- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Autosomal dominant Alzheimer’s disease (ADAD) is genetically determined, but variability in age of symptom onset suggests additional factors may influence cognitive trajectories.
Although apolipoprotein E (_APOE_) genotype and educational attainment both influence dementia onset in sporadic AD, evidence for these effects in ADAD is limited. To investigate the effects
of _APOE_ and educational attainment on age-related cognitive trajectories in ADAD, we analyzed data from 675 Presenilin-1 E280A mutation carriers and 594 non-carriers. Here we show that
age-related cognitive decline is accelerated in ADAD mutation carriers who also have an _APOE_ e4 allele compared to those who do not and delayed in mutation carriers who also have an _APOE_
e2 allele compared to those who do not. Educational attainment is protective and moderates the effect of _APOE_ on cognition. Despite ADAD mutation carriers being genetically determined to
develop dementia, age-related cognitive decline may be influenced by other genetic and environmental factors. SIMILAR CONTENT BEING VIEWED BY OTHERS EFFECTS OF POLYGENIC RISK FOR ALZHEIMER’S
DISEASE ON RATE OF COGNITIVE DECLINE IN NORMAL AGING Article Open access 24 July 2020 GENETIC EFFECTS ON LONGITUDINAL COGNITIVE DECLINE DURING THE EARLY STAGES OF ALZHEIMER’S DISEASE
Article Open access 06 October 2021 ASSOCIATION OF GENETIC RISK OF ALZHEIMER’S DISEASE AND COGNITIVE FUNCTION IN TWO EUROPEAN POPULATIONS Article Open access 21 February 2025 INTRODUCTION
Presence of the e4 allele of the apolipoprotein E (_APOE_) gene is associated with increased risk for developing sporadic Alzheimer’s disease (AD) and an earlier age of clinical onset than
for individuals without an e4 allele1,2. However, evidence for an effect of _APOE_ e4 genotype on cognitive function in autosomal dominant AD (ADAD) has been limited and inconclusive. ADAD
is genetically determined by mutations on the amyloid precursor protein (_APP_), Presenilin-1 (_PSEN1_), and Presenilin-2 (_PSEN2_) genes3. The largest known kindred with ADAD due to a
single mutation (_PSEN1_ E280A) resides in Antioquia, Colombia. Carriers of this mutation have a median age of onset of mild cognitive impairment at 44 years and of dementia at 49 years4.
Despite the group’s well-characterized trajectory, there is individual variability in disease progression, highlighting the need to identify other genetic and environmental factors which may
influence age-related cognitive decline. The role of _APOE_ e4 in this kindred has been inconclusive. In a previous study of 109 _PSEN1_ E280A mutation carriers, those who had an _APOE_ e4
allele had an earlier age of dementia onset than those who did not5. A subsequent study of 71 carriers in the same kindred found no effect of _APOE_ e4, but found that the presence of the e2
allele was associated with delayed clinical onset by approximately eight years6. Broader investigations including ADAD carriers from multiple families have reported differences between
_APP_ and _PSEN1_ mutations, which may mask _APOE_ effects in combined analyses, but indicate detrimental effects on cognitive performance and decline in _PSEN1_ carriers7,8, demonstrating
the importance of additional investigation in a large single kindred. In addition, environmental factors (such as lifestyle, health, and socioeconomic conditions) may influence age-related
cognitive trajectories and mitigate genetic risk9,10,11. One such factor is education (often defined as years of formal educational attainment), which has been identified as an important
modifiable factor for dementia delay and prevention12. Higher educational attainment has been associated with slowed cognitive decline in older adults13 and lower dementia incidence14,
indicating educational attainment promotes cognitive resilience in the face of pathology15. The reported impact of educational attainment in ADAD, however, is inconsistent. Lower educational
attainment was a predictor of cognitive decline in ADAD due to various mutations7 and of earlier clinical onset in _PSEN1_ E280A carriers16. Unexpectedly, however, lower educational
attainment (less than three years) has also been associated with later onset of dementia in carriers of the _PSEN1_ E280A mutation5. Of note, ruralness was independently associated with both
lower educational attainment and later age of onset in that sample5, which may have contributed to the observed relationship. There have been no reported significant interactive effects
between _APOE_ genotype and educational attainment in ADAD to date. Additional investigation in this kindred is required to clarify these discrepant findings. The role of genetic and
environmental factors impacting age-related cognitive decline in ADAD are critical to further understand disease progression and to support future prevention and treatment goals. In this
study, we aimed to evaluate the influence of _APOE_ genotype on cognitive function in 675 _PSEN1_ E280A carriers and 594 non-carrier family members, and secondarily explore whether
educational attainment may be protective and moderate the relationship between _APOE_ and cognitive function. We hypothesized that presence of the e4 allele would be associated with
accelerated onset of cognitive impairment, presence of the e2 allele would be associated with delayed cognitive impairment, and that higher educational attainment would be protective against
cognitive impairment. Consistent with our hypotheses, in this work we show that the onset of age-related cognitive decline is accelerated in _PSEN1_ E280A mutation carriers who are also
_APOE_ e4+ compared to those who are _APOE_ e4− and delayed in those who are _APOE_ e2+ compared to _APOE_ e2−. Further, we find that educational attainment is protective and moderates the
effect of _APOE_ on cognition in _PSEN1_ carriers. RESULTS SAMPLE CHARACTERISTICS Sample characteristics are presented in Table 1. Of the 675 _PSEN1_ E280A mutation carriers, 141 were _APOE_
e4+ and 534 were _APOE_ e4−. The _APOE_ e4+ and e4− _PSEN1_ E280A mutation carriers did not differ in age (_p_ = 0.64) or sex (_p_ = 0.89), and they did not differ in MMSE score (_p_ =
0.52) when collapsing across age. _PSEN1_ E280A mutation carriers who were also _APOE_ e4+ had on average more years of educational attainment than those who were _APOE_ e4− (_p_ = 0.02). Of
the 594 _PSEN1_ E280A mutation non-carriers, 148 were _APOE_ e4+ and 446 were _APOE_ e4−. Within these non-carriers, _APOE_ e4+ and e4− individuals did not differ in age (_p_ = 0.42), sex
(_p_ = 0.23), educational attainment (_p_ = 0.42), or MMSE score (_p_ = 0.82). AGE-RELATED COGNITIVE FUNCTION BY _PSEN1_ AND _APOE_ E4 GENOTYPE We first estimated the age-related trajectory
of cognitive impairment, measured through MMSE total score, using the Hamiltonian Markov chain Monte Carlo method in _PSEN1_ E280A mutation carriers and non-carriers, irrespective of APOE
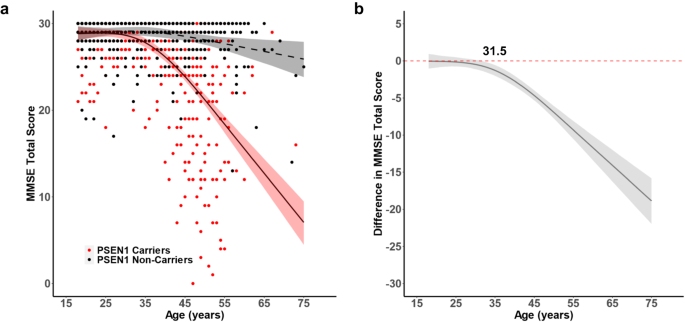
genotype. MMSE was negatively associated with age in _PSEN1_ carriers and significantly differentiated carriers from non-carriers at 31.5 years (Fig. 1). We then estimated the age-related
trajectory of cognitive impairment as a function of _APOE_ e4 genotype separately in _PSEN1_ E280A mutation carriers and non-carriers. The cognitive trajectories of _APOE_ e4+ and e4−
_PSEN1_ E280A mutation carriers diverged at 44.3 years, approximately the median age of onset of mild cognitive impairment in this kindred4 (Fig. 2A, B). In contrast, the age-related
cognitive trajectories of _APOE_ e4+ and e4− _PSEN1_ E280A mutation non-carriers did not diverge (Fig. 2C, D). To supplement these analyses, age of clinical onset was compared in a subset of
_PSEN1_ mutation carriers who had converted to MCI or dementia. Consistent with the prior findings, _PSEN1_ mutation carriers who were also _APOE_ e4+ had earlier ages of clinical onset
compared to those who were _APOE_ e4− (Supplementary Table 1). ROLE OF EDUCATIONAL ATTAINMENT ON COGNITION IN _PSEN1_ E280A CARRIERS Years of educational attainment was examined as a
protective and potentially modifying factor of the relationship between _APOE_ e4 and cognitive function using separate linear regressions for _PSEN1_ E280A mutation carriers and
non-carriers. Within _PSEN1_ E280A mutation carriers, being _APOE_ e4+ was associated with lower MMSE scores compared to _APOE_ e4− (_β_ = −3.37, _p_ = 0.001). Irrespective of _APOE_
genotype, higher educational attainment was associated with higher MMSE scores (_β_ = 0.41, _p_ < 0.001). There was also a significant interaction between _APOE_ e4 and years of
educational attainment (_β_ = 0.32, _p_ = 0.005; Fig. 3a) such that the negative effect of _APOE_ e4+ was attenuated as years of educational attainment increased. In other words, higher
levels of educational attainment mitigated the additional risk conferred by the presence of at least one e4 allele in _PSEN1_ E280A carriers. In non-carriers of the _PSEN1_ E280A mutation,
there was no significant effect of _APOE_ e4 on MMSE score (_β_ = −0.17, _p_ = 0.66), but there was a significant main effect of educational attainment (_β_ = 0.16, _p_ < 0.001) such that
higher educational attainment was associated with higher MMSE scores. There was no interaction between _APOE_ e4 and educational attainment (_β_ = −0.003, _p_ = 0.94). AGE-RELATED COGNITIVE
FUNCTION BY _PSEN1_ AND _APOE_ E2 GENOTYPE Our findings suggest that within _PSEN1_ E280A carriers, age-related cognitive decline begins earlier in those who are _APOE_ e4+ than for those
with other _APOE_ genotypes, including those who are homozygous e3 and e2+. Because presence of the e2 allele has been associated with delayed clinical onset in this kindred6, we sought to
examine the association between _APOE_ e2 and cognition in our current sample. Of the 675 _PSEN1_ E280A mutation carriers, 102 were _APOE_ e2+ and 573 were _APOE_ e2− (Supplementary Table
2). The _APOE_ e2+ and e2− _PSEN1_ E280A mutation carriers did not differ in age (_p_ = 0.20), sex (_p_ = 0.65), educational attainment (_p_ = 0.69), or MMSE score (_p_ = 0.37). Of the 594
_PSEN1_ E280A mutation non-carriers, 73 were _APOE_ e2+ and 521 were _APOE_ e2−, and _APOE_ e2+ and e2− individuals did not differ in age (_p_ = 0.47), sex (_p_ = 0.12), educational
attainment (_p_ = 0.70), or MMSE score (_p_ = 0.66). We first estimated the age-related trajectory of cognitive impairment as a function of _APOE_ e2 genotype separately in _PSEN1_ E280A
mutation carriers and non-carriers (Supplementary Fig. 1a, b). The cognitive trajectories of _APOE_ e2+ and e2− _PSEN1_ E280A mutation carriers diverged at 41.1 years, such that _APOE_ e2+
_PSEN1_ carriers had delayed age-related global cognitive decline. Age of clinical onset of the MCI and dementia converters did not significantly differ between _APOE_ e2+ and e2− genotypes,
but the trends were in the hypothesized direction such that _APOE_ e2+ mutation carriers had on average later clinical onset (Supplementary Table 1). In _PSEN1_ E280A mutation non-carriers,
the age-related cognitive trajectories of _APOE_ e2+ and e2− individuals did not diverge (Supplementary Fig. 1c, d). We then assessed the relationships between _APOE_ e2 genotype and
educational attainment on global cognitive function. Within _PSEN1_ E280A mutation carriers, there was a main effect of genotype, such that those who were _APOE_ e2− had lower MMSE scores
than those who were _APOE_ e2+ (_ß_ = −2.78, _p_ = 0.007). Across _APOE_ genotype, higher educational attainment was associated with higher MMSE scores (_ß_ = 0.26, _p_ = 0.018).
Additionally, there was a significant interaction between _APOE_ e2 genotype and educational attainment, ß = 0.24, _p_ = 0.046 (Fig. 3b), such that higher educational attainment attenuated
the negative effect of being _APOE_ e2−. DISCUSSION _APOE_ e4 has long been associated with increased risk and earlier age of onset for sporadic AD1,2,17,18,19,20. A rare variant on the
_APOE_ e3 allele was found to delay onset of MCI by three decades in a _PSEN1_ E280A carrier21, yet the evidence linking the more common e2 and e4 variants has been mixed5,6,22. Our findings
in over 1,000 participants from a single kindred show an added effect of _APOE_ genotype in carriers of the _PSEN1_ E280A mutation for ADAD, such that _APOE_ e4+ _PSEN1_ mutation carriers
had accelerated onset of age-related cognitive decline compared to _APOE_ e4− _PSEN1_ mutation carriers. The age-related trajectory of clinical impairment diverged between _APOE_ e4+ and e4−
_PSEN1_ mutation carriers around age 44, approximately the median age of onset of mild cognitive impairment in this kindred4. Our results are consistent with a prior study reporting a
detrimental effect of _APOE_ e4+ genotype in _PSEN1_ E280A mutation carriers5. A subsequent study of 71 _PSEN1_ E280A mutation carriers found no effect of _APOE_ e4 genotype, but the
relationship was in the expected negative direction6, suggesting the study may have been underpowered to detect an effect. Conversely, we found that _PSEN1_ E280A mutation carriers who were
_APOE_ e2+ had delayed onset of age-related cognitive decline, replicating a prior finding from this kindred6. The _APOE_ e2 allele has also been associated with delayed onset and protection
against cognitive decline in older adults20,23,24. More research is needed to determine how these genetic risk factors contribute to earlier cognitive decline in ADAD. Both _APOE_ e4 and
_PSEN1_ mutations influence accumulation of β-amyloid (Aβ) pathology in the brain25,26,27. This genetic combination may result in earlier or higher pathological burden, but future studies
will need to consider age-related trajectories of brain pathology in addition to clinical impairment. Since other mutations causing ADAD also alter Aβ production, similar results might be
expected in other ADAD mutations. However, the extent to which these results generalize to ADAD caused by other mutations is uncertain. A study of _APP_ and _PSEN1_ mutation carriers from
six families found no overall effect of _APOE_ e4 on cognitive decline, but in a direct comparison of age-related cognitive decline of _APOE_ e4+ _APP_ and _PSEN1_ carriers, the _APOE_ e4+
_PSEN1_ carriers had a steeper decline than the _APOE_ e4+ _APP_ carriers7. Additional questions remain about the nuances of _APOE_ genotype in ADAD. It is currently unknown whether the risk
is greater in homozygous _APOE_ e4 ADAD mutation carriers than in heterozygous _APOE_ e4 ADAD mutation carriers, as is observed in sporadic AD2. This question is particularly challenging to
answer in ADAD given the small percentage of homozygous _APOE_ e4 carriers coupled with the small population of ADAD mutation carriers. Observational evidence from a sample of 17 carriers
of an _APP_ mutation supports this notion, such that homozygous _APOE_ e4 mutation carriers had the earliest age of onset, followed by the heterozygous _APOE_ e4 mutation carriers, and
finally the _APOE_ e2 mutation carriers with the latest onset28. In a comparison of early- and late-onset AD, _APOE_ e4 genotype was associated with accelerated cognitive decline in both
groups29. Coupled with the current results, these findings provide converging evidence that _APOE_ may have similar effects in sporadic and autosomal dominant AD. Furthermore, in sporadic
AD, _APOE_ e4 has been associated with better cognitive performance and differing neural activity during young adulthood30,31,32, reflecting possible antagonistic pleiotropy of this genetic
risk factor33,34. Structurally, _APOE_ e4+ adults show greater parahippocampal thickness than _APOE_ e4− adults35 and differences in white matter integrity that may relate to observed
cognitive benefits36. Our study only considered age-related cognitive trajectories in adult carriers; however, it is possible that _APOE_ e4 may provide some biological or cognitive benefit
in younger _PSEN1_ carriers (i.e., childhood, akin to young adults in sporadic AD). In fact, _PSEN1_ carriers in this kindred have greater cortical thickness in childhood than non-carriers,
followed by atrophy in adulthood37. Studying the influence of both _APOE_ and _PSEN1_ across the lifespan will enhance our knowledge of the effects of these genes, and, hopefully, provide
mechanisms for disease prevention and treatment in the future. Despite the additional risk conferred by _APOE_ e4, our results suggest that educational attainment may be a critical mechanism
of cognitive reserve in ADAD, as previously shown in sporadic AD13,14,15. Higher educational attainment was related to higher global cognition in _PSEN1_ carriers and mitigated the
cognitive impairment associated with _APOE_ e4. Prior studies in this kindred have found opposing results, including one in which more years of educational attainment was associated with
delayed clinical onset16, and one in which higher educational attainment (defined categorically as greater than three years) was associated with lower cognition5 in _PSEN1_ mutation
carriers. The conflicting results may arise from differences in the samples’ average years of educational attainment, the treatment of educational attainment as a continuous versus
categorical variable, or from confounding variables influencing these relationships. In the prior study reporting a detrimental effect of educational attainment, low educational attainment
was highly correlated with ruralness, which similarly was associated with later clinical onset5. Ruralness, then, may reflect protective factors (e.g., physical activity, environment) that
explain the reported positive association of low educational attainment. We similarly found that ruralness was associated with fewer years of educational attainment in our sample, but we did
not find an association between ruralness and global cognition (see Supplementary Analysis). The _PSEN1_ carriers and non-carriers of our sample are members of the same families, providing
a high degree of environmental matching, although additional variables explaining quality rather than quantity of years of educational attainment may contribute to our findings and will be
important to consider in future work. Many studies examining education effects in sporadic AD include older adult populations with high levels of educational attainment, with averages often
greater than high school or college. This is not representative of the broader population, and there are many conflicting findings in the role of educational attainment on cognitive reserve
in older adults13,14,38,39. In contrast, our sample included a broad range of educational attainment. Our results indicate that low levels of formal educational attainment, in particular,
confers greater risk. As such, programs to increase early years of education may be particularly important as preventative measures, supported by the inclusion of education as one of the 12
modifiable risk factors for dementia in the most recent Lancet commission12. The factors contributing to higher versus lower educational attainment (e.g., socioeconomic status, occupational
attainment) as well as the underlying biological mechanisms of this _APOE_-educational attainment interaction require further study, particularly since higher levels of educational
attainment have been associated with lower Aβ in ADAD40. A primary limitation of this study is the reliance of single time-point data. Although our participants spanned a broad range of
ages, and age is highly linked to clinical progression in this kindred, we cannot speak to individual trajectories of decline with these data. Analysis of longitudinal cognitive decline will
further clarify whether _APOE_ influences age of onset, rate of decline, or both. Additionally, despite having one of the largest sample sizes compared to prior literature in ADAD, our
study was underpowered to assess potential gene dose-dependent effects of the _APOE_ e4 or e2 alleles (see Supplementary Fig. 2 for visualization of age-related cognitive trajectories).
Finally, educational attainment is not the sole environmental factor influencing cognition and clinical progression. More work is needed to further understand the impact of other lifestyle
and modifiable factors along with their interactions with genetic makeup. Together, our results highlight the importance of studying additional genetic and environmental risk factors in ADAD
populations, with critical implications for future disease prevention and interventions. Future studies can push these questions forward by investigating the biological basis for the
additive risk of _APOE_ e4 and _PSEN1_ mutations and for the protective role of _APOE_ e2, and for the aspects and length of educational attainment that can support cognitive function or
reduce the risk of dementia. Inclusion of blood-based biomarkers in such studies characterizing disease progression is necessary to increase access in these populations at risk and to
understand the biological mechanisms underlying these findings41,42,43. The answers to these questions will inform how to best implement educational interventions in various communities and
whether continuing late life education may provide additional protection. These answers are critical as Aβ- and _APOE_-based treatments for AD are being investigated44,45. In conclusion, our
results demonstrate that (1) age-related changes in global cognitive function may be accelerated in ADAD mutation carriers who are also _APOE_ e4+ compared to those who are _APOE_ e4−; (2)
age-related changes in global cognitive function may be delayed in ADAD mutation carriers who are also _APOE_ e2+ compared to those who are _APOE_ e2−; and (3) higher educational attainment
may have a protective effect against cognitive impairment, even in the presence of strong genetic risk factors. METHODS Study procedures were approved by the Institutional Review Board of
the University of Antioquia in Colombia (21-10-605) and were performed in accordance with the ethical standards of the Declaration of Helsinki. All participants provided informed consent
prior to the initiation of study procedures. Participants were compensated for their participation in accordance with the approved guidelines. STUDY DESIGN AND PARTICIPANTS This
cross-sectional study included participants over the age of 18 recruited from the Alzheimer’s Prevention Initiative (API) registry of ADAD, which includes more than 6000 living members of a
kindred with a high prevalence of carriers of the _PSEN1_ E280A mutation (approximately 1200 individuals)46. All members of the registry reside in Colombia and have a parent with the _PSEN1_
E280A mutation but are blind to their own genetic status. In total, 675 _PSEN1_ E280A mutation carriers (370 female, 305 male) and 594 mutation non-carriers (332 female, 262 male) were
included in these analyses. Neuropsychological assessments were performed at the University of Antioquia in Colombia. Participants completed a clinical interview and the Mini Mental State
Examination (MMSE), administered in Spanish, used as a proxy for cognitive impairment. Cognitive data were stored using REDCap (v. 13.1.29). Investigators were blind to participant genetic
status during data collection. GENOTYPING Genomic DNA was extracted from the blood by standard protocols, and _PSEN1_ E280A characterization was done at the University of Antioquia using
methods previously described47. Genomic DNA was amplified with the primers _PSEN1_-S 5′ AACAGCTCAGGAGAGGAATG 3′ and PSEN1-AS 5′ GATGAGACAAGTNCCNTGAA 3′. We used the restriction enzyme _Bsm_I
for restriction fragment length polymorphism analysis. Each participant was classified as a _PSEN1_ E280A carrier or non-carrier. _APOE_ genotyping was performed using a Kompetitive Allele
Specific PCR–KASPTM assay48 (LGV Genomics, Beverly, MA). Due to low numbers of homozygous e4 carriers and homozygous e2 carriers (see Table 2), each participant was classified based on the
presence of at least one e4 allele (e4+) or no e4 alleles (e4−), and separately, based on the presence of at least one e2 allele (e2+) or no e2 alleles (e2−). Twenty-nine participants (13
_PSEN1_ carriers and 16 non-carriers) were _APOE_ e2/e4 and, therefore, included in both _APOE_ e4+ and _APOE_ e2+ groups. STATISTICAL ANALYSIS All analyses were conducted in R version
4.2.0, modeled separately for _PSEN1_ E280A carriers and non-carriers. Group differences in continuous variables were assessed using Mann–Whitney _U_ tests due to non-normality, and
dichotomous variables were compared using chi-square tests. _APOE_ genotype was included in analyses as a dichotomous variable. Cognitive impairment was measured through MMSE total score
(maximum score = 30). Age-related trajectories were derived from cross-sectional MMSE scores modeled using a restricted cubic spline model. Model parameters were estimated using a
Hamiltonian Markov chain Monte Carlo method to compare group trajectories (prior = Cauchy distribution, chains = 8, iterations = 10,000, thin = 10). Linear regression was used to estimate
the effect of educational attainment on cognition, with MMSE total score as the dependent variable and _APOE_genotype, educational attainment, and their interaction term as predictors.
Educational attainment was included as a continuous variable, representing self-reported total years of formal educational attainment. Self-reported sex was collected for each participant
and is presented in demographic tables. Sex was not included in statistical analyses due to no a priori hypotheses about sex differences and sample size limitations of subdividing the
participants by _PSEN1_ genotype, _APOE_ genotype, and sex; however, the proportions of males and females were roughly similar, and there were no differences in sex distributions in the
comparison groups of interest, thus we believe results are generalizable to both males and females. REPORTING SUMMARY Further information on research design is available in the Nature
Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Anonymized clinical, cognitive and genetic data are available upon request, subject to an internal review by F.L., and
Y.T.Q. to ensure that participant confidentiality and _PSEN1_ E280A carrier or non-carrier status are protected, completion of a data sharing agreement and in accordance with the University
of Antioquia’s and MGH’s institutional review board and institutional guidelines. Please submit requests for participant-related data to Y.T.Q. ([email protected]). Source data are
provided with this paper. REFERENCES * Roses, A. D. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. _Annu. Rev. Med._ 47, 387–400 (1996). CAS PubMed Google Scholar *
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. _Science_ 261, 921–923 (1993). ADS CAS PubMed Google Scholar
* Bagyinszky, E., Youn, Y. C., An, S. & Kim, S. The genetics of Alzheimer’s disease. _Clin. Interv. Aging_ 9, 535 (2014). PubMed PubMed Central Google Scholar * Acosta-Baena, N. et
al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. _Lancet Neurol._ 10, 213–220 (2011). CAS PubMed Google
Scholar * Pastor, P. et al. Apolipoprotein Eε4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. _Ann. Neurol._ 54, 163–169 (2003). CAS PubMed Google Scholar * Vélez, J. I. et
al. APOE∗E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. _Mol. Psychiatry_ 21, 916–924 (2016). PubMed Google Scholar * Almkvist, O., Johansson, C., Laffita‐Mesa, J.,
Thordardottir, S. & Graff, C. APOE ε4 influences cognitive decline positively in APP and negatively in PSEN1 mutation carriers with autosomal‐dominant Alzheimer’s disease. _Eur. J.
Neurol._ 29, 3580–3589 (2022). PubMed PubMed Central Google Scholar * Almkvist, O. & Graff, C. The APOE ε4 allele affects cognitive functions differently in carriers of APP mutations
compared to carriers of PSEN1 mutations in autosomal-dominant Alzheimer’s disease. _Genes_ 12, 1954 (2021). CAS PubMed PubMed Central Google Scholar * Van Duijn, C. M. et al. Interaction
between genetic and environmental risk factors for Alzheimer’s disease: A reanalysis of case-control studies. _Genet. Epidemiol._ 11, 539–551 (1994). PubMed Google Scholar * Majoka, M. A.
& Schimming, C. Effect of social determinants of health on cognition and risk of Alzheimer disease and related dementias. _Clin. Ther._ 43, 922–929 (2021). PubMed Google Scholar *
Edwards III, G. A., Gamez, N., Escobedo Jr., G., Calderon, O. & Moreno-Gonzalez, I. Modifiable risk factors for Alzheimer’s disease. _Front. Aging Neurosci_. 11,
https://doi.org/10.3389/fnagi.2019.00146 (2019). * Livingston, G. et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. _Lancet_ 396, 413–446 (2020).
PubMed PubMed Central Google Scholar * Christensen, H. et al. Education and decline in cognitive performance: compensatory but not protective. _Int. J. Geriatr. Psychiatry_ 12, 323–330
(1997). CAS PubMed Google Scholar * Meng, X. & D’Arcy, C. Education and dementia in the context of the cognitive reserve hypothesis: a systematic review with meta-analyses and
qualitative analyses. _PLoS ONE_ 7, e38268 (2012). ADS CAS PubMed PubMed Central Google Scholar * Tucker, A. M. & Stern, Y. Cognitive reserve in aging. _Curr. Alzheimer Res._ 8,
354–360 (2011). CAS PubMed PubMed Central Google Scholar * Aguirre-Acevedo, D. C. et al. Cognitive decline in a colombian kindred with autosomal dominant Alzheimer disease. _JAMA
Neurol._ 73, 431 (2016). PubMed PubMed Central Google Scholar * William Rebeck, G., Reiter, J. S., Strickland, D. K. & Hyman, B. T. Apolipoprotein E in sporadic Alzheimer’s disease:
allelic variation and receptor interactions. _Neuron_ 11, 575–580 (1993). Google Scholar * Poirier, J. et al. Apolipoprotein E polymorphism and Alzheimer’s disease. _Lancet_ 342, 697–699
(1993). CAS PubMed Google Scholar * Ashford, J. W. APOE genotype effects on Alzheimer’s disease onset and epidemiology. _J. Mol. Neurosci._ 23, 157–166 (2004). CAS PubMed Google Scholar
* Reiman, E. M. et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. _Nat. Commun._ 11, 667 (2020). ADS CAS
PubMed PubMed Central Google Scholar * Arboleda-Velasquez, J. F. et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. _Nat.
Med._ 25, 1680–1683 (2019). CAS PubMed PubMed Central Google Scholar * Van Broeckhoven, C. et al. APOE genotype does not modulate age of onset in families with chromosome 14 encoded
Alzheimer’s disease. _Neurosci. Lett._ 169, 179–80 (1994). PubMed Google Scholar * Suri, S., Heise, V., Trachtenberg, A. J. & Mackay, C. E. The forgotten APOE allele: a review of the
evidence and suggested mechanisms for the protective effect of APOE ɛ2. _Neurosci. Biobehav. Rev._ 37, 2878–2886 (2013). CAS PubMed Google Scholar * Corder, E. H. et al. Protective effect
of apolipoprotein E type 2 allele for late onset Alzheimer disease. _Nat. Genet._ 7, 180–184 (1994). CAS PubMed Google Scholar * Lemere, C. A. et al. The E280A presenilin 1 Alzheimer
mutation produces increased Aβ42 deposition and severe cerebellar pathology. _Nat. Med._ 2, 1146–1150 (1996). CAS PubMed Google Scholar * Wildsmith, K. R., Holley, M., Savage, J. C.,
Skerrett, R. & Landreth, G. E. Evidence for impaired amyloid β clearance in Alzheimer’s disease. _Alzheimer’s Res. Ther_. 5, 33 (2013). * Ye, S. et al. Apolipoprotein (apo) E4 enhances
amyloid β peptide production in cultured neuronal cells: ApoE structure as a potential therapeutic target. _Proc. Natl Acad. Sci. USA_ 102, 18700–18705 (2005). ADS CAS PubMed PubMed
Central Google Scholar * Sorbi, S. et al. Epistatic effect of APP717 mutation and apolipoprotein E genotype in familial Alzheimer’s disease. _Ann. Neurol._ 38, 124–127 (1995). CAS PubMed
Google Scholar * Polsinelli, A. J. et al. APOE ε4 carrier status and sex differentiate rates of cognitive decline in early‐ and late‐onset Alzheimer’s disease. _Alzheimer’s Dement_.
https://doi.org/10.1002/alz.12831 (2022). * Evans, S. et al. Cognitive and neural signatures of the APOE E4 allele in mid-aged adults. _Neurobiol. Aging_ 35, 1615–1623 (2014). CAS PubMed
PubMed Central Google Scholar * Rusted, J. M. et al. APOE e4 polymorphism in young adults is associated with improved attention and indexed by distinct neural signatures. _Neuroimage_ 65,
364–373 (2013). CAS PubMed Google Scholar * Dennis, N. A. et al. Temporal lobe functional activity and connectivity in young adult APOE ɛ4 carriers. _Alzheimer’s Dement._ 6, 303–311
(2010). Google Scholar * Tuminello, E. R. & Han, S. D. The apolipoprotein e antagonistic pleiotropy hypothesis: review and recommendations. _Int. J. Alzheimers Dis._ 2011, 726197
(2011). PubMed PubMed Central Google Scholar * Gharbi-Meliani, A. et al. The association of APOE ε4 with cognitive function over the adult life course and incidence of dementia: 20 years
follow-up of the Whitehall II study. _Alzheimers Res. Ther._ 13, 5 (2021). CAS PubMed PubMed Central Google Scholar * Dowell, N. G. et al. Structural and resting-state MRI detects
regional brain differences in young and mid-age healthy APOE-e4 carriers compared with non-APOE-e4 carriers. _NMR Biomed._ 29, 614–624 (2016). CAS PubMed Google Scholar * Dowell, N. G. et
al. MRI of carriers of the apolipoprotein E e4 allele-evidence for structural differences in normal-appearing brain tissue in e4+ relative to e4- young adults. _NMR Biomed._ 26, 674–82
(2013). PubMed Google Scholar * Fox‐Fuller, J. T. et al. Cortical thickness across the lifespan in a Colombian cohort with autosomal‐dominant Alzheimer’s disease: a cross‐sectional study.
_Alzheimer’s Dement._ 13, e12233 (2021). Google Scholar * Wilson, R. S. et al. Education and cognitive reserve in old age. _Neurology_ 92, e1041–e1050 (2019). PubMed PubMed Central Google
Scholar * Sharp, E. S. & Gatz, M. Relationship between education and dementia. _Alzheimer Dis. Assoc. Disord._ 25, 289–304 (2011). PubMed PubMed Central Google Scholar * Gonneaud,
J. et al. Association of education with Aβ burden in preclinical familial and sporadic Alzheimer disease. _Neurology_ 95, e1554–e1564 (2020). CAS PubMed PubMed Central Google Scholar *
Leuzy, A., Cullen, N. C., Mattsson-Carlgren, N. & Hansson, O. Current advances in plasma and cerebrospinal fluid biomarkers in Alzheimer’s disease. _Curr. Opin. Neurol._ 34, 266–274
(2021). CAS PubMed Google Scholar * Telser, J., Risch, L., Saely, C. H., Grossmann, K. & Werner, P. P-tau217 in Alzheimer’s disease. _Clin. Chim. Acta_ 531, 100–111 (2022). CAS
PubMed Google Scholar * Aguillon, D. et al. Plasma p-tau217 predicts in vivo brain pathology and cognition in autosomal dominant Alzheimer’s disease. _Alzheimers Dement_.
https://doi.org/10.1002/alz.12906 (2022). * Wisniewski, T. & Drummond, E. APOE-amyloid interaction: therapeutic targets. _Neurobiol. Dis._ 138, 104784 (2020). CAS PubMed PubMed Central
Google Scholar * Williams, T., Borchelt, D. R. & Chakrabarty, P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer’s disease. _Mol. Neurodegener._ 15, 8 (2020).
CAS PubMed PubMed Central Google Scholar * Reiman, E. M. et al. Alzheimer’s prevention initiative: a plan to accelerate the evaluation of presymptomatic treatments. _J. Alzheimer’s Dis._
26, 321–329 (2011). Google Scholar * Lendon, C. L. et al. E280A PS-1 mutation causes Alzheimer’s disease but age of onset is not modified by ApoE alleles. _Hum. Mutat._ 10, 186–195 (1997).
CAS PubMed Google Scholar * He, C., Holme, J. & Anthony, J. SNP genotyping: the KASP Assay BT - crop breeding: methods and protocols. in _Methods in Molecular Biology_ (eds. Fleury,
D. & Whitford, R.) 75–86 (Springer New York, 2014). Download references ACKNOWLEDGEMENTS The authors thank the _PSEN1_ Colombian families for contributing their valuable time and effort,
without which this study would not have been possible. We thank the research staff of the Group of Neuroscience of Antioquia for their help coordinating study visits for the Colombian API
Registry. We thank Geidy Serrano from the Banner institute for her help quantifying DNA samples. This study was supported by grant DP5OD019833 from the National Institutes of Health Office
of the Director (Y.T.Q.), the Massachusetts General Hospital Executive Committee on Research (Y.T.Q.), and grant R01AG054671 from the National Institute on Aging (Y.T.Q.). The funders had no
role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the
manuscript for publication. AUTHOR INFORMATION Author notes * These authors jointly supervised this work: Eric M. Reiman, Francisco Lopera, Yakeel T. Quiroz. AUTHORS AND AFFILIATIONS *
Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA Stephanie Langella, N. Gil Barksdale, Gabriel Oliveira, Clara Vila-Castelar & Yakeel T. Quiroz * Grupo de
Neurociencias de Antioquia, Facultad de Medicina, Universidad de Antioquia, Medellin, Colombia Daniel Vasquez, David Aguillon, Natalia Acosta-Baena, Juliana Acosta-Uribe, Ana Y. Baena,
Gloria Garcia-Ospina, Margarita Giraldo-Chica, Victoria Tirado, Claudia Muñoz, Silvia Ríos-Romenets, Claudia Guzman-Martínez, Francisco Lopera & Yakeel T. Quiroz * Banner Alzheimer’s
Institute, Phoenix, AZ, USA Yinghua Chen, Yi Su, Jeremy J. Pruzin, Valentina Ghisays & Eric M. Reiman * Neuroscience Research Institute and Department of Molecular, Cellular and
Developmental Biology, University of California Santa Barbara, Santa Barbara, CA, USA Juliana Acosta-Uribe & Kenneth S. Kosik * Brigham and Women’s Hospital, Harvard Medical School,
Boston, MA, USA Hyun-Sik Yang * Schepens Eye Research Institute of Mass Eye and Ear, Harvard Medical School, Boston, MA, USA Joseph F. Arboleda-Velasquez Authors * Stephanie Langella View
author publications You can also search for this author inPubMed Google Scholar * N. Gil Barksdale View author publications You can also search for this author inPubMed Google Scholar *
Daniel Vasquez View author publications You can also search for this author inPubMed Google Scholar * David Aguillon View author publications You can also search for this author inPubMed
Google Scholar * Yinghua Chen View author publications You can also search for this author inPubMed Google Scholar * Yi Su View author publications You can also search for this author
inPubMed Google Scholar * Natalia Acosta-Baena View author publications You can also search for this author inPubMed Google Scholar * Juliana Acosta-Uribe View author publications You can
also search for this author inPubMed Google Scholar * Ana Y. Baena View author publications You can also search for this author inPubMed Google Scholar * Gloria Garcia-Ospina View author
publications You can also search for this author inPubMed Google Scholar * Margarita Giraldo-Chica View author publications You can also search for this author inPubMed Google Scholar *
Victoria Tirado View author publications You can also search for this author inPubMed Google Scholar * Claudia Muñoz View author publications You can also search for this author inPubMed
Google Scholar * Silvia Ríos-Romenets View author publications You can also search for this author inPubMed Google Scholar * Claudia Guzman-Martínez View author publications You can also
search for this author inPubMed Google Scholar * Gabriel Oliveira View author publications You can also search for this author inPubMed Google Scholar * Hyun-Sik Yang View author
publications You can also search for this author inPubMed Google Scholar * Clara Vila-Castelar View author publications You can also search for this author inPubMed Google Scholar * Jeremy
J. Pruzin View author publications You can also search for this author inPubMed Google Scholar * Valentina Ghisays View author publications You can also search for this author inPubMed
Google Scholar * Joseph F. Arboleda-Velasquez View author publications You can also search for this author inPubMed Google Scholar * Kenneth S. Kosik View author publications You can also
search for this author inPubMed Google Scholar * Eric M. Reiman View author publications You can also search for this author inPubMed Google Scholar * Francisco Lopera View author
publications You can also search for this author inPubMed Google Scholar * Yakeel T. Quiroz View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
E.M.R., F.L., and Y.T.Q. initiated this work, directed and supervised conduction of the study. S.L, N.G.B and Y.T.Q. drafted the manuscript. Clinical information was collected and analyzed
by F.L., D.V., D.A., N.A-B, A.B., M.G-C, V.T., C.M., S.R-R., and C.V.-C. Genetic data was collected and analyzed by G.G., C.G-M., J.A.-U., and K.K. Statistical analyses were conducted by
Y.C., V.G, and Y.S. All authors revised and contributed to finalize the manuscript. CORRESPONDING AUTHOR Correspondence to Yakeel T. Quiroz. ETHICS DECLARATIONS COMPETING INTERESTS S.L. is
supported by a grant from the Alzheimer’s Association (AARF-22-920754). Y.S. reports grants from The Alzheimer’s Association, The BrightFocus Foundation, NIH/NIA, State of Arizona, outside
the submitted work. C.V.-C. reports grants from the Alzheimer’s Association (AARF 2019A005859) and the National Institute on Aging (K99AG073452). K.S.K. is on the Board of Directors for the
Tau Consortium, receives funding from the NIA, the Alzheimer Association, and the Alzheimer’s Drug Discovery Foundation. H-S.Y. reports a grant from the National Institute of Aging (K23
AG062750). E.M.R. reports grants from National Institute on Aging (P30 AG072980, R01 AG069453, R01 AG055444), Banner Alzheimer’s Foundation and the NOMIS Foundation during the conduct of the
study. E.M.R. is a compensated scientific advisor for Alzheon, Aural Analytics, Denali, Retromer Therapeutics, and Vaxxinity, an uncompensated scientific advisor for Lilly, and a cofounder,
advisor and shareholder of AlzPATH, which is involved in the development of blood-based biomarkers for Alzheimer’s disease outside the scope of the submitted. In addition, E.M.R. is the
inventor of a patent issued to Banner Health, which involves the use of biomarker endpoints in at-risk persons to accelerate the evaluation of Alzheimer’s disease prevention therapies and is
outside the submitted work. F.L. was supported by an Anonymous Foundation, and the Administrative Department of Science, Technology and Innovation (Colciencias Colombia;111565741185).
E.M.R. and F.L. are principal investigators of the Alzheimer’s Prevention Initiative (API) Autosomal Dominant AD Trial, which is supported by NIA, philanthropy, Genentech, and Roche. Y.T.Q.
was supported by grants from the National Institute on Aging (R01 AG054671, RF1AG077627), the Alzheimer’s Association, and Massachusetts General Hospital ECOR. Y.T.Q. serves as consultant
for Biogen. The remaining authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Yadong Huang, and the other, anonymous, reviewer(s) for
their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional
claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Langella, S., Barksdale,
N.G., Vasquez, D. _et al._ Effect of apolipoprotein genotype and educational attainment on cognitive function in autosomal dominant Alzheimer’s disease. _Nat Commun_ 14, 5120 (2023).
https://doi.org/10.1038/s41467-023-40775-z Download citation * Received: 09 February 2023 * Accepted: 09 August 2023 * Published: 23 August 2023 * DOI:
https://doi.org/10.1038/s41467-023-40775-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative