- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Metformin, a diabetes drug with anti-aging cellular responses, has complex actions that may alter dementia onset. Mixed results are emerging from prior observational studies. To address this
complexity, we deploy a causal inference approach accounting for the competing risk of death in emulated clinical trials using two distinct electronic health record systems. In
intention-to-treat analyses, metformin use associates with lower hazard of all-cause mortality and lower cause-specific hazard of dementia onset, after accounting for prolonged survival,
relative to sulfonylureas. In parallel systems pharmacology studies, the expression of two AD-related proteins, APOE and SPP1, was suppressed by pharmacologic concentrations of metformin in
differentiated human neural cells, relative to a sulfonylurea. Together, our findings suggest that metformin might reduce the risk of dementia in diabetes patients through mechanisms beyond
glycemic control, and that SPP1 is a candidate biomarker for metformin’s action in the brain.
Repurposing drugs affords a route to therapeutic development that is shorter, less expensive, and more likely to succeed1. However, with fewer economic incentives for drug repurposing than
for bringing new drugs to market, combined evidence from real-world data and mechanistic studies that supports the therapeutic hypothesis might justify a Randomized Clinical Trial (RCT).
Alzheimer’s disease (AD), with one to two decades of accumulating pathology prior to symptom onset, brings another challenge: a preclinical period so long that it is often not economically
feasible for RCTs and presents ethical problems. Observational studies in Electronic Health Records (EHR) allow longer follow-up times than RCTs and offer the possibility of evaluating drugs
already approved by the Food and Drug Administration (FDA) and/or the European Medicines Agency (EMA) within the preclinical period of dementia. Using the target trial method2,3 of
conducting observational studies that aim to mimic RCTs, we emulated the same target trial in two distinct EHR systems. We reasoned that replicating results across two samples from vastly
different settings—one a healthcare system anchored in two large tertiary care hospitals and another a nation-wide primary care network—would provide a more robust estimate of the
generalizability of the drug’s effect4,5. Moreover, differences in medical practice, data collection, timing and length of follow-up, patterns of missingness, and known and unknown sources
of bias between two EHR databases bolster any signal observed in both samples.
In this study, we emulated a target trial to estimate the effects of metformin compared to sulfonylureas on the risk of death and dementia. Metformin is a first-line antidiabetic drug with
additional properties that may slow biological aging6,7, including some evidence of increased survival8. Since the risk of dementia rises very steeply with age9, it has been hypothesized
that metformin would reduce such risk. Clinical studies of metformin, however, have had mixed results in association with dementia risk in older adults10,11. To address the potentially
opposing influences of metformin on dementia—that it might reduce the hazard of death and therefore put more people at risk of developing dementia while reducing the hazard of dementia by
slowing biological aging, we used a competing risks analysis framework12. We used the target trial method2 to emulate a trial in the Research Patient Data Registry (US RPDR13) at Mass
General Brigham (formerly Partners) Health Care system in the US and the UK Clinical Practice Research Datalink (UK CPRD14) database among initiators of metformin vs. the other first-line
therapy for diabetes, the sulfonylureas (reference group).
In parallel, we conducted an in vitro systems pharmacology evaluation of both drugs on differentiated human neural cells in culture to identify genes whose expression is differentially
altered in neural cells with metformin treatment relative to the vehicle and to glyburide, one of the sulfonylureas. The secreted products from these differentially expressed genes are
candidate pharmacodynamic markers of metformin’s actions in the brain, which can be quantified in the cerebrospinal fluid (CSF). Our EHR-based results may serve as an example of the use of
real world data (RWD) to inform the design of clinical trial eligibility criteria15 for a trial of metformin with the primary outcome of dementia onset. Further, our systems pharmacology
studies may suggest a pharmacodynamic CSF biomarker for metformin’s anti-aging actions in the human brain beyond its hypoglycemic actions.
We emulated the target trial in cohorts from the US RPDR and UK CPRD EHR databases (Table 1) with a 1-year run-in period. Our target trial outcomes were time to first diagnosis of dementia
or death in type 2 diabetics over age 50, starting on metformin- or sulfonylurea-monotherapy, and followed for at least 1 year. Of note, the 1-year run-in period was selected to ensure
sufficient drug exposure before measuring outcomes. While the duration of a clinical trial is usually fixed, the duration of follow-up in the emulated trial is often much longer (US RPDR
median: 5.0 years (max 12 years); UK CPRD median: 6.0 years (max 16 years)).
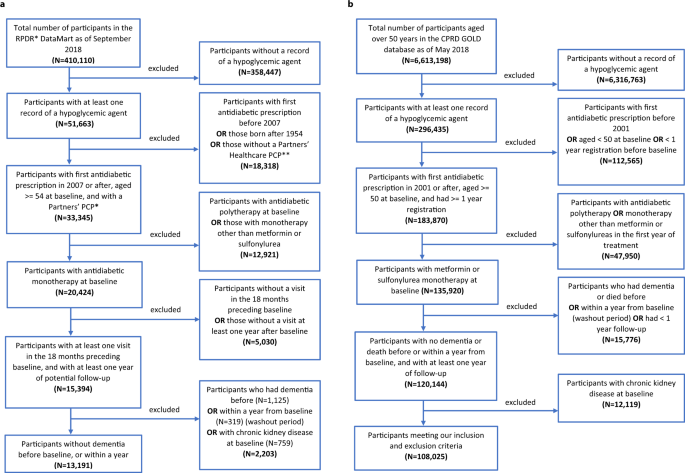
The US RPDR cohort, which was drawn from patients receiving primary care at an academic health care system, included 13,191 patients who started on metformin- (11,229; 85%) or sulfonylurea-
monotherapy (1962; 15%) (Fig. 1a). Patients who had a diagnosis of dementia, or died within the first year of follow-up, were excluded from the study population to emulate the standard
exclusion criterion in clinical trials of patients with baseline cognitive impairment or a high morbidity index. In addition, patients with chronic kidney disease (CKD; see Extended Data
Table 1 for definitions) at treatment initiation—a contraindication for metformin, but not for sulfonylureas—were excluded from the cohort. Metformin initiators were younger than their
sulfonylurea counterparts (Table 2). Among the metformin initiators, there were more hypertensives and fewer missing values for baseline body mass index (BMI) than among the sulfonylurea
initiators (Table 2). The baseline glycosylated hemoglobin (HbA1C) levels and other baseline characteristics, however, were comparable between the two groups (Table 2).
a Flowchart of inclusion and exclusion criteria of study population in RPDR. b Flowchart of inclusion and exclusion criteria of study population in CPRD.
The UK CPRD cohort, which was drawn from primary care practices across 13 regions in the UK, included 108,025 patients in total with 94,208 (87%) metformin initiators and 13,817 (13%)
sulfonylurea initiators (Fig. 1b). Patients who were diagnosed with dementia or died within the first year of follow-up were excluded from the study population (Fig. 1b). Those with CKD at
treatment initiation (Extended Data Table 2) were also excluded. As in the US RPDR cohort, we found that patients treated with metformin were younger than patients treated with sulfonylureas
in the UK CPRD cohort (Table 2). They were also more likely to have entered the cohort more recently and to have lower HbA1C and higher BMI at baseline. Further, they included more
cardiovascular disease (CVD) and hypertension cases, but fewer cancer cases at baseline than the sulfonylurea group (Table 2).
No piece of information that identifies individual patients is presented in this paper.
First, we compared the effect of metformin vs. sulfonylureas on all-cause mortality in both cohorts, since metformin use has previously been reported to improve survival relative to the
sulfonylureas in distinct US16 and UK17 cohorts of type 2 diabetics.
In the US RPDR cohort, 3.7% (n = 415) of metformin initiators and 7.8% (n = 154) of sulfonylurea initiators died during follow-up (median: 5.0 years; total: 74,107 person-years; range of age
at death: 57–104 years). Using a Cox proportional hazards (PH) regression model with inverse probability of treatment weighting (IPTW) to emulate randomization, the estimated hazard ratio
for all-cause mortality was 0.57 (95% CI: [0.48;0.67]) for metformin initiators relative to sulfonylurea initiators (Fig. 2a). Next, we examined metformin’s effects by age (≤70 vs. >70),
sex, and BMI strata. Overall, there was no evidence for heterogenous treatment effects across baseline age, sex, or BMI levels in the US RPDR cohort (Fig. 2b). Similar results were obtained
using age strata defined as ≤65 vs. >65 and ≤75 vs. >75 (Extended Data Table 3).
a, c Kaplan–Meier survival curves for metformin and sulfonylurea initiators with shaded areas representing 95% Confidence Intervals (CI), based on pointwise 0.025- and 0.975-quantiles of
sample bootstrap distributions. N = # of patients at baseline, D = # of deaths during follow-up. Hazard ratios (HR) were estimated using the Cox Proportional Hazards (PH) model, with only
treatment as a covariate, and baseline covariate distributions between treatment arms balanced by Inverse Propensity score of Treatment Weighting (IPTW). b, d Forest plots presenting
all-cause mortality HRs overall and stratified by age, sex, and BMI level at baseline, with sulfonylurea initiators as the reference group. Covariate balancing using IPTW was conducted in
each stratum independently. Error bars represent 95% CIs for hazard ratios. A two-sided Wald test of whether the hazard ratio associated with metformin treatment initiation is 1, with robust
variance estimator, was used. No further correction for multiple hypothesis testing was applied.
In the larger UK CPRD cohort, 13.7% (n = 12,941) of metformin initiators and 37.4% (n = 5173) of sulfonylurea initiators died during follow-up (median: 6.0 years; total: 696,725
person-years; range of age at death: 51–107 years). The UK CPRD had similar results for the effect of metformin vs. sulfonylureas on all-cause mortality in the full study population (Fig.
2c), with an overall hazard ratio of 0.66 (95% CI: 0.61;0.71). Results of subgroup analyses revealed evidence for a stronger effect of metformin among patients with a younger age at
treatment initiation (≤70 years), and patients with higher baseline BMI, but there was no difference by sex (Fig. 2d). The age-stratified analysis described above yielded similar results
(Extended Data Table 4).
Our harmonized Drug Repurposing in Alzheimerʼs Disease (DRIAD)-EHR approach—with analyses conducted in two very different patient populations and carefully adjusted for baseline differences
in age and other risk factors—demonstrates a robust reduction in the hazard of death in patients treated with metformin compared to those treated with sulfonylureas, consistent with previous
reports18,19. In both cohorts, we note that the survival curves between the two treatment groups separate ~3 years after treatment initiation, and that this separation persists for a long
time (12 years observed in US RPDR and 16 in UK CPRD).
Death is a competing event that precludes the development of dementia, but the use of a competing risks analysis in previous studies8,17 has been limited: death has been considered as a
competing event for dementia only in a proportional hazards model where the hazard ratio is the measure of treatment effect. Here, we emulated a target trial of metformin vs. sulfonylureas
in cognitively asymptomatic type 2 diabetics, estimating both the time-invariant hazard ratio and the time-dependent cumulative incidence function (CIF) for dementia, using a causal
competing risks framework. We defined the average treatment effect (ATE) as the difference between risk functions corresponding to two potential outcomes (for definitions, see “Methods”).
In the US RPDR cohort, 7.7% (n = 869) of metformin initiators and 12.3% (n = 241) of sulfonylurea initiators were diagnosed with dementia during follow-up (median: 5.0 years; total: 71,191
person-years; range of dementia onset age: 57–113 years). In a cause-specific Cox PH regression model with IPTW for emulation of baseline randomization, the estimated cause-specific hazard
ratio for dementia was 0.81 (95% CI: [0.69;0.94]) for metformin initiators relative to sulfonylurea initiators (Fig. 3a). In the UK CPRD cohort, 5.9% (n = 5561) of metformin initiators and
12.3% (n = 1699) of sulfonylurea initiators were diagnosed with dementia during follow-up (median: 6.0 years; total: 695,281 person-years; range of dementia onset age: 51–114 years). The
estimated cause-specific hazard ratio for dementia was 0.86 (95% CI: [0.77;0.96]) for metformin initiators, relative to sulfonylurea initiators (Fig. 3b), very similar to the US RPDR cohort.
HRs were estimated using the Cox PH model for the cause-specific hazards of dementia, with only treatment as a covariate and baseline covariate distributions between treatment arms balanced
by IPTW. a, b Forest plots present HRs overall and stratified by age, sex, and BMI level at baseline, with sulfonylureas as the reference group. N = # of patients at baseline, Onsets = # of
patients with dementia onset during follow-up and prior to death. Covariate balancing using IPTW was conducted in each stratum independently. Error bars represent 95% CIs for hazard ratios.
A two-sided Wald test of whether the hazard ratio associated with metformin treatment initiation is 1, with robust variance estimator, was used. No further correction for multiple hypothesis
testing was applied.
In the time-dependent CIF analysis, the 5-year risk (for definition, see “Methods”) of developing dementia in the US RPDR cohort was 7.2% (95% CI: [6.7;7.8]%) among metformin initiators and
8.8% (95% CI: [7.5;10]%) among sulfonylurea initiators, yielding a risk difference (for definition, see “Methods”) of −1.6% (95% CI: [−3.1;−0.17]%) (Fig. 4a, c). In the UK CPRD cohort, the
5-year risk difference was smaller at −0.35% (95% CI: [−0.68;−0.031]%) (Fig. 4b, d). Although the hazard ratios for dementia in the UK CPRD and US RPDR were similar, the risk differences
over time for both death and dementia were strikingly dissimilar in the two cohorts (Fig. 4).
Cumulative incidence functions (CIF) or risk curves, along with their 95% CIs (represented by shaded areas), based on pointwise 0.025- and 0.975-quantiles of sample bootstrap distributions,
were estimated using the Cox model for the cause-specific hazards, with only treatment as a covariate and baseline covariate distributions between treatment arms balanced by IPTW. a, b CIF
curves for dementia onset (in blue hues) and competing death (in orange hues) for metformin vs. sulfonylurea initiators. Follow-up times are up to 12 and 16 years in the US RPDR (a) and the
UK CPRD (b) cohorts, respectively. c, d Risk difference curves for dementia onset (in blue) and competing death (in orange), in the US RPDR (c) and the UK CPRD (d) cohorts, respectively. A
negative risk difference value during certain time periods indicates that initiation of metformin is beneficial, as compared to sulfonylureas.
First, while the dementia risk difference between metformin and sulfonylureas was minimal in the US RPDR, it always showed a slight benefit for metformin over sulfonylureas in this cohort.
However, the risk difference observed in the UK CPRD changed over time, and the point of no risk difference between the two drugs was reached at about 7.5 years (Fig. 4). The seemingly
discordant hazards ratio and CIF results in the UK CPRD sample are likely because metformin has a protective effect on both the hazard of dementia (HR = 0.86, 95% CI: [0.77;0.96]) and the
hazard of competing death (HR = 0.64, 95% CI: [0.59;0.69]), yielding more “survivors” over time in the metformin group, and thus more individuals at risk of developing dementia.
Second, the risk differences for both death and dementia were much closer to each other in the US RPDR than in the UK CPRD cohort. These differences between the two cohorts could potentially
be explained by different population structures, particularly their baseline age distribution (Extended Data Fig. 1). The UK CPRD cohort had a higher death rate than the US RPDR one,
affecting the total number of patients at risk over time (Extended Data Fig. 2). The comparison of the absolute cumulative hazards of dementia and death in a competing risks approach
revealed additional differences between the UK CPRD and US RPRD cohorts, in terms of both the magnitude and trajectory of diagnosed dementia (Extended Data Fig. 3). In the US RPDR cohort,
the rates of diagnosed dementia in both treatment arms were higher than the death rates, whereas the opposite pattern was observed in the UK CPRD. These underlying differences likely explain
the differing risk curves for death and dementia between the two cohorts.
Overall, the benefits of metformin observed here can be interpreted in terms of both delaying dementia onset (Fig. 4, blue curves) and prolonging life without dementia (Fig. 4, orange
curves). Notably, the risk difference for death was of larger magnitude in the UK CPRD than in the US RPDR cohort (respectively, a 10% vs. 5% reduction in risk after an average of 12 years
of follow-up, Fig. 4c, d). This can be interpreted as the result of a higher overall death rate in the UK CPRD cohort, relative to the rate of dementia onset.
To assess the robustness of our results to modeling choices, we conducted a sensitivity analysis by using a nonparametric approach, thereby relaxing the proportional hazards assumption. Of
note, the PH assumption held for both cause-specific hazards in the US RPDR cohort, while there was evidence of a deviation from this assumption in the UK CPRD cohort in terms of the hazards
of death. However, this deviation was not large enough to affect our conclusions (Extended Data Figs. 4, 5).
Since age is the principal risk factor for dementia, we further investigated the effect modification of metformin as compared to sulfonylureas by the age at treatment initiation. Stratifying
the US RPDR cohort into two groups (age ≤ 70 and age >70), we found that the ATE of metformin vs. sulfonylureas on dementia onset observed in the full sample was mainly driven by the
younger stratum (Fig. 3a), i.e., treatment initiation at age ≤70 (HR = 0.69, 95% CI: [0.54;0.88]). Conversely, the effect of metformin on dementia onset was reduced for patients who started
antidiabetic treatment at age >70 (HR = 0.94, 95% CI: [0.79;1.13]). However, there were fewer patients who started antidiabetic treatment at age >70 than earlier (38% vs. 62%) and the older
stratum had a shorter length of follow-up (median: 4.1 vs. 5.6 years; total: 22,960 vs. 48,231 person-years). Nevertheless, the age-specific finding in the US RPDR cohort suggests that
metformin may be especially beneficial—relative to sulfonylureas—for those who initiate treatment at a younger age.
The difference in treatment effect between age groups was less clear in the larger UK CPRD cohort, with a HR of 0.82 (95% CI: [0.67;0.99]) in patients aged ≤70, and of 0.88 (95% CI:
[0.77;0.99]) in those aged >70 (Fig. 3b). Similar results were obtained using the risk difference: a stronger effect of metformin on dementia onset was observed in the US RPDR cohort, as
compared to the UK CPRD, in patients who initiated treatment before age 70 (Extended Data Fig. 6).
Since baseline HbA1C levels did not modify the effect of metformin, we also explored whether the drug acted primarily by a better control of blood sugar. For this, we applied a repeated
measures mixed effects model on all HbA1C values recorded three months after treatment initiation and beyond. In the US RPDR cohort, 10,180 (77%) patients had HbA1C data available: 8794
(78%) and 1386 (71%) among metformin and sulfonylurea initiators, respectively. Interestingly, we found that although the average level of HbA1C was lower (p