- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
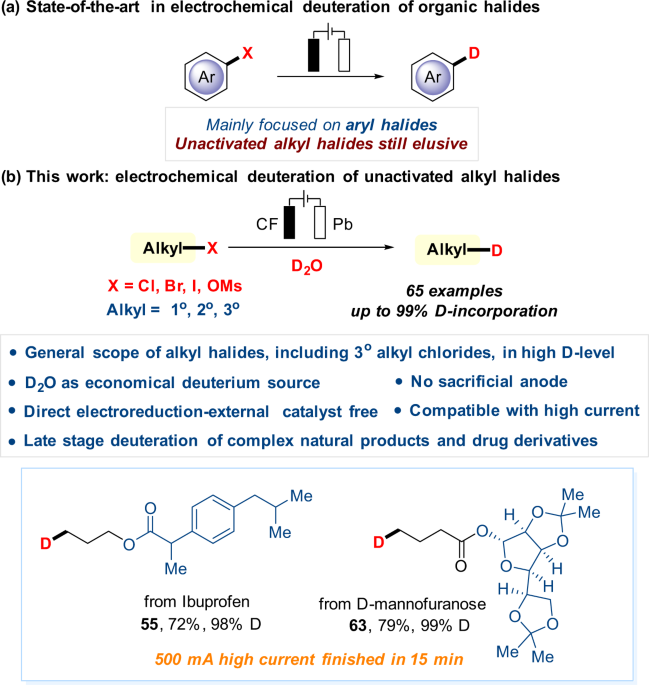
ABSTRACT Herein, a facile and general electroreductive deuteration of unactivated alkyl halides (X = Cl, Br, I) or pseudo-halides (X = OMs) using D2O as the economical deuterium source was
reported. In addition to primary and secondary alkyl halides, sterically hindered tertiary chlorides also work very well, affording the target deuterodehalogenated products with excellent
efficiency and deuterium incorporation. More than 60 examples are provided, including late-stage dehalogenative deuteration of natural products, pharmaceuticals, and their derivatives, all
with excellent deuterium incorporation (up to 99% D), demonstrating the potential utility of the developed method in organic synthesis. Furthermore, the method does not require external
catalysts and tolerates high current, showing possible use in industrial applications. SIMILAR CONTENT BEING VIEWED BY OTHERS PD-CATALYZED DEUTERATION OF ARYL HALIDES WITH DEUTERIUM OXIDE
Article Open access 16 March 2025 ORGANOPHOTOCATALYTIC SELECTIVE DEUTERODEHALOGENATION OF ARYL OR ALKYL CHLORIDES Article Open access 17 May 2021 ELECTROCHEMICAL HALOGEN-ATOM TRANSFER
ALKYLATION VIA Α-AMINOALKYL RADICAL ACTIVATION OF ALKYL IODIDES Article Open access 26 October 2023 INTRODUCTION Compounds with D-labeling play an important role in the realm of chemistry,
mechanistic studies, and pharmaceutical science1,2,3,4,5,6,7,8,9. Dehalogenative deuteration from organic halides is a straightforward way to obtain deuterated target compounds. However,
major studies focused on dehalogenative deuteration of aryl halides and activated alkyl halides10,11,12,13,14,15,16,17,18,19,20. In contrast, corresponding reports on unactivated alkyl
halides, particularly on unactivated alkyl chlorides, have not been well elucidated. Several realizations have been reported but suffer from a number of problems, such as limiting to alkyl
iodides using radical initiators21,22, the usage of a stoichiometric metal reductant23, long reaction time, and low reactivity of tertiary alkyl halides using photocatalysis24, etc.
Therefore, the development of simple, efficient, and environmentally-friendly approaches to access deuterated compounds from unactivated alkyl halides with high D-incorporation is highly
desirable. Meanwhile, organic electrochemistry has become a powerful tool for sustainable synthesis by employing electron instead of stoichiometric amounts of redox reagents in the past
decade25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44. Organic halides are important players in traditional organic chemistry as well as promising electrochemistry, owing to
their versatile reactivity and readily available properties. Thus, impressive electrochemical progress of organic halides has been made, such as reductive bifunctionalization of
alkenes45,46,47,48, reductive coupling with halides49,50,51,52,53,54,55,56, and others57,58,59,60,61,62,63,64,65,66,67,68. Hence, electrochemical deuteration of organic halides could be a
straightforward and promising route for valuable deuterated compounds, which has gained increasing attention69,70,71. However, current research and success in electrochemical deuteration
have been focused on aryl halides. In sharp contrast, electrochemically driven dehalogenative deuteration of unactivated halides has proven elusive (Fig. 1a). Direct electrochemical
deuteration of unactivated alkyl halides is challenging due to their extremely negative reduction potential19,72,73,74, and likelihood of competitive undesired side reactions50. Herein, we
report our effort in developing this dehalogenative deuteration of unactivated alkyl halides, making it a general, economically and environmentally approach through electrochemistry (Fig.
1b). Notable features of this strategy include: (a) broad scope and excellent D-incorporation, including unsatisfactory tertiary chlorides in previous studies, (b) D2O as an economical
deuterium source, (c) external catalysts-free and the use of electricity as an environmentally-friendly reductant, (d) catalytic amounts of electrolyte and no sacrificial anode, (e)
tolerance with high current (up to 500 mA), making it potentially applicable in industrial production, (f) late-stage deuteration of complex natural products and drug derivatives. RESULT
OPTIMIZATION OF ELECTROREDUCTIVE DEHALOGENATION DEUTERATION Initially, we started our investigation using D2O as the economical deuterium source, unactivated alkyl bromide 1 as the
substrate, and DMF as the solvent. After careful optimization, the corresponding product 2 was finally obtained in excellent yield and D-incorporation under 30 mA constant current with
carbon felt (CF) and Pb as electrodes, in the presence of DIPEA as well as the catalytic amount of TBAI as the electrolyte in an undivided cell at room temperature for 10 hours (Table 1,
entry 1). The control experiment revealed that the electricity was essential for the observed reactivity (entry 2). The reaction still worked well in the absence of DIPEA with 60% yield
(entry 3), which showed that DMF could also be oxidized on anode. In addition, reducing the current to 20 mA led to lower yield and D-incorporation (entry 4). Surprisingly, the desired
transformation proceeded smoothly with a higher current at 100 mA in 60 min (entry 5). It is notable that even under an extremely high current at 500 mA, the reaction still proceeded well to
give the target product in only 15 min, without diminishing the yield and D-labeling level, showing potential usage in industrial amplification (entry 6). DMSO was proven to be a poor
solvent for this transformation (entry 7), while MeCN gave a comparable yield but lower D-incorporation (entry 8). The reaction also worked well when _n_Bu4NBF4 was used as the electrolyte,
albeit in a slightly lower yield (entry 9). Finally, different electrodes were tested but none of the results surpassed entry 1 (entries 10-13). SUBSTRATE SCOPE With the optimized reaction
conditions in hand, we probed the generality of this electroreductive deuteration reaction. Firstly, various alkyl bromides were screened (Fig. 2A). Alkyl bromides with different lengths of
carbon chains worked well to afford the corresponding products with excellent D-incorporation (2-7). Substituents with electron-withdrawing or donating properties were compatible and showed
little effect on the reaction (8-13). Besides, other aromatic rings, including naphthalene, indole, and dihydrobenzofuran (15-17), were all well tolerated. The success of this procedure
could be confirmed by the excellent compatibility of a wide range of functionalities, such as terminal ether (18), ester (19), and thioether (20), unprotected lactam (21-23), Boc-protected
amine (24), terminal olefin (25), tetrahydropyran (26), as well as internal ester (27). Interestingly, for substrates containing both Br and Cl, it was found that the C-Br bond was easier to
cleave (28). Besides, alkyl iodides also worked smoothly in this protocol, affording the corresponding products with excellent deuterium incorporation (Fig. 2B, 29-31). We were pleased to
find that OMs could be a suitable leaving group to give the target product in 64% yield and 99% D-incorporation (32). Next, we turned our attention to a more challenging target, unactivated
alkyl chlorides (Fig. 2C). To our delight, primary alkyl chlorides took part in the reaction with a broad range of functionalities and showed similar results to alkyl bromides, yielding the
corresponding products in good efficiency and D-incorporation (33-45). Furthermore, other alkyl chlorides, especially tertiary substrates that did not work well in previous studies24,
yielded the desired products unquestionably in satisfactory D-incorporation using this method (46-49). To further demonstrate the generality, and environmentally-friendly nature of the
developed method, as well as its application in bioactive molecules and drug discovery, the late-stage deuteration was conducted using a series of natural products, pharmaceuticals, and
their derivatives (Fig. 2D). The dehalogenative deuteration of pharmaceuticals including Estrone (50), Pregnenolone (52), DL-Menthol (53), Phytol (57), and Triclosan (58) was successfully
achieved in 77–83% yield with 97-98% D-incorporation. Pharmaceutical intermediates ethynyl estradiol (51) and dihydro cuminyl alcohol (54) containing unsaturated bonds can also be used in
this protocol. Ibuprofen (55) and naproxen (56), which are commonly used for the ease of pain, afforded the corresponding deuterated compounds in good yields (67%, 88%) with excellent
deuterium incorporation (both 98% D). Furthermore, amino acid and glucose derivatives that widely exist in organisms can also be deuterated smoothly with high levels of D-incorporation
(59−64, 97−99%). In addition, the tolerance of the reaction system to tertiary chlorides can be extended to complex substrates (65, 66). ELECTROCHEMICAL DEHALOGENATIVE DEUTERATION OF ALKYL
BROMIDES WITH 500 MA CURRENT As high current density has a key impact on the amplification of reaction scales, we further explored the practicability of some representative substrates at a
current of 500 mA. To our delight, the reactions were complete during a much shorter reaction time (in only 15 minutes). Simple alkyl bromides with a variety of functional groups (2, 16, 23,
and 26) as well as complex alkyl bromides derived from ibuprofen (55), L-phenylalanine (59), Diacetonefructose (61), and D-mannofuranose (63) were all amenable. level of D-incorporation has
not changed at all (Fig. 3). Alkyl chloride was also well tolerated and the corresponding product 37 was obtained in 15 min. In addition, this electrochemical reduction protocol could be
applied to the gram-scale preparation of deuterated product 8 under 500 mA constant current (Fig. 4a). MECHANISTIC STUDIES Several mechanistic studies were conducted to gain insight into the
reaction mechanism. Firstly, 2 equiv. of TEMPO was added to the reaction system and in the absence of D2O, the TEMPO adduct was obtained in 64% yield (67), together with hydrodehalogenated
product 2-H in 28% yield (Fig. 4b, eq 1). Then, repeat the reaction with 2 equiv. of D2O resulted in the TEMPO adduct in 21% yield and deuterodehalogenated product 2 in 75% yield with 55%
D-incorporation (Fig. 4b, eq 2). These results indicated that the alkyl radical might be involved in the electrochemical system. Then, we sought to gain further insight into the reaction
mechanism through cyclic voltammetry (CV) experiments. A reduction peak of alkyl bromides 1 at −3.40 V (vs Ag/Ag+ in CH3CN) was observed (Fig. 5a, the red line), and no obvious reduction
peak of D2O was observed (Fig. 5a). At the same time, the oxidation potential of TBAI (Fig. 5b, pink line, 1.0 V) was very close to that of DIPEA (Fig. 5b, blue line, 1.1 V). Thus we
inferred that both TBAI and DIPEA might act as the electron donors in this reaction. Notably, the anodic oxidation of alkyl bromide 1 was also indicated with higher potential than that of
DIPEA (green line). This might explain the improved yield once external DIPEA was added by inhibiting the undesired oxidation or decomposition of the starting material (Table 1, entries 1
and 3). In addition, during the reaction system without TBAI, an increase in current can be clearly observed, indicating the electrolysis of DIPEA incorporated with formed bromo anion may
further generate conductive substances (Fig. 5c). Based on the mechanistic studies and previous literature5,48, a plausible mechanism is proposed (Fig. 6). The reaction was initiated by the
anodic oxidation of DIPEA and/or TBAI or DMF and the direct reduction of alkyl halides on the cathode to form the corresponding alkyl radical I and halide anions. The radical I continued to
be reduced to an alkyl anion II at the cathode. Finally, intermediate II reacted with deuterated water to provide the expected product III. At the same time, iminium ion intermediate IV
generated from DIPEA on the anode might combine with the released halogen anions to form ammonium salt, a possible conductive species. DISCUSSION In conclusion, we have developed a general
dehalogenative deuteration of unactivated alkyl (pseudo)halides (X = Cl, Br, I, OMs) driven by electrochemical force with high reactivity, selectivity, and excellent D-incorporation. D2O was
used as an efficient and economical deuterium source. A wide range of unactivated alkyl halides containing diverse functionalities are well tolerated, as well as the extension to late stage
deuteration of natural products, pharmaceuticals, and their derivatives. We envisage that the native features, including tolerance of high current and environmentally-friendly conditions,
will bring further opportunity in its application to organic synthesis, drug discovery and modification, and even industrial production. Further electrochemical transformations of
unactivated alkyl halides are currently ongoing in our laboratory. METHODS GENERAL PROCEDURE OF THE ELECTROCHEMICAL DEHALOGENATIVE DEUTERATION OF ALKYL BROMIDE, IODIDE OR PSEUDO-HALIDES The
electrocatalysis was carried out in an undivided cell with a carbon felt anode (10 mm × 15 mm × 5 mm) and a lead cathode (10 mm × 15 mm × 0.3 mm). To a 15 mL pre‐dried undivided
electrochemical cell (15 mL) equipped with a magnetic bar were added alkyl bromide, iodide or pseudo-halides (0.5 mmol, 1 equiv), TBAI (36.9 mg, 0.1 mmol, 20 mol%), and DMF (5.0 mL). Then
D2O (25 mmol, 50 equiv) and DIPEA (1.5 mmol, 3.0 equiv) were added via a syringe. The electrocatalysis was performed at room temperature with a constant current of 30 mA maintained for 10 h.
The carbon felt anode was washed with EtOAc (3 × 5 mL) in an ultrasonic bath. H2O (20 mL) was added to the system, and the resulting mixture was extracted with EtOAc (3 ×20 mL). The
combined organic phase was dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography to furnish the desired product. GENERAL
PROCEDURE OF THE ELECTROCHEMICAL DEHALOGENATIVE DEUTERATION OF ALKYL CHLORIDES The electrocatalysis was carried out in an undivided cell with a graphite felt anode (10 mm × 15 mm × 5 mm) and
a lead cathode (10 mm × 15 mm × 0.3 mm). To a 15 mL pre‐dried undivided electrochemical cell (15 mL) equipped with a magnetic bar were added alkyl chloride (0.5 mmol, 1 equiv), TBAI (36.9
mg, 0.1 mmol, 20 mol%) and DMF (5.0 mL). Then D2O (25 mmol, 50 equiv) and DIPEA (1.5 mmol, 3.0 equiv) were added via a syringe. The electrocatalysis was performed at room temperature with a
constant current of 50 mA maintained for 10 h. The carbon felt anode was washed with EtOAc (3 × 5 mL) in an ultrasonic bath. H2O (20 mL) was added to the system, and the resulting mixture
was extracted with EtOAc (3 × 20 mL). The combined organic phase was dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography
to furnish the desired product. GENERAL PROCEDURE OF THE ELECTROCHEMICAL DEHALOGENATIVE DEUTERATION OF ALKYL BROMIDES AND CHLORIDE WITH 500 MA CURRENT The electrocatalysis was carried out
in an undivided cell with a carbon felt anode (or graphite felt) (10 mm × 15 mm × 5 mm) and a lead cathode (10 mm × 15 mm × 0.3 mm). To a 15 mL pre‐dried undivided electrochemical cell (15
mL) equipped with a magnetic bar were added alkyl bromide or chloride (0.5 mmol, 1 equiv), TBAI (36.9 mg, 0.1 mmol, 20 mol%) and DMF (5.0 mL). Then D2O (25 mmol, 50 equiv) and DIPEA (1.5
mmol, 3.0 equiv) were added via a syringe. The electrocatalysis was performed at room temperature with a constant current of 500 mA maintained for 15 min (reaction system exothermic). The
carbon felt anode was washed with EtOAc (3 × 5 mL) in an ultrasonic bath. H2O (20 mL) was added to the system, and the resulting mixture was extracted with EtOAc (3 × 20 mL). The combined
organic phase was dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography to furnish the desired product. GRAM-SCALE
SYNTHESIS OF 8 To an undivided reaction flask (diameter: 40 mm, length: 130 mm, volume: 200 mL) equipped with a teflon-coated magnetic stirring bar and teflon cap, a carbon felt anode (25 mm
× 50 mm × 5 mm), and a lead cathode (25 mm × 50 mm × 0.3 mm) were added (4-bromobutoxy)benzene (3.44 g, 15 mmol), TBAI (1.11 g, 3.0 mmol), DMF (100 mL), D2O (15 mL, 0.75 mol) and DIPEA
(5.80 g, 45 mmol). The electrocatalysis was performed at room temperature with a constant current of 500 mA maintained for 4 h (the reaction system was exothermic). The carbon felt anode was
washed with EtOAc (3 × 20 mL) in an ultrasonic bath. H2O (200 mL) was added to the system, and the resulting mixture was extracted with EtOAc (3 × 200 mL). The combined organic phase was
dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography to furnish the desired product. DATA AVAILABILITY The authors
declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. Extra data are available from the author upon request.
REFERENCES * Wiberga, K. B. The Deuterium Isotope Effect. _Chem. Rev._ 55, 713–743 (1995). Article Google Scholar * Gómez-Gallego, M. & Sierra, M. A. Kinetic Isotope Effects in the
Study of Organometallic Reaction Mechanisms. _Chem. Rev._ 111, 4857–4963 (2011). Article PubMed CAS Google Scholar * Kushner, D. J., Baker, A., & Dunstall, T. G. C. Pharmacological
Uses and Perspectives of Heavy Water and Deuterated Compounds. _J. Physiol. Pharmacol_. 77, 79−88 (1999). * Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of
Deuterium in Medicinal Chemistry. _J. Med. Chem._ 62, 5276–5297 (2019). Article CAS PubMed Google Scholar * Liu, X., Liu, R., Qiu, J., Cheng, X. & Li, G. Chemical-Reductant-Free
Electrochemical Deuteration Reaction using Deuterium Oxide. _Angew. Chem. Int. Ed._ 59, 13962–13967 (2020). Article CAS Google Scholar * Li, W. et al. Scalable and Selective Deuteration
of (Hetero)arenes. _Nat. Chem._ 14, 334–341 (2022). Article CAS PubMed PubMed Central Google Scholar * Norcott, P. L. Current electrochemical approaches to selective deuteration. _Chem.
Commun._ 58, 2944–2953 (2022). Article CAS Google Scholar * Liao, L.-L. et al. Electrochemical Ring-Opening Dicarboxylation of Strained Carbon–Carbon Single Bonds with CO2: Facile
Synthesis of Diacids and Derivatization into Polyesters. _J. Am. Chem. Soc._ 144, 2062–2068 (2022). Article CAS PubMed Google Scholar * Kopf, S. et al. Recent Developments for the
Deuterium and Tritium Labeling of Organic Molecules. _Chem. Rev._ 122, 6634–6718 (2022). Article CAS PubMed Google Scholar * Cockrell, J. R. & Murray, R. W. Deuterium labeling by
electrochemical reactions. _J. Electrochem. Soc._ 119, 849–851 (1972). Article ADS CAS Google Scholar * Renaud, R. N. Electrochemical Synthesis of Deuterio Organic Compounds. I.
Electrochemical Reduction of 1-Halonaphthalenes and Synthesis of 1-Chloro-4-methylnaphthalene. _Can. J. Chem._ 52, 376–380 (1974). Article CAS Google Scholar * Grimshaw, J. &
Trocha-Grimshaw, J. Electrochemical reactions. Part XVIII. Reductive cleavage of aromatic carbon–halogen bonds in the presence of deuterium oxide. _J. Chem. Soc. Perkin Trans._ 2, 215–218
(1975). Article Google Scholar * Tashiro, M., Iwasaki, A. & Fukata, G. Studies on Selective Preparation of Aromatic Compounds. 14. An Attempt to Prepare All Possible Deuterated Phenols
by the Reductive Dehalogenation of the Corresponding Halophenols with Raney Alloys in an Alkaline Deuterium Oxide Solution. _J. Org. Chem._ 43, 196–199 (1978). Article CAS Google Scholar
* Janni, M. & Peruncheralathan, S. Catalytic Selective Deuteration of Halo(hetero)Arenes. _Org. Biomol. Chem._ 14, 3091–3097 (2016). Article CAS PubMed Google Scholar * Kuriyama,
M. et al. Deuterodechlorination of Aryl/Heteroaryl Chlorides Catalyzed by a Palladium/Unsymmetrical NHC System. _J. Org. Chem._ 81, 8934–8946 (2016). Article CAS PubMed Google Scholar *
Li, W. et al. Copper-catalysed Low-temperature Water–gas Shift Reaction for Selective Deuteration of Aryl Halides. _Chem. Sci._ 12, 14033–14038 (2012). Article Google Scholar * Wang, X. et
al. General and Practical Potassium Methoxide/Disilane-Mediated Dehalogenative Deuteration of (Hetero)Arylhalides. _J. Am. Chem. Soc._ 140, 10970–10974 (2018). Article CAS PubMed Google
Scholar * Liu, C. et al. Controllable Deuteration of Halogenated Compounds by Photocatalytic D2O splitting. _Nat. Commun._ 9, 80 (2018). Article ADS PubMed PubMed Central CAS Google
Scholar * Zhou, Z.-Z., Zhao, J.-H., Gou, X.-Y., Chen, X.-M. & Liang, Y.-M. Visible-Light-Mediated Hydrodehalogenation and Br/D Exchange of Inactivated Aryl and Alkyl Halides with a
Palladium Complex. _Org. Chem. Front._ 6, 1649–1654 (2019). Article CAS Google Scholar * Ling, X. et al. A Visible-light-photocatalytic Water-splitting Strategy for Sustainable
Hydrogenation/deuteration of Aryl Chlorides. _Sci. China Chem._ 63, 386–392 (2020). Article CAS Google Scholar * Soulard, V., Villa, G., Vollmar, D. P. & Renaud, P. Radical
Deuteration with D2O: Catalysis and Mechanistic Insights. _J. Am. Chem. Soc._ 140, 155–158 (2018). Article CAS PubMed Google Scholar * Song, Z. et al. Water Compatible Hypophosphites-d2
Reagents: Deuteration Reaction via Deutero-deiodination in Aqueous Solution. _Org. Lett._ 22, 1736–1741 (2020). Article CAS PubMed Google Scholar * Xia, A., Xie, X., Hu, X., Xu, W. &
Liu, Y. Dehalogenative Deuteration of Unactivated Alkyl Halides Using D2O as the Deuterium Source. _J. Org. Chem._ 84, 13841–13857 (2019). Article CAS PubMed Google Scholar * Li, Y. et
al. Organophotocatalytic Selective Deuterodehalogenation of Aryl or Alkyl Chlorides. _Nat. Commun._ 12, 2894 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Yoshida, J.,
Kataoka, K., Horcajada, R. & Nagaki, A. Modern Strategies in Electroorganic Synthesis. _Chem. Rev._ 108, 2265–2299 (2008). Article CAS PubMed Google Scholar * Francke, R. &
Little, R. D. Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. _Chem. Soc. Rev._ 43, 2492–2521 (2014). Article CAS PubMed Google Scholar * Yan, M.,
Kawamata, Y. & Baran, P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. _Chem. Rev._ 117, 13230–13319 (2017). Article CAS PubMed PubMed
Central Google Scholar * Sauermann, N., Meyer, T. H., Qiu, Y. & Ackermann, L. Electrocatalytic C−H Activation. _ACS Catal._ 8, 7086–7103 (2018). Article CAS Google Scholar * Yuan,
Y. & Lei, A. Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions. _Acc. Chem. Res._ 52, 3309–3324 (2019). Article CAS PubMed Google Scholar * Röckl, J. L.,
Pollok, D., Franke, R. & Waldvogel, S. R. A Decade of Electrochemical Dehydrogenative C, C-Coupling of Aryls. _Acc. Chem. Res._ 53, 45–61 (2020). Article PubMed CAS Google Scholar *
Jiao, K.-J., Xing, Y.-K., Yang, Q.-L., Qiu, H. & Mei, T.-S. Site-Selective C–H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis. _Acc. Chem. Res._
53, 300–310 (2020). Article CAS PubMed Google Scholar * Qiu, Y. et al. Rhodaelectro-Catalyzed C–H and C–C Activation. _CCS Chem._ 2, 1529–1552 (2020). Google Scholar * Novaes, L. F. T.
et al. Electrocatalysis as an Enabling Technology for Organic Synthesis. _Chem. Soc. Rev._ 50, 7941–8002 (2021). Article CAS PubMed PubMed Central Google Scholar * Cheng, X., et al.
Recent Applications of Homogeneous Catalysis in Electrochemical Organic Synthesis. _CCS Chem_. https://doi.org/10.31635/ccschem.021.202101451 (2022). * Bäckvall, J.-E., Gogoll, A.
Palladium-Hydroquinone Catalysed Electrochemical 1,4-Oxidation of Conjugated Dienes. _J. Chem. Soc. Chem. Commun_. 1236–1238 (1987). * Horn, E. J. et al. Scalable and Sustainable
Electrochemical Allylic C–H oxidation. _Nature_ 533, 77–81 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Qiu, Y., Tian, C., Massignan, L., Rogge, T. & Ackermann,
L. Electrooxidative Ruthenium-Catalyzed C−H/O−H Annulation by Weak O-Coordination. _Angew. Chem. Int. Ed._ 57, 5818–5822 (2018). Article CAS Google Scholar * Tang, S., Liu, Y. & Lei,
A. Electrochemical Oxidative Cross-coupling with Hydrogen Evolution: A Green and Sustainable Way for Bond Formation. _Chem_ 4, 27–45 (2018). Article CAS Google Scholar * Shrestha, A.,
Lee, M., Dunn, A. L. & Sanford, M. S. Palladium-Catalyzed C−H Bond Acetoxylation via Electrochemical Oxidation. _Org. Lett._ 20, 204–207 (2018). Article CAS PubMed Google Scholar *
Xiang, J. et al. Hindered Dialkyl Ether Synthesis with Electrogenerated Carbocations. _Nature_ 573, 398–402 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Shen, T.
& Lambert, T. H. Electrophotocatalytic Diamination of Vicinal C–H Bonds. _Science_ 371, 620–626 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Liang, Y. et al.
Electrochemically Induced Nickel Catalysis for Oxygenation Reactions with Water. _Nat. Catal._ 4, 116–123 (2021). Article CAS Google Scholar * Ma, C. et al. Recent advances in organic
electrosynthesis employing transition metal complexes as electrocatalysts. _Sci. Bull._ 66, 2412–2429 (2021). Article CAS Google Scholar * Zhang, B., et al. Ni-Electrocatalytic
C(sp3)–C(sp3) Doubly Decarboxylative Coupling. _Nature_, https://doi.org/10.1038/s41586-022-04691-4. (2022). * Kurono, N., Honda, E., Komatsu, F., Orito, K. & Tokuda, M. Regioselective
Synthesis of Substituted 1-Indanols, 2,3-Dihydrobenzo-furans and 2,3-Dihydroindoles by Electrochemical Radical Cyclization Using an Arene Mediator. _Tetrahedron_ 60, 1791–1801 (2004).
Article CAS Google Scholar * Mitsudo, K. et al. Electro-Reductive Cyclization of Aryl Halides Promoted by Fluorene Derivatives. _Electrochim. Acta_ 82, 444–449 (2012). Article CAS
Google Scholar * Zhang, W. & Lin, S. electroreductive Carbofunctionalization of Alkenes with Alkyl Bromides via a Radical-Polar Crossover Mechanism. _J. Am. Chem. Soc._ 142, 20661–20670
(2020). Article CAS PubMed PubMed Central Google Scholar * Dong, X., Roeckl, J. L., Waldvogel, S. R. & Morandi, B. Merging Shuttle Reactions and Paired Electrolysis for Reversible
Vicinal Dihalogenations. _Science_ 371, 507–514 (2021). Article ADS CAS PubMed Google Scholar * Kuroboshi, M., Shiba, T. & Tanaka, H. Viologen as Catalytic Organic Reductant:
Electro-Reductive Dimerization of Aryl Bromides in a Pd/Viologen Double Mediatory System. _Tetrahedron Lett._ 54, 3666–3668 (2013). Article CAS Google Scholar * Jiao, K. et al.
Nickel-Catalyzed Electrochemical Reductive Reley Cross-Coupling of Alkyl Halides to Aryl Halides. _Angew. Chem. Int. Ed._ 59, 6520–6524 (2020). Article CAS Google Scholar * Qiu, H. et al.
Enantioselective Ni-Catalyzed Electrochemical Synthesis of Biaryl Atropisomers. _J. Am. Chem. Soc._ 142, 9872–9878 (2020). Article CAS PubMed Google Scholar * Li, Z.-J. et al.
Electrochemically Enabled, Nickel-Catalyzed Dehydroxylative Cross-Coupling of Alcohols with Aryl Halides. _J. Am. Chem. Soc._ 143, 3536–3543 (2021). Article CAS PubMed Google Scholar *
Kumar, G. S. et al. Nickel-Catalyzed Chain-Walking Cross-Electrophile Coupling of Alkyl and Aryl Halides and Olefin Hydroarylation Enabled by Electrochemical Reduction. _Angew. Chem. Int.
Ed._ 59, 6513–6519 (2020). Article CAS Google Scholar * Truesdell, B. L., Hamby, T. B. & Sevov, C. S. General C(sp2)−C(sp3) Cross-Electrophile Coupling Reactions Enabled by Overcharge
Protection of Homogeneous Electrocatalysts. _J. Am. Chem. Soc._ 142, 5884–5893 (2020). Article CAS PubMed Google Scholar * Zhang, W., et al. Electrochemically Driven Cross-Electrophile
Coupling of Alkyl Halides. See, ChemRxiv preprint https://doi.org/10.26434/chemrxiv-2021-c2hd6-v2 (2021). * Zhang, W. et al. Electrochemically driven cross-electrophile coupling of alkyl
halides. _Nature_ 604, 292–297 (2022). Article ADS CAS PubMed Google Scholar * Kim, H., Kim, H., Lambert, T. H. & Lin, S. Reductive Electrophotocatalysis: Merging Electricity and
Light to Achieve Extreme Reduction Potentials. _J. Am. Chem. Soc._ 142, 2087–2092 (2020). Article CAS PubMed PubMed Central Google Scholar * Sun, L., Sahloul, K. & Mellah, M. Use of
Electrochemistry to Provide Efficient SmI2 Catalytic System for Coupling Reactions. _ACS Catal._ 3, 2568–2573 (2013). Article CAS Google Scholar * Gao, Y. et al. Electrochemical
Nozaki−Hiyama−Kishi Coupling: Scope, Applications, and Mechanism. _J. Am. Chem. Soc._ 143, 9478–9488 (2021). Article CAS PubMed PubMed Central Google Scholar * Liu, D., Ma, H.-X., Fang,
P. & Mei, T.-S. Nickel-Catalyzed Thiolation of Aryl Halides and Heteroaryl Halides through Electrochemistry. _Angew. Chem. Int. Ed._ 58, 5033–5037 (2019). Article CAS Google Scholar
* Kawamata, Y. et al. Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications. _J. Am. Chem. Soc._ 141, 6392–6402 (2019). Article CAS PubMed PubMed
Central Google Scholar * Zhang, H.-J. et al. Chemoselective, Scalable Nickel-Electrocatalytic O-Arylation of Alcohols. _J. Am. Chem. Soc._ 143, 9478–9488 (2021). Article CAS Google
Scholar * Liu, D. et al. Nickel-Catalyzed N-Arylation of NH-Sulfoximines with Aryl Halides via Paired Electrolysis. _Angew. Chem. Int. Ed._ 60, 9444–9449 (2021). Article CAS Google
Scholar * Senboku, H., Kanaya, H., Fujimura, Y. & Tokuda, M. Stereo-chemical Study on Electrochemical Carboxylation of Vinyl Triflates. J. _Electroanal. Chem._ 507, 82–88 (2001).
Article CAS Google Scholar * Ang, N. W. J., Oliveira, J. C. A. & Ackermann, L. Electroreductive Cobalt-Catalyzed Carboxylation: Cross-Electrophile Electrocoupling with Atmospheric
CO2. _Angew. Chem. Int. Ed._ 59, 12842–12847 (2020). Article CAS Google Scholar * Sun, G.-Q. et al. Nickel-catalyzed electrochemical carboxylation of unactivated aryl and alkyl halides
with CO2. _Nat. Commun._ 12, 7086 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Parrish, J. D. & Little, R. D. Electrochemical Formation of Glycals in THF.
_Tetrahedron Lett._ 42, 7371–7374 (2001). Article CAS Google Scholar * Huang, J.-M., Wang, X.-X. & Dong, Y. Electrochemical Allylation Reactions of Simple Imines in Aqueous Solution
Mediated by Nanoscale Zinc Architectures. _Angew. Chem., Int. Ed._ 50, 924–927 (2001). Article CAS Google Scholar * Mitsudo, K., Okada, T., Shimohara, S., Mandai, H. & Suga, S.
Electro-reductive Halogen-Deuterium Exchange and Methylation of Aryl Halides in Acetonitrile. _Electrochem_ 81, 362–364 (2013). Article CAS Google Scholar * Liu, C., Han, S., Li, M.,
Chong, X. & Zhang, B. Electrocatalytic Deuteration of Halides with D2O as the Deuterium Source over a Copper Nanowire Arrays Cathode. _Angew. Chem. Int. Ed._ 59, 18527–18531 (2020).
Article CAS Google Scholar * Lu, L., Li, H., Zheng, Y., Bu, F. & Lei, A. Facile and Economical Electrochemical Dehalogenative Deuteration of (Hetero)Aryl Halides. _CCS Chem._ 3,
2669–2675 (2020). Article CAS Google Scholar * Lambert, F. L. & Ingall, G. B. Voltammetry of organic halogen compounds. IV. _Reduct. Org. chlorides vitreous (Glass.) carbon electrode
Tetrahedron Lett._ 15, 3231–3234 (1974). Google Scholar * Cybularczyk-Cecotka, M., Szczepanik, J. & Giedyk, M. Photocatalytic strategies for the activation of organic chlorides. _Nat.
Catal._ 3, 872–886 (2020). Article CAS Google Scholar * Wang, B. et al. Electrochemical Borylation of Alkyl Halides: Fast, Scalable Access to Alkyl Boronic Esters. _J. Am. Chem. Soc._
143, 12985–12991 (2021). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS Financial support from the Fundamental Research Funds for the Central Universities (No.
63213063), Frontiers Science Center for New Organic Matter, Nankai University (Grant No. 63181206) and Nankai University are gratefully acknowledged. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * State Key Laboratory and Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, 94 Weijin Road,
Tianjin, 300071, China Pengfei Li, Chengcheng Guo, Siyi Wang, Dengke Ma, Tian Feng, Yanwei Wang & Youai Qiu Authors * Pengfei Li View author publications You can also search for this
author inPubMed Google Scholar * Chengcheng Guo View author publications You can also search for this author inPubMed Google Scholar * Siyi Wang View author publications You can also search
for this author inPubMed Google Scholar * Dengke Ma View author publications You can also search for this author inPubMed Google Scholar * Tian Feng View author publications You can also
search for this author inPubMed Google Scholar * Yanwei Wang View author publications You can also search for this author inPubMed Google Scholar * Youai Qiu View author publications You can
also search for this author inPubMed Google Scholar CONTRIBUTIONS Y.Q. and P.L. conceived and designed the study and wrote the manuscript. P.L, C.C.G, S.W, D.M, T.F, Y.W, Y.Q. performed the
experiments, mechanistic studies and revised the manuscript. All authors contributed to the analysis and interpretation of the data. CORRESPONDING AUTHOR Correspondence to Youai Qiu. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing interest. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewers for their contribution
to the peer review of this work. Peer reviewer reports are available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Li, P., Guo, C., Wang, S. _et al._ Facile and general
electrochemical deuteration of unactivated alkyl halides. _Nat Commun_ 13, 3774 (2022). https://doi.org/10.1038/s41467-022-31435-9 Download citation * Received: 29 January 2022 * Accepted:
16 June 2022 * Published: 30 June 2022 * DOI: https://doi.org/10.1038/s41467-022-31435-9 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative






