Lister, N.L. _et al._ Low-dose carboplatin modifies the tumor microenvironment to augment CAR T cell efficacy in human prostate cancer models. _Nat Commun_ 14, 5346 (2023).
https://doi.org/10.1038/s41467-023-40852-3 Download citation * Received: 06 September 2022 * Accepted: 11 August 2023 * Published: 02 September 2023 * DOI:
https://doi.org/10.1038/s41467-023-40852-3 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative