- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT A potential therapeutic target to curb obesity and diabetes is thermogenic beige adipocytes. However, beige adipocytes quickly transition into white adipocytes upon removing
stimuli. Here, we define the critical role of _cyclin dependent kinase inhibitor 2A (Cdkn2a)_ as a molecular pedal for the beige-to-white transition. Beige adipocytes lacking _Cdkn2a_
exhibit prolonged lifespan, and male mice confer long-term metabolic protection from diet-induced obesity, along with enhanced energy expenditure and improved glucose tolerance.
Mechanistically, _Cdkn2a_ promotes the expression and activity of beclin 1 (BECN1) by directly binding to its mRNA and its negative regulator BCL2 like 1 (BCL2L1), activating autophagy and
accelerating the beige-to-white transition. Reactivating autophagy by pharmacological or genetic methods abolishes beige adipocyte maintenance induced by _Cdkn2a_ ablation. Furthermore,
hyperactive BECN1 alone accelerates the beige-to-white transition in mice and human. Notably, both _Cdkn2a_ and _Becn1_ exhibit striking positive correlations with adiposity. Hence, blocking
_Cdkn2a_-mediated BECN1 activity holds therapeutic potential to sustain beige adipocytes in treating obesity and related metabolic diseases. SIMILAR CONTENT BEING VIEWED BY OTHERS _EGR1_
LOSS-OF-FUNCTION PROMOTES BEIGE ADIPOCYTE DIFFERENTIATION AND ACTIVATION SPECIFICALLY IN INGUINAL SUBCUTANEOUS WHITE ADIPOSE TISSUE Article Open access 28 September 2020 DEPLETION OF JUNB
INCREASES ADIPOCYTE THERMOGENIC CAPACITY AND AMELIORATES DIET-INDUCED INSULIN RESISTANCE Article 08 January 2024 BLOCKAGE OF PPARΓ T166 PHOSPHORYLATION ENHANCES THE INDUCIBILITY OF BEIGE
ADIPOCYTES AND IMPROVES METABOLIC DYSFUNCTIONS Article Open access 03 November 2022 INTRODUCTION Obesity and its associated metabolic diseases, resulting from chronic energy imbalance, have
rapidly prevailed over the past decades worldwide and have become a global public health issue1. In mammals, adipocytes are an important regulator of energy homeostasis by storing and
releasing energy. White adipocytes store energy as one large lipid droplet, while brown adipocytes with multilocular lipid droplets and abundant mitochondria can dissipate energy to produce
heat mediated by uncoupling protein (UCP1)2. Notably, beige adipocytes are a subset of “brown-like” cells in white adipose tissue (WAT) that also express UCP1 and contain plentiful
mitochondria, thereby possessing thermogenic capability3. Although brown adipocytes are limited to infants, beige adipocytes can be recruited throughout WAT by various mechanisms in adult
humans, which confers metabolic benefits, including reduced blood glucose and increased energy expenditure4,5,6. Thus, beige adipocytes have emerged as a promising cellular target as a
metabolic sink for glucose and free fatty acids to combat obesity and diabetes. In both rodents and humans, the biogenesis of beige adipocytes is highly inducible by a list of physiological
and pharmacological stimuli, such as chronic cold exposure or β3 adrenergic receptor agonists7. However, beige adipocytes are short-lived; after removing external stimuli, UCP1+ multilocular
beige adipocytes convert into dormant unilocular “white” adipocytes, expressing several white adipocyte-enriched genes8,9,10,11. Thus, constant stimuli are needed to maintain the activity
of beige adipocytes, which has become one of the major clinical hurdles for using beige adipocytes as a sustainable therapy for chronic metabolic diseases. Compared to beige adipocyte
triggering, relatively little research has been conducted to investigate the fates of beige adipocytes after discontinuing beiging stimuli. Therefore, it is critical to elucidate the precise
mechanism(s) by which beiging is maintained and to examine whether maintaining beiging can improve metabolic fitness. The genomic _Cdkn2a_ locus encodes two crucial cell cycle regulators,
p16Ink4a and p14ARF, which regulate the p16Ink4a/RB and p14ARF/p53 pathways, respectively. The role of _Cdkn2a_ as a tumor suppressor gene is well known12,13,14. Recently, _Cdkn2a_ has been
linked to obesity and type 2 diabetes in both rodent and human genome-wide association studies15,16. Studies have demonstrated that _Cdkn2a_ expression is increased in WAT in obese mouse
models and obese humans, inducing senescence and inflammation and aggravating systemic insulin resistance17,18,19. Whole-body _Cdkn2a_ knockout mice gained less weight and fat under a
high-fat diet, along with increased beige fat formation in WAT and energy expenditure19. Consistent with these studies, our recent study showed that the deletion of _Cdkn2a_ in UCP1-positive
cells within inguinal WAT (IGW) stimulated beige adipocyte proliferation20. These findings suggest that _Cdkn2a_ plays a crucial role in obesity and diabetes. However, whether _Cdkn2a_
regulates beige fat maintenance is unknown. In this study, we investigated the function of _Cdkn2a_ in beige fat maintenance by employing our defined _Cdkn2a_ mouse models and in vitro beige
maintenance assays. We found that loss of _Cdkn2a_ in beige adipocytes results in prolonged beige adipocyte maintenance, enhanced energy expenditure, improved glucose tolerance, and
increased resistance to high-fat, high-sucrose (HFHS) diet-induced obesity. Mechanistically, _Cdkn2a_ deficiency promotes beige adipocyte maintenance by inhibiting BECN1-mediated autophagy.
Importantly, reactivating autophagy in _Cdkn2a_-ablated beige adipocytes reversed beige adipocyte maintenance phenotypes. Consistent with a role in energy expenditure, both _Cdkn2a_ and
_Becn1_ exhibit striking positive correlations with adiposity in mice and humans. Collectively, our findings unveil insights into the molecular mechanisms by which _Cdkn2a_ regulates beige
adipocyte maintenance via BECN1-mediated autophagy and provide a potential therapeutic strategy to lock into a stable beige identity and treat obesity and related metabolic diseases. RESULTS
_CDKN2A_ DEFICIENCY MAINTAINS THE MORPHOLOGICAL AND MOLECULAR CHARACTERISTICS OF BEIGE ADIPOCYTES AFTER WITHDRAWAL OF COLD EXPOSURE To investigate the function of _Cdkn2a_ in beige fat
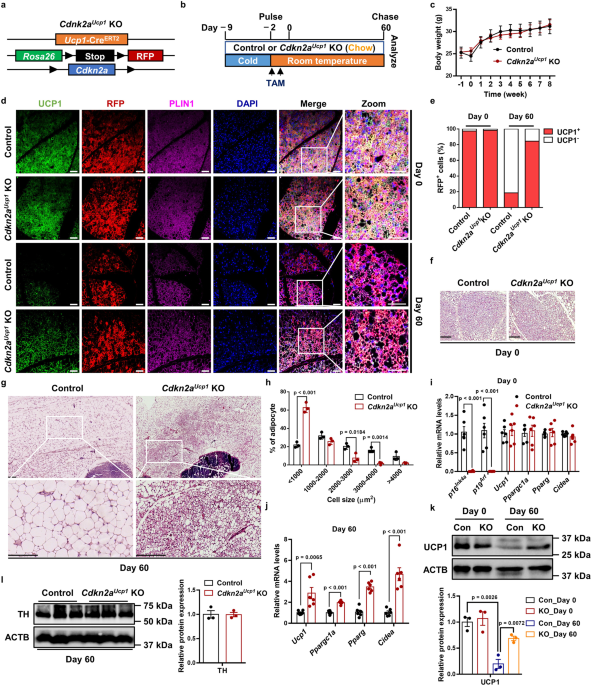
maintenance in vivo, we crossed tamoxifen (TAM)-inducible _Ucp1_-CreERT2 mice with a _Rosa_26RRFP indelible labeling reporter and _Cdkn2a_ floxed mice (_Cdkn2a__Ucp1_ KO, Fig. 1a). Using
this fate-mapping model, we first induced beige adipocyte biogenesis in IGW for 7 days by cold exposure (6 °C) to 2-month-old male mice. After cold exposure, we deleted _Cdkn2a_ in all newly
induced and preexisting UCP1-expressing beige adipocytes through TAM administration (1.5 mg/kg) for two consecutive days. We housed the mice at room temperature (22 °C) for 60 days
(rewarming period) to trace beige adipocyte maintenance (Fig. 1b). No significant difference was found in body weight or body composition between control and _Cdkn2a__Ucp1_ KO mice after the
rewarming period following cold exposure (Fig. 1c and Supplementary Fig. 1a). As expected, at day 0, all RFP+ beige adipocytes within IGW expressed endogenous UCP1 following cold exposure,
as assessed by RFP, UCP1, and PLIN immunofluorescence staining (Fig. 1d, e). Consistent with a previous study that showed that beige adipocytes transition back to their white adipose
state10, we found that at day 60, nearly 80% of beige adipocytes lost UCP1 expression, as quantified by RFP+, UCP1−, and PLIN+ immunofluorescence staining (Fig. 1d, e). In contrast,
_Cdkn2a__Ucp1_ KO mice contained a higher percentage of UCP1-expressing RFP+ beige adipocytes in IGW compared to control mice at 60 days post withdrawal of cold stimulus and TAM
administration (Fig. 1d, e). Consistent with UCP1 immunostaining, hematoxylin and eosin (H&E) staining showed that beige fat biogenesis, determined by multilocular lipids, was similar at
day 0 (Fig. 1f), but _Cdkn2a__Ucp1_ KO mice contained more beige adipocytes at day 60 (Fig. 1g). Moreover, _Cdkn2a_ deletion reduced adipocyte size in IGW at day 60 (Fig. 1g, h). At the
molecular level, we first confirmed that _p16__Ink4_a and _p19__Arf_, two transcript variants of _Cdkn2a_, were deleted in IGW beige fat by qPCR analysis (Fig. 1i). There was no significant
difference in the basal expression levels of _Ucp1_ and other thermogenic genes, such as _Ppargc1a_, _Pparg_, and _Cidea_, in IGW between control and _Cdkn2a__Ucp1_ KO mice at day 0 (Fig.
1i). In agreement with beige adipocyte maintenance, loss of _Cdkn2a_ increased thermogenic gene expression in IGW at day 60 compared to the control group (Fig. 1j). Furthermore, we observed
a similar pattern of protein abundance of UCP1: no difference at day 0 but significantly higher in IGW from _Cdkn2a__Ucp1_ KO mice after the rewarming period (Fig. 1k). Similar to the
observations in male mice, female _Cdkn2a_Ucp1 KO mice also exhibited increased beige adipocyte content (Supplementary Data Fig. 1b) and higher expression levels of thermogenic genes at day
60 compared to control mice (Supplementary Data Fig. 1c). This suggests that Cdkn2a knockout promotes beige adipocyte maintenance in both male and female mice. To investigate whether
increased sympathetic neural signaling was responsible for the prolonged beige fat maintenance in _Cdkn2a__Ucp1_ KO mice, we performed western blot against tyrosine hydroxylase (TH), which
is a marker of norepinephrine turnover. However, no significant difference in TH expression was observed in IGW between control and _Cdkn2a__Ucp1_ KO mice at day 60 (Fig. 1l). We next
evaluated whether deleting _Cdkn2a_ in UCP1+ cells impacted the morphological and molecular characteristics of brown adipocytes and other organs. We observed no differences in interscapular
brown adipose tissue (BAT) morphology between control and _Cdkn2a__Ucp1_ KO mice based on H&E analysis (Supplementary Fig. 1d). In contrast to beige adipocytes, brown adipocytes from
both control and _Cdkn2a__Ucp1_ KO mice expressed constitutively high levels of UCP1 even at 60 days after the rewarming period following cold exposure (Supplementary Fig. 1e–g), which was
consistent with a previous study10. Moreover, with qPCR analysis, we confirmed that _Cdkn2a_ (_p16__Ink4_a and _p19__Arf_) was largely ablated in BAT as expected, while thermogenic gene
expression in BAT did not change compared to the control and _Cdkn2a__Ucp1_ KO mice at both day 0 and day 60 (Supplementary Fig. 1h). Furthermore, morphology and adipocyte size of
perigonadal WAT (PGW) showed no significant difference between control and _Cdkn2a__Ucp1_ KO mice (Supplementary Fig. 1i, j). In addition, no significant difference was observed in liver
histology (Supplementary Fig. 1k) and other tissue weights (Supplementary Fig. 1l) between control and _Cdkn2a__Ucp1_ KO mice. Overall, these results indicate that _Cdkn2a_ promotes beige
adipocyte maintenance but does not affect brown adipocytes or other organs after the rewarming period following cold exposure. DELETING _CDKN2A_ PREVENTS HFHS-INDUCED OBESITY AND GLUCOSE
INTOLERANCE BY ENHANCING ENERGY EXPENDITURE AND THERMOGENESIS In both mice and humans, beige adipocytes can reduce body fat and glucose21, presenting a key therapeutic promise. Our data thus
far suggest that deleting _Cdkn2a_ in beige adipocytes can extend their lifespan but without significant metabolic benefits under a standard chow diet. Chronic exposure to the HFHS diet,
which mimics the human diet in many countries, can cause obesity, T2DM, and associated metabolic syndrome22. To further understand the functional relevance of _Cdkn2a_-mediated beige
adipocyte maintenance in obesity and its related diabetes, we challenged the control and _Cdkn2a__Ucp1_ KO mice with a HFHS diet for 120 days during the rewarming period following cold
exposure (Fig. 2a). Deleting _Cdkn2a_ prevented HFHS-induced weight gain (Fig. 2b, c), while food intake showed no significant difference between control and _Cdkn2a__Ucp1_ KO mice
(Supplementary Fig. 2a). Compared to control mice, _Cdkn2a__Ucp1_ KO mice exhibited markedly reduced body fat content and increased lean mass content (Fig. 2d). Furthermore, _Cdkn2a_
deficiency mainly decreased the weight of WAT depots (IGW, PGW, retroperitoneal WAT (rpWAT)) but not the weight of other tissues, including BAT, muscle, kidney, spleen, pancreas, heart, and
liver (Supplementary Fig. 2b). _Cdkn2a_ knockout decreased adipocyte size in IGW (Fig. 2e, f) and PGW (Supplementary Fig. 2c, d). Furthermore, we found that _Cdkn2a_ knockout ameliorated
HFHS-induced whitening of BAT (Supplementary Fig. 2e) and fatty liver (Supplementary Fig. 2e). Compared to control mice, _Cdkn2a__Ucp1_ KO mice exhibited significantly decreased blood
glucose levels (Fig. 2g) and serum insulin concentrations (Fig. 2h) under HFHS conditions. Importantly, loss of _Cdkn2a_ improved glucose metabolism and insulin sensitivity compared to
control mice, as assessed by the glucose tolerance test (Fig. 2i, j) and insulin tolerance test (Fig. 2k, l). Taken together, these results indicate that prolonged maintenance of beige
adipocytes by deleting _Cdkn2a_ prevents HFHS-induced body weight and fat mass gain and its associated impaired glucose metabolism. To further investigate the metabolic significance of
retaining beige adipocytes in vivo, we next measured gas exchange and whole-body energy expenditure of control and _Cdkn2a__Ucp1_ KO mice under HFHS in metabolic cages. We found that
_Cdkn2a__Ucp1_ KO mice showed increases in oxygen consumption (Supplementary Fig. 2f), carbon dioxide generation (Supplementary Fig. 2g), and energy expenditure (Fig. 2m) compared to control
mice during both light and dark cycles. No significant difference was found in locomotor activity between control and _Cdkn2a__Ucp1_ KO mice (Supplementary Fig. 2h). Consistent with the
increased metabolic rate, _Cdkn2a__Ucp1_ KO mice also exhibited a higher core body temperature at room temperature (Fig. 2n). As expected, the H&E staining results showed that _Cdkn2a_
deletion preserved beige fat identity in IGW (Fig. 2e). Furthermore, _Cdkn2a__Ucp1_ KO mice contained a higher percentage of UCP1-expressing RFP+ cells in IGW than control mice (Fig. 2o, p).
Consistently, loss of _Cdkn2a_ maintained higher levels of thermogenic gene expression and UCP1 protein expression and decreased mRNA levels of white adipocyte marker genes, such as
_Adipoq_, _Fabp4_, and _Lep_, in IGW (Fig. 2q, r), indicating that _Cdkn2a_ knockout prevented beige-to-white adipocyte transition. In contrast, there was no significant difference in the
expression of UCP1 and other thermogenic genes in BAT between control and _Cdkn2a__Ucp1_ KO mice (Supplementary Fig. 2i–l). These data indicate that prolonged maintenance of beige adipocytes
by _Cdkn2a_ deletion preserves energy expenditure and promotes thermogenesis under HFHS challenges. _CDKN2A_ NEGATIVELY REGULATES BEIGE ADIPOCYTE MAINTENANCE IN VITRO To delineate the
underlying cellular and molecular mechanisms, we established a cellular system to study beige adipocyte maintenance in vitro. We isolated stromal vascular fraction (SVF) cells from the IGW
of TAM-uninduced control and _Cdkn2a__Ucp1_ KO mice and cultured them to produce beige adipocytes in vitro. After _Cdkn2a_ deletion and RFP labeling by 4-hydroxy-tamoxifen (4-OHT) treatment,
we withdrew external stimuli by replacing the differentiation medium with DMEM containing 5% bovine serum albumin (BSA) at the initial stage (day 0). Then we determined how many RFP+ cells
were still beige adipocytes at the final stage (day 4) (Fig. 3a). We found that the mRNA levels of _p16__Ink4_a and _p19__Arf_ were inversely associated with UCP1 levels among brown, beige,
and white adipocytes (Supplementary Fig. 3a). Furthermore, the mRNA levels of _p16__Ink4_a and _p19__Arf_ gradually increased after withdrawing external stimuli (Supplementary Fig. 3b).
Together, these data suggest a negative regulatory role of _Cdkn2a_ in beige adipocyte maintenance. At the initial stage, both control and _Cdkn2a__Ucp1_ KO cells exhibited similar amounts
of multilocular lipid droplet formation and UCP1 expression (Fig. 3b–d). At the final stage, the number and size of lipid droplets and UCP1 expression were significantly decreased in control
cells (Fig. 3b–d), recapitulating our in vivo system in which RFP+ beige adipocytes lose UCP1 expression. In contrast to control cells, _Cdkn2a__Ucp1_ KO beige adipocytes contained more
lipid droplets (Fig. 3b) and a higher percentage of UCP1-expressing RFP+ cells (Fig. 3c, d) at the final stage. Consistently, no difference was found in the basal expression levels of
thermogenic genes between control and _Cdkn2a__Ucp1_ KO beige adipocytes at the initial stage (Supplementary Fig. 3c). In contrast, at the final stage, _Cdkn2a__Ucp1_ KO cells expressed
higher levels of thermogenic genes, such as _Ucp1_, _Ppargc1a_, _Pparg_, _Prdm16_, and _Cidea_, than control cells (Fig. 3e). Furthermore, the UCP1 protein level was higher in _Cdkn2a__Ucp1_
KO cells after withdrawing external stimuli than in control cells (Fig. 3f). Consistent with the higher amount of UCP1+ beige adipocytes in _Cdkn2a__Ucp1_ KO, we found that _Cdkn2a_
deficiency promoted basal mitochondrial respiration and maximal mitochondrial respiratory capacity of beige adipocytes at the final stage but not at the initial stage, as assessed by using a
seahorse extracellular flux analyzer (Fig. 3g). Next, we investigated the role of _Cdkn2a_ in regulating brown adipocyte maintenance in vitro by using the same cellular system
(Supplementary Fig. 4a). In contrast to beige maintenance, the expression levels of _p16__Ink4_a and _p19__Arf_ were dramatically decreased during brown adipocyte maintenance (Supplementary
Fig. 4b). Interestingly, the size and number of lipid droplets were decreased in both control and _Cdkn2a__Ucp1_ KO brown adipocytes at the final stage (Supplementary Fig. 4c). No
significant change was found between control and _Cdkn2a__Ucp1_ KO brown adipocytes at either the initial or the final stage, based on morphology and UCP1 immunostaining (Supplementary Fig.
4c–e). Additionally, thermogenic gene expression was similar between the control and _Cdkn2a__Ucp1_ KO cells during brown fat maintenance (Supplementary Fig. 4f, g). Collectively, these
results indicate that loss of _Cdkn2a_ facilitates the maintenance of beige adipocytes, but not brown adipocytes, in vitro. _CDKN2A_ REGULATES BEIGE ADIPOCYTE MAINTENANCE IN A CELL
CYCLE-INDEPENDENT MANNER Since _Cdkn2a_ is a well-known cell cycle inhibitor gene, we examined whether _Cdkn2a_ regulated beige adipocyte maintenance in a cell cycle-dependent manner. Our
previous study showed that deleting _Cdkn2a_ in UCP1+ cells in IGW promotes beige adipocyte proliferation20. To examine directly if the higher number of UCP1-expressing RFP+ cells in
_Cdkn2a__Ucp1_ KO was derived from increased proliferative events, we first assessed the proliferation of control and _Cdkn2a__Ucp1_ KO cells during beige fat maintenance by
bromodeoxyuridine (BrdU) staining. We observed that _Cdkn2a_ knockout did not affect beige adipocyte proliferation in our in vitro cellular system (Supplementary Fig. 5a). To further rule
out the possibility that beige adipocyte maintenance is regulated in a cell cycle-dependent manner, we examined the role of a downstream gene of _Cdkn2a_, _Ccnd1_, which also plays a key
role in the regulation of cell cycle. We found that the mRNA level of _Ccnd1_ was unchanged during the beige-to-white adipocyte transition (Supplementary Fig. 5b). To further investigate the
effect of _Ccnd1_-mediated cell cycle progression on beige-to-white adipocyte transition in vitro, we constructed an adipocyte-specific conditional _Ccnd1_ overexpression (OE) mouse model
(Supplementary Fig. 5c, d). After doxycycline (Dox) treatment, we confirmed that the _Ccnd1_ expression was significantly higher in _Ccnd1_ OE beige adipocytes compared to control cells
(Supplementary Fig. 5e). Overexpression of _Ccnd1_ did not influence beige adipocyte maintenance, as assessed by immunostaining and western blot analysis of UCP1 (Supplementary Fig. 5f, g).
Together, these findings strongly suggest that beige adipocyte maintenance in _Cdkn2a__Ucp1_ KO mice is not primarily mediated by the activation of cell proliferation. KNOCKOUT OF _CDKN2A_
PROMOTES BEIGE FAT MAINTENANCE BY INHIBITING AUTOPHAGY A previous study showed that autophagy-induced mitochondrial clearance (mitophagy) plays a key role in regulating the beige-to-white
adipocyte transition10. Since _Cdkn2a_ has been strongly associated with autophagy in various cell types23,24,25, we examined the relationship between _Cdkn2a_ and autophagy during beige fat
maintenance. Compared to control cells, the number of LC3 puncta, a marker of autophagy, was significantly decreased in _Cdkn2a__Ucp1_ KO beige adipocytes after withdrawing external stimuli
(Fig. 4a, b). We also found that autophagy was activated in control beige adipocytes during the beige-to-white fat transition, as determined by an increased LC3-II/LC3-I ratio and decreased
SQSTM1/p62 (a protein specifically degraded in lysosomes) (Fig. 4c). In contrast, _Cdkn2a_ deficiency inhibited autophagy activation at the final stage (Fig. 4c), and this autophagy
inhibition was further boosted by treatment with the lysosomal inhibitor chloroquine (CQ) (Fig. 4d). By co-staining with Mitotracker and anti-LC3 antibody, we found that a lower level of
mitophagy contributed to the higher mitochondrial content in _Cdkn2a__Ucp1_ KO beige adipocytes compared to control cells (Fig. 4e). In agreement with this result, _Cdkn2a_ knockout led to
an increased mtDNA copy number compared to controls (Fig. 4f). At the molecular level, _Cdkn2a__Ucp1_ KO beige adipocytes exhibited higher protein abundance of mitochondrial markers VDAC and
TOMM20 (Fig. 4g). In addition, the LC3-II/LC3-I ratio was similar in control and _Ccnd1_ OE beige adipocytes after withdrawing external stimuli (Supplementary Fig. 5f), suggesting that
_Ccnd1_ does not affect autophagy during beige adipocyte maintenance. These results indicate that loss of _Cdkn2a_ suppresses autophagy during beige adipocyte maintenance. To test whether
_Cdkn2a_ regulated beige adipocyte maintenance in an autophagy-dependent manner, we treated control and _Cdkn2a__Ucp1_ KO mice with rapamycin, an autophagy inducer (Fig. 4h). We found that
rapamycin treatment accelerated the beige-to-white adipocyte transition in control mice (Fig. 4i), indicating a negative correlation between autophagy activation and beige adipocyte
maintenance. Moreover, rapamycin treatment blocked the prolonged beige adipocyte maintenance in IGW from the _Cdkn2a__Ucp1_ KO mice (Fig. 4i). Consistent with the in vivo data, rapamycin
treatment in vitro also largely hindered _Cdkn2a__Ucp1_ KO-mediated beige adipocyte maintenance based on the number of UCP1-expressing RFP+ cells (Fig. 4j, k). Furthermore, the upregulated
expression of UCP1 and other thermogenic genes in _Cdkn2a__Ucp1_ KO beige adipocytes was restored to the levels of control cells by rapamycin treatment (Fig. 4l, m). We confirmed that
rapamycin treatment induced autophagy as expected in both control and _Cdkn2a__Ucp1_ KO cells, as assessed by an increased LC3-II/LC3-I ratio (Fig. 4m). Additionally, rapamycin treatment
reversed the enhanced oxygen consumption rate induced by _Cdkn2a_ knockout (Fig. 4n). Together, these in vivo and in vitro results suggest that _Cdkn2a_ deficiency prevents beige-to-white
adipocyte transition by suppressing autophagy. _CDKN2A_ PROMOTES AUTOPHAGY BY INCREASING _BECN1_ EXPRESSION To investigate how CDKN2A affects autophagy, we tested the effect of _Cdkn2a_
knockout on the mRNA levels of autophagy-related genes in vivo and in vitro. We found that _Cdkn2a_ deficiency reduced the gene expression of _Becn1_ in both IGW and beige adipocytes, while
the expression of other autophagy-related genes, including _Atg5_, _Atg12_, _Pink1_, _Bnip3_, _Bnip3l_, and _Fundc1_, was unchanged (Fig. 5a and Supplementary Fig. 6a). The protein
expression of BECN1 was increased in the control group after withdrawing external stimuli (Fig. 5b and Supplementary Fig. 6b). Compared to the control group, the _Cdkn2a_ knockout group
exhibited lower protein levels of BECN1 in vivo and in vitro (Fig. 5b and Supplementary Fig. 6b). Furthermore, we found that the mRNA level of _Becn1_ was significantly higher in IGW than in
BAT (Supplementary Fig. 6c). Consistently, thermogenic brown and beige adipocytes expressed lower _Becn1_ (Supplementary Fig. 6d), indicating a negative correlation between _Becn1_ and
thermogenesis. Similar to the expression pattern of _p16__Ink4_a and _p19__Arf_, the gene expression of _Becn1_ was increased after withdrawing external stimuli (Fig. 5c). Interestingly, the
deletion of _Cdkn2a_ in brown adipocytes also inhibited _Becn1_ expression in vivo and in vitro (Supplementary Fig. 6e, f). To test whether _Cdkn2a_ within beige adipocytes regulated
autophagy via _Becn1_, we performed in vitro beige maintenance assays using a previously established hyperactive BECN1 mouse model induced by a single knock-in mutation (F121A) that
decreases its interaction with the negative regulator BCL226 (Supplementary Fig. 6g). We examined whether _Cdkn2a__Ucp1_ KO beige adipocytes could still be maintained in a hyperactive BECN1
setting (Fig. 5d). After a short 20-day rewarming period following cold exposure, we found that hyperactive BECN1 blunted _Cdkn2a__Ucp1_ knockout--mediated beige adipocyte maintenance based
on H&E analysis (Fig. 5e). We further performed RFP and UCP1 immunostaining on IGW sections and confirmed that upon hyperactive BECN1 (_Cdkn2a__Ucp1_ KO + _Becn1_F121A), _Cdkn2a__Ucp1_
KO beige adipocytes started to lose UCP1 expression at day 20 of the rewarming period, with a similar percentage of UCP1-expressing RFP+ cells (Fig. 5f, g) and comparable levels of
thermogenic gene expression within IGW (Fig. 5h, i) as the control group. In agreement with these mouse models, we confirmed that the autophagy level was restored to the control level in
_Cdkn2a__Ucp1_ KO+_Becn1_F121A mice compared to control and _Cdkn2a__Ucp1_ KO mice (Fig. 5i), as determined by western blot analysis of the LC3-II/LC3-I ratio. Consistent with our in vivo
data, hyperactive BECN1 also impaired _Cdkn2a_ knockout-mediated beige adipocyte maintenance in vitro, as assessed by quantification of UCP1-expressing RFP+ cells (Fig. 5j, k). Additionally,
the mRNA expression of UCP1 and other thermogenic genes in the _Cdkn2a__Ucp1_ KO beige adipocytes was restored to the levels in the control mice after hyperactive BECN1 (Fig. 5l, m).
Together, these findings suggest that _Cdkn2a_ regulates beige adipocyte maintenance through BECN1-mediated autophagy. HYPERACTIVE BECN1 ACCELERATES THE LOSS OF BEIGE ADIPOCYTE
CHARACTERISTICS UPON REMOVAL OF EXTERNAL STIMULI IN VITRO To investigate the role of BECN1 in beige fat maintenance, we isolated SVF cells from _Becn1_F121A mice and performed in vitro
studies (Fig. 6a). We first confirmed that hyperactive BECN1 increased the number of LC3 puncta and the LC3-II/LC3-I ratio, hallmarks of enhanced autophagy, in beige adipocytes compared to
control cells (Fig. 6b–d). We found that hyperactive BECN1 did not affect beige adipogenesis (Fig. 6e, f) or UCP1 expression at the initial stage (day 0) (Fig. 6g, h). At the final stage
(day 2), hyperactive BECN1 impaired beige adipocyte maintenance and decreased thermogenic gene expression (Fig. 6g–j). We also investigated the function of BECN1 in brown adipocyte
maintenance (Supplementary Fig. 7a). Hyperactive BECN1 also enhanced autophagy in brown adipocytes (Supplementary Fig. 7b, c) but did not affect brown fat biogenesis (Supplementary Fig. 7d,
e). Similar to _Cdkn2a_ knockout in beige adipocytes, no significant difference was observed in maintenance or thermogenic gene expression between control and _Becn1_F121A brown adipocytes
at the final stage (Supplementary Fig. 7f–h), suggesting that BECN1-promoted autophagy was not a regulator of brown adipocyte maintenance. Together, these findings suggest that hyperactive
BECN1 impairs the maintenance of beige adipocytes but not brown adipocytes. To further investigate whether BECN1-mediated autophagy regulates human beige adipocyte maintenance, we isolated
SVF cells from human subcutaneous WAT and performed in vitro beige maintenance assays. We induced human beige adipogenesis and treated cells with Tat-Becn1, a peptide known to stimulate
autophagy through the mobilization of endogenous BECN127, after the withdrawal of stimuli. We validated that Tat-Becn1 enhanced autophagy in human beige adipocytes, as assessed by an
increased LC3-II/LC3-I ratio (Fig. 6k). As expected, Tat-BENC1 accelerated the decrease of expression of UCP1 and other thermogenic genes in human beige adipocytes after removing stimuli
(Fig. 6k, l). These results indicate that BECN1-mediated autophagy plays a conserved role in regulating beige adipocyte maintenance between humans and mice. P16INK4A FACILITATES _BECN1_
EXPRESSION BY PREVENTING ITS MRNA DECAY To explore the underlying mechanisms by which _Cdkn2a_ regulates BECN1-mediated autophagy in beige adipocytes, we first examined whether there were
any changes in the cellular distributions of p16INK4a and p19ARF during the beige-to-white transition. Intriguingly, the cytoplasmic accumulation of p16INK4a in beige adipocytes was
significantly increased 6 h post withdrawing external stimuli, while the cellular distribution of p19ARF was unaltered (Fig. 7a), suggesting that p16INK4a and p19ARF might play distinct
roles in mediating _Becn1_. Since a previous mRNA-bound proteome study identified p16INK4a and p19ARF as potential RNA-binding proteins28, we decided to investigate whether p16INK4a and/or
p19ARF regulate _Becn1_ expression by binding to its mRNA. We performed RNA immunoprecipitation (RIP)-qPCR with antibodies against endogenous p16INK4a or p19ARF (Fig. 7b). We found that
p16INK4a interacted with the mRNA of _Becn1_, but not _Atg5_, _Atg7_, _Pink1_, or _Ucp1_, in beige adipocytes upon withdrawal of external stimuli (Fig. 7c). In addition, no interaction was
observed between p19ARF and _Becn1_ mRNA (Fig. 7d). Together, these results indicate that p16INK4a regulates _Becn1_ by specifically targeting its transcript. Since p16INK4a positively
modulated _Becn1_ gene expression and p16INK4a was involved in the regulation of mRNA stability29, we investigated the _Becn1_ mRNA stability in control and _Cdkn2a__Ucp1_ KO beige
adipocytes by using an RNA decay assay. As expected, _Cdkn2a_ deficiency markedly decreased the mRNA stability of _Becn1_, but not _Ucp1_, in beige adipocytes (Fig. 7e). Collectively, these
results demonstrate that p16INK4a serves as an RNA-binding protein during the beige-to-white transition and regulates _Becn1_ expression by mediating its mRNA stability. P19ARF PROMOTES
BECN1-MEDIATED AUTOPHAGY BY INTERACTING WITH BCL2L1 A previous study reported that p19ARF reduced the formation of the protein complex between BECN1 and its negative regulator, BCL2L1, and
then induced autophagy in cancer cells23. Here, we asked whether p19ARF might interfere with the ability of BCL2L1 to complex with BECN1 protein, leading to enhanced autophagy during the
beige-to-white transition. To test this hypothesis, we first detected the interaction among p19ARF, BECN1 and BCL2L1 in beige adipocytes. Using a co-IP assay, we found that p19ARF could
interact with BECN1 and BCL2L1 (Fig. 7f). In contrast, p16INK4a was unable to interact with BECN1 or BCL2L1 (Fig. 7g). In the reciprocal IP, p19ARF was also pulled down by an antibody
against BCL2L1 (Fig. 7h). Importantly, the complex formation between BECN1 and BCL2L1 was markedly increased in _Cdkn2a__Ucp1_ KO beige adipocytes compared to control cells (Fig. 7h). Taken
together, these results indicate that p19ARF enhances BECN1-mediated autophagy by interacting with BCL2L1. _CDKN2A_ AND _BECN1_ EXPRESSION ARE POSITIVELY ASSOCIATED WITH OBESITY IN MICE AND
HUMANS We further investigated the correlation between _Cdkn2a_ and _Becn1_ mRNA levels and obesity. Upon HFHS feeding, the mRNA levels of _p16__Ink4_a, _p19__Arf_, and _Becn1_ in IGW were
significantly higher than in mice fed a standard chow diet (Fig. 8a). Most remarkably, the _p16__INK4_a, _p14__ARF_, and _BECN1_ mRNA levels in subcutaneous adipose tissues of obese human
individuals were significantly higher than that of non-obese individuals (Fig. 8b), linking _p16__INK4_a, _p14__ARF_, and _BECN1_ expression to obesity in human. Furthermore, we found that
higher mRNA levels of _p16__INK4_a and _p14__ARF_ were associated with higher body weight, BMI, and fat mass in humans (Fig. 8c, d). _BECN1_ expression was positively correlated with body
weight, BMI, fat mass, and blood glucose levels (Fig. 8e). Thus, these data indicate that _CDKN2A and BECN1_ mRNA levels could be indicators of human obesity and T2DM. DISCUSSION Since the
rediscovery of brown and beige adipocytes in adults, tremendous efforts have been made to find signals to induce beige adipocyte formation within WAT. In this study, we used unique mouse
models to demonstrate that beige adipocytes can be formed and maintained by pharmacological and genetic methods. The main finding of this study is that _Cdkn2a_ is a crucial negative
regulator of beige adipocyte maintenance through direct interaction with _Becn1_. We report that genetically deleting _Cdkn2a_ in beige adipocytes promotes their lifespan and is associated
with an improved metabolic profile upon HFHS diet challenge. Our data reveal a previously unknown function of _Cdkn2a_ in beige fat biology and suggest that inhibitors of the _Cdkn2a_
pathway would be beneficial in reversing the deleterious effects of metabolic syndromes. Therefore, it serves as a potential therapeutic target in treating obesity and its related metabolic
diseases. Activation of beige fat biogenesis is accompanied by improved metabolic benefits. There is, however, little evidence that maintaining beige adipocyte phenotype can be beneficial.
In this study, we investigated whether sustained beige adipocytes can improve metabolic fitness under diet-induced obesity, thereby achieving anti-obesity and anti-diabetes effects. We found
that _Cdkn2a_ deletion promotes beige adipocyte maintenance under both a standard chow and a HFHS diet. Importantly, the retained beige adipocytes, induced by UCP1+-cell-specific _Cdkn2a_
deletion, are adequate to prevent body weight gain, fat expansion, and ectopic lipid storage in the liver under HFHS feeding. Along with the presence of preserved beige adipocytes, the mice
also have improved glucose tolerance. These results are intriguing, given the recent clinical observation that a strong association exists between greater brown fat activity and lower
cardiovascular risk, such as high blood pressure and coronary artery disease30. Several recent studies have shown that beige and brown fat can influence systemic metabolism through releasing
endocrine signals, including peptides, lipids, miRNAs, and secreted proteins, independent of non-shivering thermogenesis31,32. Our data suggest that signals released from _Cdkn2a__Ucp1_ KO
beige adipocytes may communicate with other organs to maintain whole-body energy metabolism. One critical obstacle in beige adipocyte research is that there are no specific Cre lines to
precisely target brown vs. beige adipocytes in mouse models. Deletion of _Cdkn2a_ by UCP1-CreERT2 inevitably creates a model with gene knockout in brown fat, which may confound the
phenotype. However, in our _Cdkn2a__Ucp1_ KO model, we did not observe BAT phenotypes based on morphology and thermogenic gene expression profile, indicating that the resulting metabolic
benefits may largely come from beige adipocyte maintenance. Interestingly, although our data suggest that BAT maintenance is independent of _Cdkn2a_ and _Becn1_, the regulation of _Becn1_
expression by _Cdkn2a_ is essentially conserved in brown and beige adipocytes both in vivo and in vitro. It is thus conceivable that, in addition to _Cdkn2a_ and _Becn1_, multiple complex
mechanisms may exist to retain BAT identity and their physiological roles. Combined with the prior knowledge that beige adipocytes are developmentally, molecularly, and functionally
different from brown adipocytes33, our finding substantiates this important concept that brown and beige adipocyte identities are different and are also distinctly regulated.
Mechanistically, we found that _Cdkn2a_ deficiency mediates beige adipocyte maintenance by regulating autophagy and is independent of _Ccnd1_ activity and likely independent of cell cycle
control. During the beige-to-white transition, autophagy genes have been shown to contribute to beige adipocyte “whitening”10,11,34,35,36. Interestingly, we found that rapamycin further
decreased _Ucp1_ and _Cidea_ expression compared to the control mice. As rapamycin is an inhibitor of mTORC1, which is essential for promoting beige adipogenesis37,38, it is possible that
rapamycin repressed beige adipocyte activity by inhibiting the mTORC1 pathway. Consistently, early studies using adipose-specific deletion of autophagy-related genes show that mice
accumulate more mitochondria in WAT, producing a browning of WAT phenotype with reduced body weight and WAT mass39,40. Among the autophagy pathway, BECN1 is a scaffolding protein that is key
for the initiation of autophagosome biogenesis41, and therefore, we can target and activate autophagy at early steps; the role of BECN1 in adipose tissue has been recently appreciated:
adipocyte-specific BECN1 knockout mice develop severe lipodystrophy and metabolic dysfunctions42,43. However, it is not clear what role BECN1 plays in UCP1+ cells and whether BECN1 mediates
beige activity downstream of the _Cdkn2a_ pathway. In this study, we identified the Cdkn2a pathway as an upstream mechanism that cooperates with autophagy regulation to govern beige-to-white
adipocyte transition. More specifically, p16INK4a facilitates _Becn1_ expression by its function in preventing its mRNA decay, and p19ARF promotes BECN1-mediated autophagy by interacting
with its negative regulator BCL2L1. Thus, our findings provide mechanistic insights into key players in beige fat maintenance regulation. Therapeutically, the expressions of both _Cdkn2a_
and _Becn1_ are upregulated in obesity and positively associated with adiposity in mice and humans. Our findings reveal that _Cdkn2a_ and _Becn1_ function as potent positive regulators of
beige-to-white adipocyte transition, providing a potential therapeutic target for pharmacological intervention to combat the obesity epidemic and its related metabolic diseases. Since direct
manipulation of either _Cdkn2a_ or _Becn1_ could cause several downstream effects and/or potential tumor formation, we believe that defining specific upstream and downstream factors of the
_Cdkn2a_-_Becn1_ pathway will pave the way to develop more focused treatments for obesity and metabolic diseases. Previously, we and others have demonstrated that the ability to form beige
adipocytes declines when mice grow older44,45,46,47,48,49,50,51. Therefore, it will be interesting to test whether manipulating _Cdkn2a_ or _Becn1_ still maintains their beiging capacity and
whether beige adipocytes can be maintained in the older stage. The possibility that the beiging potential can be retained in obese and aged humans and animals suggests a potential
therapeutic approach to use beige adipocytes against obesity in older adults. In summary, we have identified _Cdkn2a_ as an important player in beige adipocyte maintenance via Becn1-mediated
autophagy. Our work creates a concept that beige adipocytes can stay long-lived after discontinuing the stimuli and could be developed as an alternative therapeutic target for treating
obesity by improved energy expenditure and metabolic fitness. METHODS MOUSE MODELS Mice were housed at 22 ± 2 °C with a humidity of 35 ± 5% and a 14:10 light:dark cycle with a standard
rodent chow diet and water unless otherwise indicated. _Rosa26R_RFP (Stock No. 007914) mice were obtained from the Jackson Laboratory. _Ucp1_-CreERT2 and _Cdkn2a_fl/fl mice were generously
provided by Dr. Eric N. Olson (University of Texas Southwestern Medical Center). _Becn1_F121A mice were generously provided by Dr. Congcong He (Northwestern University). All mice were
maintained on mixed C57BL6/J-129SV background. All animal experiments were performed according to procedures reviewed and approved by the Institutional Animal Care and Use Committee of the
University of Illinois at Chicago. Mice were euthanized by carbon dioxide asphyxiation (CO2) inhalation and cervical dislocation was performed as a secondary euthanasia procedure. GENERATION
OF TRE-CCND1 MICE Mice were generated as previously described52. Briefly, we introduced p2Lox-_Ccnd1_ into ZX1 ES cells and selected for G418-resistant recombinant colonies, in which Ccnd1
was recombined downstream of a tetracycline-responsive promoter (TRE). ZX1 ES cell clones were then validated in culture, and three clones were used to generate three chimeric mouse lines.
All three-mouse strains (TRE-_Ccnd1_) behaved similarly and had similar induction of _Ccnd1_ expression in response to doxycycline. CELL CULTURE The isolated cells were cultured in DMEM/F12
media (Sigma-Aldrich) supplemented with 10% FBS (Sigma-Aldrich) and 1% Penicillin/Streptomycin (Gibco) at 37 °C in a 5% CO2 humidified incubator. For beige and brown adipocyte
differentiation, post-confluent cells were induced by DMEM/F12 containing 10% FBS, 0.5 mM isobutylmethylxanthine (Sigma-Aldrich), 1 µg/ml insulin (Sigma-Aldrich), 1 µM dexamethasone
(Sigma-Aldrich), 60 µM indomethacin (Sigma-Aldrich), 1 nM triiodo-L-thyronine (Sigma-Aldrich), and 1 µM rosiglitazone (Sigma-Aldrich) for 2 days and with DMEM/F12 containing 10% FBS and 1
µg/ml insulin, 1 nM triiodo-L-thyronine, and 1 µM rosiglitazone every 2 days. Fresh medium was replaced every 2 days until ready for harvest. To withdraw external stimuli, the
differentiation medium was changed to DMEM containing 5% BSA for 4 days. TAMOXIFEN TREATMENT To induce Cre recombination, mice were intraperitoneally injected with 1.5 mg/kg body weight of
tamoxifen (TAM; Cayman Chemical) dissolved in sunflower oil (Sigma-Aldrich) for 2 consecutive days. For Cre induction in vitro, cells were treated with 2 µM 4-hydroxy-Tamoxifen (4-OHT,
Sigma-Aldrich). COLD EXPOSURE EXPERIMENT For cold exposure experiments, mice were placed in a 6 °C environmental chamber (Environmental & Temperature Solutions) for 7 days. Body
temperature was measured using a rectal probe (Physitemp). The probe was lubricated with glycerol and inserted 1.27 centimeters (0.5 inch), and the temperature was recorded when stabilized
at the indicated time points. METABOLIC CAGE STUDIES Mice were housed individually and acclimatized to the metabolic chambers (Promethion System, Sable System International) at the UIC
Biologic Resources Laboratory for 2 days before data collection was initiated. For the subsequent 3 days, food intake, VO2, VCO2, energy expenditure and physical activity were monitored over
a 12 h light/dark cycle with food provided ad libitum. For energy expenditure analysis, embedded ANCOVA tools in a web application CalR53 were used to perform regression-based indirect
calorimetric analysis. For body composition analysis, the total fat and lean mass were assessed with Bruker Minispec 10 whole body composition analyzer (Bruker). GLUCOSE AND INSULIN
TOLERANCE TESTS For the glucose tolerance test, mice fasted for 16 h were intraperitoneally injected with glucose (1.5 g/kg). For insulin tolerance test, mice fasted for 4 h were
intraperitoneally injected with human insulin (1.0 U/kg). Glucose levels were measured in tail blood at 0, 15, 30, 45, 60, 90 and 120 min after glucose injection by using a glucose meter
(Contour Next). ISOLATION OF STROMAL VASCULAR FRACTION (SVF) Isolation of SVF cells from IGW and BAT was performed as previously described20. Briefly, IGW or BAT was excised from 6-week-old
male mice, minced by scissors and then digested in isolation buffer (100 mM HEPES, 0.12 M NaCl, 50 mM KCl, 5 mM D-glucose, 1.5% BSA, 1 mM CaCl2, pH 7.3) containing 1 mg/ml type I collagenase
(Worthington) at 37 °C for 30 min. Digested tissue was filtered through 70 µm mesh to remove large pieces, and the flow-through was then centrifuged at 800 × _g_ for 5 min. Pellets
containing primary preadipocytes were resuspended in red blood cell lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) for 5 min, followed by centrifugation at 800 × _g_ for 5 min. The
pellets were resuspended and seeded. Human SVF cells were isolated from fresh subcutaneous adipose tissues. WESTERN BLOT ANALYSIS Proteins from cells and tissue were extracted with RIPA
buffer (Boston BioProducts) supplemented with protease inhibitor cocktail (MedChem Express) and centrifuged at 12,000 × _g_ for 15 min at 4 °C. The total protein amounts were detected by a
BCA assay kit (Thermo Fisher Scientific). Protein samples were separated in an SDS-PAGE gel and transferred to PVDF membranes (Millipore). The membranes were blocked with 5% non-fat milk at
room temperature and then incubated with relevant primary antibodies at 4 °C overnight, followed by HRP-conjugated secondary antibody at room temperature for 2 h. The signals were detected
by the addition of Western ECL Substrate (Thermo Fisher Scientific). The protein bands were detected using ChemiDoc MP Image system (Biorad). The primary antibodies used in this study are
listed in Supplementary Table 1. HISTOLOGICAL STAINING Hematoxylin and eosin (H&E) staining was performed on paraffin sections using standard methods. Briefly, tissues were fixed in 10%
formalin overnight, dehydrated, embedded in paraffin, and sectioned with a microtome at 4–8 μm thicknesses. Adipocyte sizes were quantified by using ImageJ. For immunohistochemistry (IHC),
sections were deparaffinized, boiled in antigen-retrieval solution, treated with primary antibodies in blocking buffer (5% donkey serum) at 4 °C overnight, treated with secondary antibody
for 2 h at room temperature in blocking buffer, and stained with Vectastain ABC KIT (Vector Laboratories) and DAB KIT (Vector Laboratories). Immunostaining images were taken using a Leica
DMi8 microscope (Leica). For immunofluorescence staining, paraffin sections were incubated with permeabilization buffer (0.3% Triton X-100 in PBS) for 30 min at room temperature, with
primary antibody at 4 °C overnight, and with secondary antibody for 2 h at room temperature, all in blocking buffer (5% donkey serum in 1X PBS). Images were taken using a confocal laser
microscope (Carl Zeiss Ltd). OIL RED O STAINING Cells were washed and fixed in 4% paraformaldehyde at room temperature for 1 h. After washing with 60% isopropanol three times, the cells were
stained with 60% filtered Oil Red O working solution (vol/vol in distilled water) of Oil red O stock solution (Sigma-Aldrich) at room temperature for 15 min. Cells were washed with ddH2O
before imaging. To quantify lipid accumulation, Oil Red O-stained lipids were eluted in 100% isopropanol, and then the optical density (OD) was measured at 500 nm. RNA ISOLATION AND
QUANTITATIVE REAL-TIME PCR (QPCR) ANALYSIS Total RNA from tissue or cells was isolated using Tripure Isolation Reagent (Sigma-Aldrich) and Bullet Blender Homogenizer (Next Advance) according
to the manufacturer’s protocol. cDNA was converted from 2 μg of total RNA by using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). qPCR was performed using 2X
Universal SYBR Green Fast qPCR Mix (Abclonal) following the manufacturer’s instructions and analyzed with a ViiA7 system (Applied Biosystems). Data were analyzed using the comparative Ct
method and the relative expression of mRNAs was determined after normalization to _Rplp0_. Primer sequences are available in Supplementary Table 2. OXYGEN CONSUMPTION RATE (OCR) ANALYSIS The
oxygen consumption rate of beige adipocytes was measured by using a Seahorse XF96 Extracellular Flux Analyzer according to the manufacturer’s instructions. SVF cells were seeded on XF96 and
induced to differentiate and maintain. The plate was subjected to a temperature-controlled (37 °C) extracellular analyzer at indicated time point. After measuring initial oxygen consumption
rate (OCR), 4 μM oligomycin, 2 μM FCCP and 1 μM rotenone/actinomycin A were then sequentially added into the plate by automatic pneumatic injection. Data were analyzed by Seahorse Wave
software. Basal respiration was calculated as [OCRinitial – OCRR&A]. The maximum respiration rate was computed as [OCRFCCP – OCRR&A]. MEASUREMENT OF MITOCHONDRIAL DNA (MTDNA) The
total DNA was isolated from beige adipocytes by using DNA isolation kit. mtDNA was amplified using primers specific for the mitochondrial cytochrome c oxidase subunit 2 (Cox2) gene and
normalized to an intron of the nuclear-encoded Hbb (β-globin) gene as described before54. The primer sequences were listed in Supplementary Table 2. RNA IMMUNOPRECIPITATION-QPCR (RIP-QPCR)
RIP was performed by using a Dynabeads Protein A immunoprecipitation kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, mature adipocytes were lysed in
lysis buffer (150 mM KCl, 10 mM HEPES, 2 mM EDTA, 0.5% NP-40, 0.5 mM DTT, protease inhibitor cocktail and RNase inhibitor) for 30 min at 4 °C. The lysates were centrifuged, and the
supernatant was collected. A small aliquot of lysate was saved as input, and the remaining sample was incubated with Protein A magnetic beads conjugated with p16INK4a, p19ARF or IgG antibody
at 4 °C for 4 h. Subsequently, the beads were washed with wash buffer and eluted in elution buffer. The input and immunoprecipitated RNAs were isolated by Tripure Isolation Reagent and
reverse transcribed into cDNA. The fold enrichment was detected by qPCR. CO-IMMUNOPRECIPITATION (CO-IP) Co-IP was performed by using a Pierce crosslink magnetic IP/Co-IP kit (Thermo Fisher
Scientific) according to the manufacturer’s instructions. Cells were rinsed with cold PBS, lysed in IP lysis buffer, cleared by centrifugation and protein concentration was estimated. A
small aliquot of lysate was saved as input, and the remaining sample was incubated with Protein A magnetic beads conjugated with p16INK4a, p19ARF, BCL2L1 or IgG antibody at 4 °C overnight.
Then, the beads were washed with wash buffer and eluted by heating in loading buffer. Immunoprecipitates and input were electrophoresed, transferred to PVDF membranes, and probed with the
indicated antibodies. RNA STABILITY ASSAY RNA stability assay was performed as previously described55. Briefly, cells were treated with 5 µg/ml actinomycin D (Sigma-Aldrich) to inhibit
global mRNA transcription. Samples were collected at the indicated time points. Total RNA was extracted for reverse transcription, and the levels of genes of interest were analyzed by qPCR.
HUMAN PARTICIPANTS Study participants were recruited from the Bariatric and General Surgery Clinics at the University of Illinois Hospital. We enrolled twelve obese subjects (8 females and 4
males) who underwent laparoscopic gastric sleeve bariatric surgery and five lean healthy controls (3 females and 2 males) who underwent elective surgeries such as hernia repair and
abdominal wall reconstruction. Subcutaneous adipose tissue samples were obtained during surgery. Adipose tissue samples were snap-frozen and stored for biological analyses. The age of the
participants ranged from 21 to 47 years old. Exclusion criteria included chronic inflammatory diseases, chronic organ failure, autoimmune diseases, cancer of any type, current smoking, or
current pregnancy. Subjects who were deemed eligible were informed about the study details, risks, and precautions taken to reduce this risk. All the study participants provided informed
written consent. Physical characteristics, including body weight and body mass index (BMI), were measured. Total fat mass was assessed using dual x-ray absorptiometry (DXA; iDXA, General
Electric Inc). Plasma concentrations of glucose were quantified in the fasting state as we previously described56. All protocols and procedures of the study followed the standards set by the
latest version of the Declaration of Helsinki and were approved by the Institutional Review Board of The University of Illinois at Chicago (protocol code 2017-1125). The details of
population characteristics are listed in Supplementary Table 3. STATISTICS AND REPRODUCIBILITY All data are presented as mean ± SEM. Two-tailed unpaired Student’s _t_ test (for comparison of
two groups) or one‐way ANOVA followed by Tukey’s test (for comparison of three or more groups) were conducted using GraphPad Prism software. All experiments were repeated two or three times
with representative data or images shown. _p_ < 0.05 was considered statistically significant in all the experiments. REPORTING SUMMARY Further information on research design is
available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The data that support the findings of this study are available in the “Methods” and
Supplementary Material of this article. Additional data are available upon request from the corresponding author. Source data are provided with this paper. REFERENCES * Bluher, M. Obesity:
global epidemiology and pathogenesis. _Nat. Rev. Endocrinol._ 15, 288–298 (2019). Article PubMed Google Scholar * Chouchani, E. T., Kazak, L. & Spiegelman, B. M. New advances in
adaptive thermogenesis: UCP1 and beyond. _Cell Metab._ 29, 27–37 (2019). Article CAS PubMed Google Scholar * Wu, J. et al. Beige adipocytes are a distinct type of thermogenic fat cell in
mouse and human. _Cell_ 150, 366–376 (2012). Article CAS PubMed PubMed Central Google Scholar * Cypess, A. M. et al. Activation of human brown adipose tissue by a beta3-adrenergic
receptor agonist. _Cell Metab._ 21, 33–38 (2015). Article CAS PubMed PubMed Central Google Scholar * Finlin, B. S. et al. Human adipose beiging in response to cold and mirabegron. _JCI
Insight_ 3 https://doi.org/10.1172/jci.insight.121510 (2018). * Sidossis, L. S. et al. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. _Cell Metab._
22, 219–227 (2015). Article CAS PubMed PubMed Central Google Scholar * Bartelt, A. & Heeren, J. Adipose tissue browning and metabolic health. _Nat. Rev. Endocrinol._ 10, 24–36
(2014). Article CAS PubMed Google Scholar * Rosenwald, M., Perdikari, A., Rulicke, T. & Wolfrum, C. Bi-directional interconversion of brite and white adipocytes. _Nat. Cell Biol._
15, 659–667 (2013). Article CAS PubMed Google Scholar * Roh, H. C. et al. Warming induces significant reprogramming of beige, but not brown, adipocyte cellular identity. _Cell Metab._
27, 1121–1137.e5 (2018). Article CAS PubMed PubMed Central Google Scholar * Altshuler-Keylin, S. et al. Beige adipocyte maintenance is regulated by autophagy-induced mitochondrial
clearance. _Cell Metab._ 24, 402–419 (2016). Article CAS PubMed PubMed Central Google Scholar * Lu, X. et al. Mitophagy controls beige adipocyte maintenance through a Parkin-dependent
and UCP1-independent mechanism. _Sci. Signal._ 11 https://doi.org/10.1126/scisignal.aap8526 (2018). * Kamijo, T. et al. Tumor suppression at the mouse INK4a locus mediated by the alternative
reading frame product p19ARF. _Cell_ 91, 649–659 (1997). Article CAS PubMed Google Scholar * Zindy, F., Quelle, D. E., Roussel, M. F. & Sherr, C. J. Expression of the p16INK4a tumor
suppressor versus other INK4 family members during mouse development and aging. _Oncogene_ 15, 203–211 (1997). Article CAS PubMed Google Scholar * Levine, A. J. & Oren, M. The first
30 years of p53: growing ever more complex. _Nat. Rev. Cancer_ 9, 749–758 (2009). Article CAS PubMed PubMed Central Google Scholar * Hannou, S. A., Wouters, K., Paumelle, R. &
Staels, B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: what have we learned from GWASs? _Trends Endocrinol. Metab._ 26, 176–184 (2015). Article CAS
PubMed Google Scholar * Morris, A. P. et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. _Nat. Genet._ 44,
981–990 (2012). Article CAS PubMed PubMed Central Google Scholar * Palmer, A. K. et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. _Aging Cell_ 18,
e12950 (2019). Article PubMed PubMed Central Google Scholar * Helman, A. et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. _Nat. Med._ 22, 412–420
(2016). Article CAS PubMed PubMed Central Google Scholar * Rabhi, N. et al. Cdkn2a deficiency promotes adipose tissue browning. _Mol. Metab._ 8, 65–76 (2018). Article CAS PubMed
Google Scholar * Park, J. et al. Progenitor-like characteristics in a subgroup of UCP1+ cells within white adipose tissue. _Dev. Cell_ 56, 985–999.e4 (2021). Article CAS PubMed PubMed
Central Google Scholar * Sidossis, L. & Kajimura, S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. _J. Clin. Invest._ 125, 478–486
(2015). Article PubMed PubMed Central Google Scholar * Liu, Y. et al. Yes-associated protein targets the transforming growth factor beta pathway to mediate high-fat/high-sucrose
diet-induced arterial stiffness. _Circ. Res._ 130, 851–867 (2022). Article CAS PubMed Google Scholar * Pimkina, J., Humbey, O., Zilfou, J. T., Jarnik, M. & Murphy, M. E. ARF induces
autophagy by virtue of interaction with Bcl-xl. _J. Biol. Chem._ 284, 2803–2810 (2009). Article CAS PubMed PubMed Central Google Scholar * Budina-Kolomets, A., Hontz, R. D., Pimkina, J.
& Murphy, M. E. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. _Autophagy_ 9, 1553–1565 (2013). Article
CAS PubMed PubMed Central Google Scholar * Hao, X. et al. ADAR1 downregulation by autophagy drives senescence independently of RNA editing by enhancing p16(INK4a) levels. _Nat. Cell
Biol._ 24, 1202–1210 (2022). Article CAS PubMed PubMed Central Google Scholar * Rocchi, A. et al. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and
improved cognition in Alzheimer’s disease. _PLoS Genet._ 13, e1006962 (2017). Article PubMed PubMed Central Google Scholar * Shoji-Kawata, S. et al. Identification of a candidate
therapeutic autophagy-inducing peptide. _Nature_ 494, 201–206 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Baltz, A. G. et al. The mRNA-bound proteome and its global
occupancy profile on protein-coding transcripts. _Mol. Cell_ 46, 674–690 (2012). Article CAS PubMed Google Scholar * Al-Mohanna, M. A., Al-Khalaf, H. H., Al-Yousef, N. & Aboussekhra,
A. The p16INK4a tumor suppressor controls p21WAF1 induction in response to ultraviolet light. _Nucleic Acids Res._ 35, 223–233 (2007). Article CAS PubMed Google Scholar * Becher, T. et
al. Brown adipose tissue is associated with cardiometabolic health. _Nat. Med._ 27, 58–65 (2021). Article CAS PubMed PubMed Central Google Scholar * Villarroya, F., Cereijo, R.,
Villarroya, J. & Giralt, M. Brown adipose tissue as a secretory organ. _Nat. Rev. Endocrinol._ 13, 26–35 (2017). Article CAS PubMed Google Scholar * Whitehead, A. et al. Brown and
beige adipose tissue regulate systemic metabolism through a metabolite interorgan signaling axis. _Nat. Commun._ 12, 1905 (2021). Article ADS PubMed PubMed Central Google Scholar *
Ikeda, K., Maretich, P. & Kajimura, S. The common and distinct features of brown and beige adipocytes. _Trends Endocrinol. Metab._ 29, 191–200 (2018). Article CAS PubMed PubMed
Central Google Scholar * Pellegrini, C. et al. Altered adipocyte differentiation and unbalanced autophagy in type 2 Familial Partial Lipodystrophy: an in vitro and in vivo study of adipose
tissue browning. _Exp. Mol. Med._ 51, 89 (2019). Article PubMed PubMed Central Google Scholar * Ro, S. H. et al. Autophagy in adipocyte browning: emerging drug target for intervention
in obesity. _Front. Physiol._ 10, 22 (2019). Article ADS PubMed PubMed Central Google Scholar * Liu, M. et al. Grb10 promotes lipolysis and thermogenesis by phosphorylation-dependent
feedback inhibition of mTORC1. _Cell Metab._ 19, 967–980 (2014). Article CAS PubMed PubMed Central Google Scholar * Liu, D. X. et al. Activation of mTORC1 is essential for
beta-adrenergic stimulation of adipose browning. _J. Clin. Investig._ 126, 1704–1716 (2016). Article ADS PubMed PubMed Central Google Scholar * Liu, D. X., Ceddia, R. P. & Collins,
S. Cardiac natriuretic peptides promote adipose ‘browning’ through mTOR complex-1. _Mol. Metab._ 9, 192–198 (2018). Article CAS PubMed PubMed Central Google Scholar * Baerga, R., Zhang,
Y., Chen, P. H., Goldman, S. & Jin, S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. _Autophagy_ 5, 1118–1130 (2009). Article
CAS PubMed Google Scholar * Zhang, Y. et al. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. _Proc. Natl Acad. Sci. USA_ 106,
19860–19865 (2009). Article ADS CAS PubMed PubMed Central Google Scholar * He, C. & Levine, B. The Beclin 1 interactome. _Curr. Opin. Cell Biol._ 22, 140–149 (2010). Article CAS
PubMed PubMed Central Google Scholar * Jin, Y. et al. Depletion of adipocyte Becn1 leads to lipodystrophy and metabolic dysregulation. _Diabetes_ 70, 182–195 (2021). Article CAS PubMed
Google Scholar * Son, Y. et al. Adipocyte-specific Beclin1 deletion impairs lipolysis and mitochondrial integrity in adipose tissue. _Mol. Metab._ 39, 101005 (2020). Article CAS PubMed
PubMed Central Google Scholar * Berry, D. C. et al. Cellular aging contributes to failure of cold-induced beige adipocyte formation in old mice and humans. _Cell Metab._ 25, 166–181
(2017). Article CAS PubMed Google Scholar * Geach, T. Adipose tissue: reversing age-related decline in beiging. _Nat. Rev. Endocrinol._ 13, 64 (2017). Article CAS PubMed Google
Scholar * Palmer, A. K. & Kirkland, J. L. Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. _Exp. Gerontol._ 86, 97–105 (2016). Article CAS
PubMed PubMed Central Google Scholar * Rogers, N. H., Landa, A., Park, S. & Smith, R. G. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose
tissue. _Aging Cell_ 11, 1074–1083 (2012). Article CAS PubMed Google Scholar * Yoneshiro, T. et al. Age-related decrease in cold-activated brown adipose tissue and accumulation of body
fat in healthy humans. _Obesity_ 19, 1755–1760 (2011). Article PubMed Google Scholar * Wang, Q. et al. Brown adipose tissue activation is inversely related to central obesity and
metabolic parameters in adult human. _PLoS ONE_ 10, e0123795 (2015). Article PubMed PubMed Central Google Scholar * Collins, S., Daniel, K. W., Petro, A. E. & Surwit, R. S.
Strain-specific response to beta 3-adrenergic receptor agonist treatment of diet-induced obesity in mice. _Endocrinology_ 138, 405–413 (1997). Article CAS PubMed Google Scholar *
Goncalves, L. F. et al. Ageing is associated with brown adipose tissue remodelling and loss of white fat browning in female C57BL/6 mice. _Int. J. Exp. Pathol._ 98, 100–108 (2017). Article
CAS PubMed PubMed Central Google Scholar * Iacovino, M. et al. Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and
primary cells. _Stem Cells_ 29, 1580–1588 (2011). Article CAS PubMed Google Scholar * Mina, A. I. et al. CalR: a web-based analysis tool for indirect calorimetry experiments. _Cell
Metab._ 28, 656–666.e1 (2018). Article CAS PubMed PubMed Central Google Scholar * Wu, R. et al. RNA-binding protein YBX1 promotes brown adipogenesis and thermogenesis via
PINK1/PRKN-mediated mitophagy. _FASEB J._ 36, e22219 (2022). Article CAS PubMed Google Scholar * Wang, X. et al. m(6)A mRNA methylation controls autophagy and adipogenesis by targeting
Atg5 and Atg7. _Autophagy_ 16, 1221–1235 (2020). Article CAS PubMed Google Scholar * Haloul, M. et al. Hyperhomocysteinemia and low folate and vitamin B12 are associated with vascular
dysfunction and impaired nitric oxide sensitivity in morbidly obese patients. _Nutrients_ 12 https://doi.org/10.3390/nu12072014 (2020). Download references ACKNOWLEDGEMENTS We thank Dr.
Lijun Rong for help with manuscript preparation and giving fruitful advice. We thank members in the Y.J. laboratory, especially Nipun Velupally and Meram Mohamed Mahmoud, for mouse
genotyping and technical help. We thank Cynthia Rose Adams and Jeanette Purcell for assistance with mouse husbandry, Stefan J. Green and the Research Resources Center for RT-PCR analysis,
Dr. Brian Layden and Metabolic Phenotyping Core for analytical and phenotypical mouse measurements, Balaji Ganesh and Flow Cytometry Core facility for FACS and members of the Y.J. laboratory
for helpful comments on the manuscript. This work was supported by grants to Y.J. from the National Institute of Diabetes and Digestive and Kidney Disease grant (NIDDK) K01 DK11177, R03
DK127149, R01 DK132398 and Pilot & Feasibility Diabetes Research & Training Center (DRTC) Award (P30DK020595). Cartoons in Figs. 3a, 6a, 7i and Supplementary Figs. 4a, 5d, 7a were
created with BioRender.com. AUTHOR INFORMATION Author notes * These authors contributed equally: Ruifan Wu, Jooman Park. AUTHORS AND AFFILIATIONS * Department of Physiology and Biophysics,
College of Medicine, University of Illinois at Chicago, Chicago, IL, 60612, USA Ruifan Wu, Jooman Park, Yanyu Qian, Zuoxiao Shi, Ruoci Hu, Yexian Yuan & Yuwei Jiang * Guangdong
Laboratory of Lingnan Modern Agriculture, Guangdong Province Key Laboratory of Animal Nutritional Regulation and National Engineering Research Center for Breeding Swine Industry, College of
Animal Science, South China Agricultural University, Guangzhou, 510642, China Ruifan Wu, Yexian Yuan, Gang Shu & Qingyan Jiang * Department of Pharmaceutical Sciences, University of
Illinois at Chicago, Chicago, IL, 60612, USA Zuoxiao Shi, Ruoci Hu & Yuwei Jiang * Department of Microbiology and Immunology, University of Illinois at Chicago, Chicago, IL, 60612, USA
Shaolei Xiong & Zilai Wang * Department of Pharmacology and Regenerative Medicine, College of Medicine, University of Illinois at Chicago, Chicago, IL, 60612, USA Gege Yan &
Sang-Ging Ong * Division of Cardiology, Department of Medicine, University of Illinois at Chicago, Chicago, IL, 60612, USA Sang-Ging Ong * Department of Kinesiology and Nutrition, University
of Illinois at Chicago, Chicago, IL, 60612, USA Qing Song & Zhenyuan Song * Division of Endocrinology, Department of Medicine, University of Illinois at Chicago, Chicago, IL, 60612, USA
Abeer M. Mahmoud, Pingwen Xu & Yuwei Jiang * Department of Cell and Developmental Biology, Feinberg School of Medicine, Northwestern University, Chicago, IL, 60611, USA Congcong He *
Lillehei Heart Institute, University of Minnesota, Minneapolis, MN, 55455, USA Robert W. Arpke & Michael Kyba Authors * Ruifan Wu View author publications You can also search for this
author inPubMed Google Scholar * Jooman Park View author publications You can also search for this author inPubMed Google Scholar * Yanyu Qian View author publications You can also search
for this author inPubMed Google Scholar * Zuoxiao Shi View author publications You can also search for this author inPubMed Google Scholar * Ruoci Hu View author publications You can also
search for this author inPubMed Google Scholar * Yexian Yuan View author publications You can also search for this author inPubMed Google Scholar * Shaolei Xiong View author publications You
can also search for this author inPubMed Google Scholar * Zilai Wang View author publications You can also search for this author inPubMed Google Scholar * Gege Yan View author publications
You can also search for this author inPubMed Google Scholar * Sang-Ging Ong View author publications You can also search for this author inPubMed Google Scholar * Qing Song View author
publications You can also search for this author inPubMed Google Scholar * Zhenyuan Song View author publications You can also search for this author inPubMed Google Scholar * Abeer M.
Mahmoud View author publications You can also search for this author inPubMed Google Scholar * Pingwen Xu View author publications You can also search for this author inPubMed Google Scholar
* Congcong He View author publications You can also search for this author inPubMed Google Scholar * Robert W. Arpke View author publications You can also search for this author inPubMed
Google Scholar * Michael Kyba View author publications You can also search for this author inPubMed Google Scholar * Gang Shu View author publications You can also search for this author
inPubMed Google Scholar * Qingyan Jiang View author publications You can also search for this author inPubMed Google Scholar * Yuwei Jiang View author publications You can also search for
this author inPubMed Google Scholar CONTRIBUTIONS Y.J. conceived and designed the experiments. R.W. and J.P. conducted most of the experiments. R.W., J.P., and Y.J. interpreted the
experiments. R.W.A. and M.K. developed the TRE-Ccnd1 transgenic mouse line. A.M.M. provided the human adipose samples. R.W. and Y.J. wrote and revised the manuscript, and J.P., Y.Q., Z.S.,
R.H., Y.Y., S.X., Z.W., G.Y., S.G.O., Q.S., Z.S., A.M.M., P.X., C.H., R.W.A., M.K., G.S., and Q.J. reviewed it. CORRESPONDING AUTHOR Correspondence to Yuwei Jiang. ETHICS DECLARATIONS
COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Jean-Sébastien Annicotte, Pamela Mattar and the other,
anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE
DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Wu, R., Park, J., Qian, Y. _et al._ Genetically prolonged beige fat in male mice confers long-lasting metabolic health. _Nat Commun_ 14, 2731 (2023).
https://doi.org/10.1038/s41467-023-38471-z Download citation * Received: 24 January 2023 * Accepted: 04 May 2023 * Published: 12 May 2023 * DOI: https://doi.org/10.1038/s41467-023-38471-z
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative




